
Explainable Machine Learning for Longitudinal Multi-Omic Microbiome


Abstract
:1. Introduction
1.1. Dynamic Longitudinal Data
- Data size: Current datasets lack large-scale data, suffering from economic and logistic constraints that limit and affect data collection standards. Further advantages could be taken once we define how to decode large-scale microbiome data in a precise and efficient manner [22].
- Comparability and reproducibility: The lack of validated clinical models and differences in methodologies is preventing the translation of valuable results into real-world clinical practice.
- Inherent characteristics of microbiome data: Sparsity, compositionality, and high variability are the main statistical properties that describe microbiome data hence leading to several computational challenges. High-throughput RNA-seq technologies used in the process of generating microbiome data from the sample often introduce technical artifacts that translate into errors and noise. Thus, the bottleneck has shifted from data generation to data analysis. Moreover, microbiome data is compositional, so instead of looking at the absolute abundances of cells, we are mapping reads, and there is a fixed sequencing depth, i.e., four reads/sample, given by the technology used to obtain the sequences.
- Interpretability: Incorporating phylogenetic and functional relationships among organisms into unified dynamic models of the human microbiome is crucial. Studies need to integrate multi-omic datasets to fully understand microbes and their interactions instead of exploring unique taxonomic composition analysis.
1.2. State of the Research Field
1.3. Interpretability
