
DenSec: Secreted Protein Prediction in Cerebrospinal Fluid Based on DenseNet and Transformer


Abstract
:1. Introduction
2. Protein Data
2.1. Data Collection
2.2. Encoding Protein
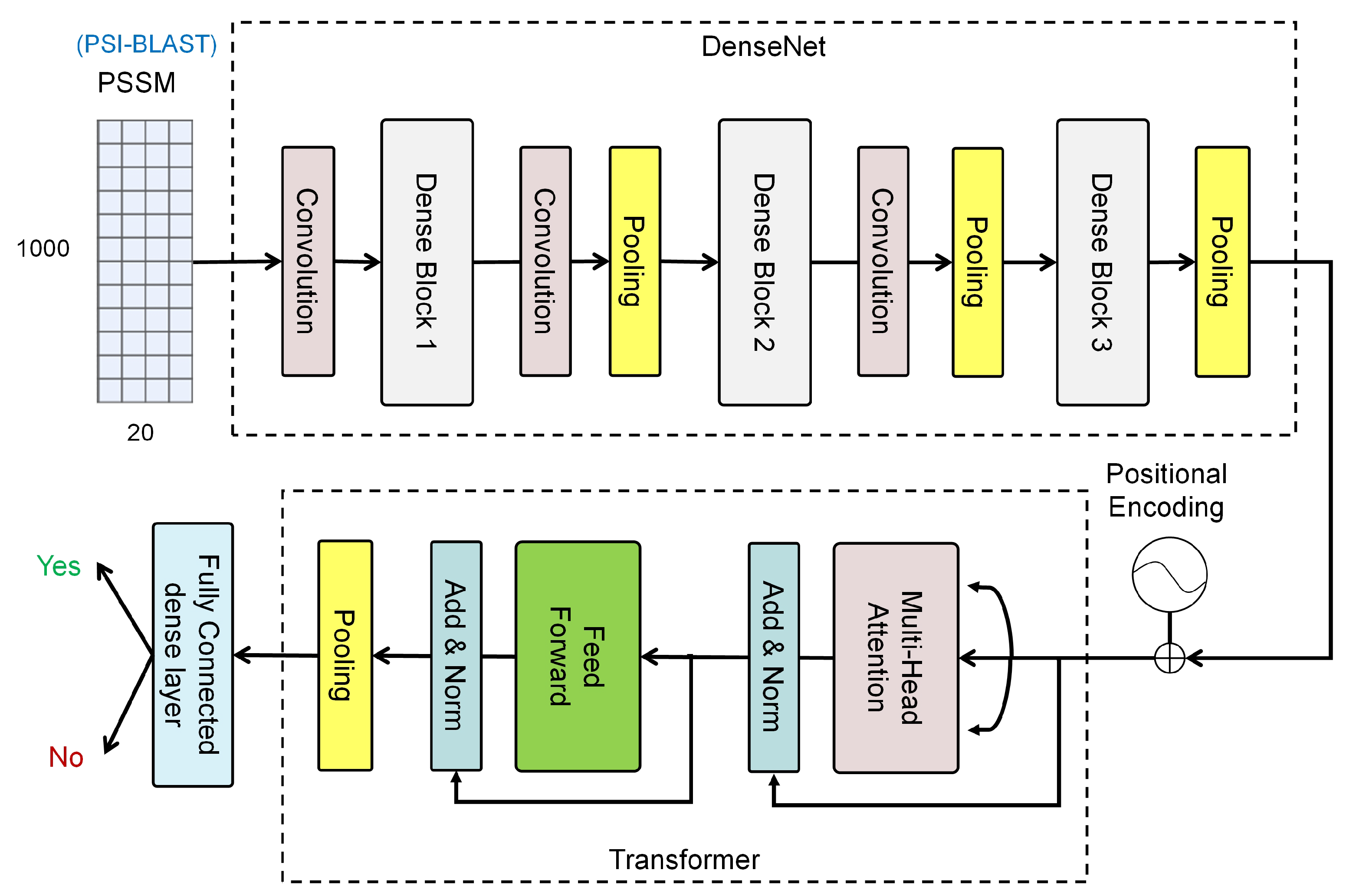
3. The Proposed Method
- A new deep learning model is proposed to predict CSF proteins based on DenseNet and Transformer architecture;
- We employ the DenseNet for feature extraction instead of traditional CNNs, which allows the model to achieve better performance with fewer parameters and computational costs;
- We propose Transformer to capture possible long-range dependencies between protein sequence and secreted status of proteins, which contributes to the improved performance.