1.4. Main Aim and Contributions of the Work
2. Materials and Methods
- Clinical features (metadata): subject identification (e.g., “Subject ID”), time steps for sample time series (e.g., “week”), phenotype/cluster of each sample (“diagnosis”), external perturbations (“antibiotic”)
- Metabolomic features: metabolic concentrations, mass-to-charge ratio (m/z) (continuous)
- Metagenomic features: taxonomic profile (continuous) corresponding to the relative abundance in percentage or counts per million
- Metatranscriptomic features: relative abundance of each metabolic pathway (continuous). The information is divided into two different datasets: HMP2 and HMP2 pilot. Data matrix (tables) will be preprocessed and expressed as abundances
Disease-State Prediction Model
Dynamic Bayesian Network
- Prior equivalent sample size ν = 10.
- Prior assumed standard deviation: σ = 1
- Maximum number of parents =3.
3. Results
3.1. DBN Model
3.2. Pre-Processing
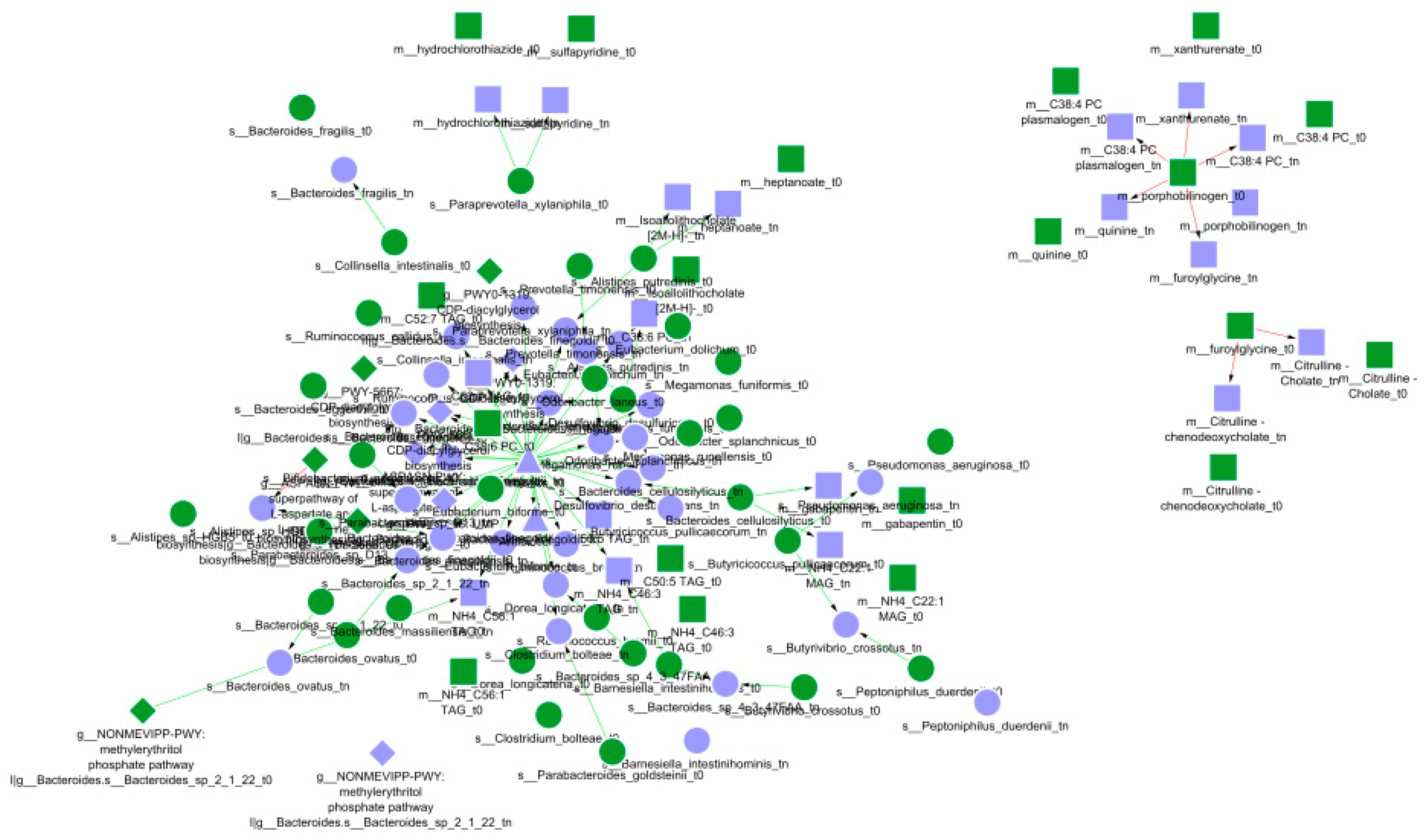
3.3. The Resulting Network
3.4. Analysis and Interpretation of Experimental Results
4. Discussion
- The pre-processing of the data set.
- The fitting of the DBN model in two steps: structure and parameter learning. The output of this step was a 2-stage dynamic Bayes net class object (DBN).
- The inference and test of the DBN on a subset of variables given the evidence on the other variables. The output of this step was the predicted values and log probabilities of observing a less likely outcome for each variable that the value assigned to that variable by the input data.
- Dynamic Bayesian network visualisation and analysis for the biological interpolation of results.