3.1. Feature Learning Using DenseNet
3.2. Classification Using Transformer
4. Results
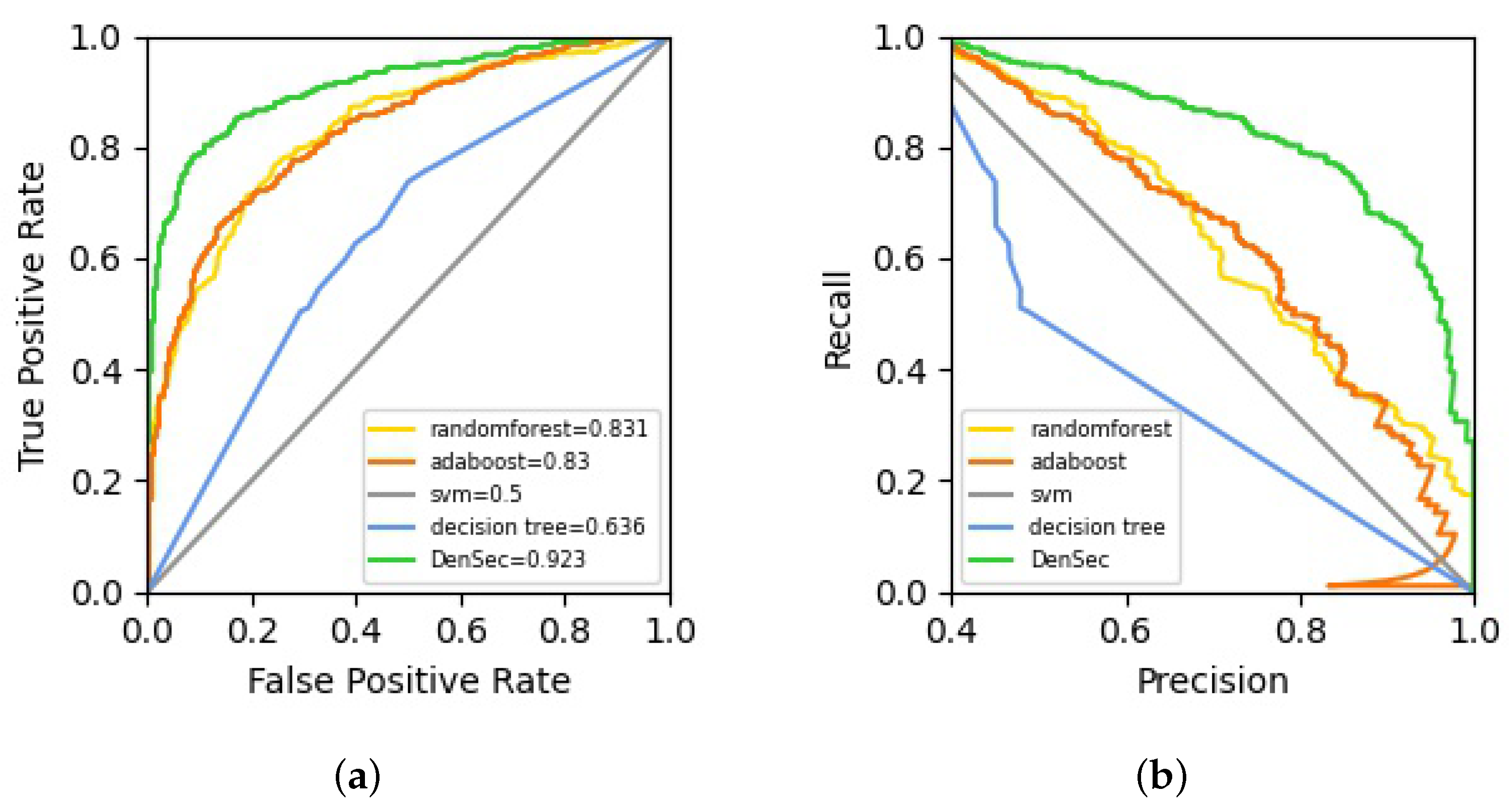
4.1. Result Analysis Method
4.2. Evaluating the Performance of Classification
4.3. Ablation Study on Classification
- Transformer
- DenseNet
- DenseNet with Transformer (DenSec)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Margolis, J.; Kenrick, K.G. Two-dimensional resolution of plasma proteins by combination of polyacrylamide disc and gradient gel electrophoresis. Nature 1969, 221, 1056–1057. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Lin, R.C. UPLC—MSE application in disease biomarker discovery: The discoveries in proteomics to metabolomics. Chem.-Biol. Interact. 2014, 215, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.J. Rays of positive electricity and their application to chemical analyses. Nature 1914, 92, 549–550. [Google Scholar]
- Huang, L.; Shao, D.; Wang, Y.; Cui, X.; Li, Y.; Chen, Q.; Cui, J. Human body-fluid proteome: Quantitative profiling and computational prediction. Brief. Bioinf. 2021, 22, 315–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khasawneh, A.H.; Garling, R.J.; Harris, C.A. Cerebrospinal fluid circulation: What do we know and how do we know it? Brain Circ. 2018, 4, 14–18. [Google Scholar]
- Shao, D.; Dai, Y.F.; Li, N.F.; Cao, X.Q.; Zhao, W.; Cheng, L.; Rong, Z.Q.; Huang, L.; Wang, Y.; Zhao, J. Artificial Intelligence in Clinical Research of Cancers. Brief. Bioinf. 2022, 23, 1–12. [Google Scholar] [CrossRef]
- Shao, D.; Huang, L.; Wang, Y.; Cui, X.T.; Li, Y.F.; Wang, Y.; Ma, Q.; Du, W.; Cui, J. HBFP: A new repository for Human Body-Fluid Proteome. Database 2021, 2021, baab065. [Google Scholar] [CrossRef]
- Cui, J.; Liu, Q.; Puett, D.; Xu, Y. Computational prediction of human proteins that can be secreted into the bloodstream. Bioinformatics 2008, 24, 2370–2375. [Google Scholar] [CrossRef]
- Wang, J.; Liang, Y.; Wang, Y.; Cui, J.; Liu, M.; Du, W.; Xu, Y. Computational prediction of human salivary proteins from blood circulation and application to diagnostic biomarker identification. PLoS ONE 2013, 8, e80211. [Google Scholar] [CrossRef]
- Sun, Y.; Du, W.; Zhou, C.; Zhou, Y.; Cao, Z.; Tian, Y.; Wang, Y. A computational method for prediction of saliva-secretory proteins and its application to identification of head and neck cancer biomarkers for salivary diagnosis. IEEE Trans. Nanobiosci. 2015, 14, 167–174. [Google Scholar] [CrossRef]
- Shao, D.; Huang, L.; Wang, Y.; He, K.; Cui, X.; Wang, Y.; Cui, J. DeepSec: A deep learning framework for secreted protein discovery in human body fluids. Bioinformatics 2022, 38, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Pang, R.; Li, G.; Cao, H.; Li, Y.; Liang, Y. DeepUEP: Prediction of urine excretory proteins using deep learning. IEEE Access 2020, 8, 100251–100261. [Google Scholar] [CrossRef]
- Du, W.; Zhao, X.; Sun, Y.; Zheng, L.; Li, Y.; Zhang, Y. SecProCT: In silico prediction of human secretory proteins based on capsule network and transformer. Int. J. Mol. Sci. 2021, 22, 9054. [Google Scholar] [CrossRef] [PubMed]
- Shome, D.; Kar, T.; Mohanty, S.N.; Tiwari, P.; Muhammad, K.; AlTameem, A.; Saudagar, A.K.J. Covid-transformer: Interpretable covid-19 detection using vision transformer for healthcare. Int. J. Environ. Res. Public Health 2021, 18, 11086. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Liu, Z.; Van Der Maaten, L.; Weinberger, K.Q. Densely connected convolutional networks. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017. [Google Scholar]
- Bahdanau, D.; Cho, K.; Bengio, Y. Neural machine translation by jointly learning to align and translate. arXiv 2014, arXiv:1409.0473. [Google Scholar]
- Zhang, Z.; Miao, C.; Liu, C.; Tian, Q.; Zhou, Y. HA-RoadFormer: Hybrid attention transformer with multi-branch for large-scale high-resolution dense road segmentation. Mathematics 2022, 10, 1915. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Finn, R.D. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, 427–432. [Google Scholar] [CrossRef]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; De La Paz, M.L.; Martins, I.C.; Reumers, J.; Rousseau, F. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Peng, J.; Ma, J.; Xu, J. Protein secondary structure prediction using deep convolutional neural fields. Sci. Rep. 2016, 6, 18962. [Google Scholar] [CrossRef] [Green Version]
- Ba, J.L.; Kiros, J.R.; Hinton, G.E. Layer normalization. arXiv 2016, arXiv:1607.06450. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention is all you need. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Casadio, R. DeepSig: Deep learning improves signal peptide detection in proteins. Bioinformatics 2018, 34, 1690–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quang, D.; Xie, X. DanQ: A hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016, 44, e107. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Sequence Length Range | Training Dataset | Testing Dataset | ||
|---|---|---|---|---|
| # of Proteins | Proportion | # of Proteins | Proportion | |
| <500 | 4763 | 61.06% | 889 | 70.56% |
| 500–1000 | 2095 | 26.86% | 263 | 20.87% |
| >1000 | 942 | 12.08% | 108 | 8.57% |
| Methods | Accuracy | Sensitivity | Specificity | MCC | AUC |
|---|---|---|---|---|---|
| SVM | 0.563 | 0.547 | 0.656 | 0.405 | 0.500 |
| [0.525, 0.596] | [0.505, 0.597] | [0.594, 0.703] | [0.362, 0.466] | [0.483, 0.527] | |
| AdaBoost | 0.738 | 0.605 | 0.848 | 0.471 | 0.830 |
| [0.694, 0.784] | [0.528, 0.701] | [0.723, 0.897] | [0.412, 0.526] | [0.774, 0.882] | |
| Decision Tree | 0.601 | 0.458 | 0.745 | 0.412 | 0.636 |
| [0.569, 0.645] | [0.393, 0.581] | [0.590, 0.796] | [0.386, 0.504] | [0.584, 0.688] | |
| RandomForest | 0.742 | 0.606 | 0.861 | 0.489 | 0.831 |
| [0.692, 0.778] | [0.543, 0.736] | [0.815, 0.883] | [0.408, 0.541] | [0.765, 0.874] | |
| DenSec | 0.860 | 0.859 | 0.870 | 0.726 | 0.923 |
| [0.844, 0.873] | [0.830, 0.885] | [0.857, 0.889] | [0.688, 0.765] | [0.858, 0.937] |
| Methods | Accuracy | Sensitivity | Specificity | MCC | AUC |
|---|---|---|---|---|---|
| DeepSig | 0.742 | 0.684 | 0.784 | 0.469 | 0.805 |
| [0.701, 0.779] | [0.627, 0.730] | [0.716, 0.862] | [0.435, 0.497] | [0.774, 0.828] | |
| DanQ | 0.759 | 0.711 | 0.792 | 0.502 | 0.829 |
| [0.712, 0.803] | [0.651, 0.758] | [0.727, 0.841] | [0.429, 0.548] | [0.797, 0.843] | |
| DeepSec | 0.823 | 0.800 | 0.846 | 0.571 | 0.858 |
| [0.808, 0.842] | [0.753, 0.833] | [0.821, 0.868] | [0.537, 0.614] | [0.826, 0.889] | |
| DenSec | 0.860 | 0.859 | 0.870 | 0.726 | 0.923 |
| [0.844, 0.873] | [0.830, 0.885] | [0.857, 0.889] | [0.688, 0.765] | [0.858, 0.937] |
| Methods | Accuracy | Sensitivity | Specificity | MCC | AUC |
|---|---|---|---|---|---|
| Transformer | 0.596 | 0.381 | 0.832 | 0.440 | 0.579 |
| [0.576, 0.615] | [0.331, 0.422] | [0.794, 0.851] | [0.425, 0.483] | [0.548, 0.604] | |
| DenseNet | 0.781 | 0.753 | 0.802 | 0.552 | 0.767 |
| [0.752, 0.803] | [0.729, 0.788] | [0.786, 0.849] | [0.534, 0.577] | [0.731, 0.804] | |
| DenSec | 0.860 | 0.859 | 0.870 | 0.726 | 0.923 |
| [0.844, 0.873] | [0.830, 0.885] | [0.857, 0.889] | [0.688, 0.765] | [0.858, 0.937] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, L.; Qu, Y.; He, K.; Wang, Y.; Shao, D. DenSec: Secreted Protein Prediction in Cerebrospinal Fluid Based on DenseNet and Transformer. Mathematics 2022, 10, 2490. https://doi.org/10.3390/math10142490
Huang L, Qu Y, He K, Wang Y, Shao D. DenSec: Secreted Protein Prediction in Cerebrospinal Fluid Based on DenseNet and Transformer. Mathematics. 2022; 10(14):2490. https://doi.org/10.3390/math10142490
Chicago/Turabian StyleHuang, Lan, Yanli Qu, Kai He, Yan Wang, and Dan Shao. 2022. "DenSec: Secreted Protein Prediction in Cerebrospinal Fluid Based on DenseNet and Transformer" Mathematics 10, no. 14: 2490. https://doi.org/10.3390/math10142490
APA StyleHuang, L., Qu, Y., He, K., Wang, Y., & Shao, D. (2022). DenSec: Secreted Protein Prediction in Cerebrospinal Fluid Based on DenseNet and Transformer. Mathematics, 10(14), 2490. https://doi.org/10.3390/math10142490