5. Conclusions
Limitations and Future Research
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Moran, M.A. The Global Ocean Microbiome. Science 2015, 350, aac8455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, U.G.; Sachs, J.L. Engineering Microbiomes to Improve Plant and Animal Health. Trends Microbiol. 2015, 23, 606–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling Function and Taxonomy in the Global Ocean Microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Kolodkin-Gal, I. Harvesting the Complex Pathways of Antibiotic Production and Resistance of Soil Bacilli for Optimizing Plant Microbiome. FEMS Microbiol. Ecol. 2020, 96, fiaa142. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Ehrlich, S.D. MetaHIT: The European Union Project on Metagenomics of the Human Intestinal Tract. In Metagenomics of the Human Body; Nelson, K.E., Ed.; Springer: New York, NY, USA, 2011; pp. 307–316. ISBN 978-1-4419-7089-3. [Google Scholar]
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.-M.; et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef] [Green Version]
- Cornejo-Pareja, I.; Ruiz-Limón, P.; Gómez-Pérez, A.M.; Molina-Vega, M.; Moreno-Indias, I.; Tinahones, F.J. Differential Microbial Pattern Description in Subjects with Autoimmune-Based Thyroid Diseases: A Pilot Study. J. Pers. Med. 2020, 10, 192. [Google Scholar] [CrossRef]
- Depner, M.; Taft, D.H.; Kirjavainen, P.V.; Kalanetra, K.M.; Karvonen, A.M.; Peschel, S.; Schmausser-Hechfellner, E.; Roduit, C.; Frei, R.; Lauener, R.; et al. Maturation of the Gut Microbiome during the First Year of Life Contributes to the Protective Farm Effect on Childhood Asthma. Nat. Med. 2020, 26, 1766–1775. [Google Scholar] [CrossRef]
- Joseph, C.L.M.; Zoratti, E.M.; Ownby, D.R.; Havstad, S.; Nicholas, C.; Nageotte, C.; Misiak, R.; Enberg, R.; Ezell, J.; Johnson, C.C. Exploring Racial Differences in IgE-Mediated Food Allergy in the WHEALS Birth Cohort. Ann. Allergy Asthma Immunol. 2016, 116, 219–224.e1. [Google Scholar] [CrossRef] [Green Version]
- Metwally, A.A.; Yu, P.S.; Reiman, D.; Dai, Y.; Finn, P.W.; Perkins, D.L. Utilizing Longitudinal Microbiome Taxonomic Profiles to Predict Food Allergy via Long Short-Term Memory Networks. PLoS Comput. Biol. 2019, 15, e1006693. [Google Scholar] [CrossRef]
- Leiva-Gea, I.; Sánchez-Alcoholado, L.; Martín-Tejedor, B.; Castellano-Castillo, D.; Moreno-Indias, I.; Urda-Cardona, A.; Tinahones, F.J.; Fernández-García, J.C.; Queipo-Ortuño, M.I. Gut Microbiota Differs in Composition and Functionality Between Children with Type 1 Diabetes and MODY2 and Healthy Control Subjects: A Case-Control Study. Diabetes Care 2018, 41, 2385–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A Metagenome-Wide Association Study of Gut Microbiota in Type 2 Diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of Fecal Microbiota for Early-Stage Detection of Colorectal Cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridenhour, B.J.; Brooker, S.L.; Williams, J.E.; Van Leuven, J.T.; Miller, A.W.; Dearing, M.D.; Remien, C.H. Modeling Time-Series Data from Microbial Communities. ISME J. 2017, 11, 2526–2537. [Google Scholar] [CrossRef] [Green Version]
- Bucci, V.; Tzen, B.; Li, N.; Simmons, M.; Tanoue, T.; Bogart, E.; Deng, L.; Yeliseyev, V.; Delaney, M.L.; Liu, Q.; et al. MDSINE: Microbial Dynamical Systems Inference Engine for Microbiome Time-Series Analyses. Genome Biol. 2016, 17, 121. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Lahti, L.; Gonze, D.; de Vos, W.M.; Raes, J. Metagenomics Meets Time Series Analysis: Unraveling Microbial Community Dynamics. Curr. Opin. Microbiol. 2015, 25, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Heshiki, Y.; Vazquez-Uribe, R.; Li, J.; Ni, Y.; Quainoo, S.; Imamovic, L.; Li, J.; Sørensen, M.; Chow, B.K.C.; Weiss, G.J.; et al. Predictable Modulation of Cancer Treatment Outcomes by the Gut Microbiota. Microbiome 2020, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Cammarota, G.; Ianiro, G.; Ahern, A.; Carbone, C.; Temko, A.; Claesson, M.J.; Gasbarrini, A.; Tortora, G. Gut Microbiome, Big Data and Machine Learning to Promote Precision Medicine for Cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 635–648. [Google Scholar] [CrossRef]
- Bodein, A.; Chapleur, O.; Droit, A.; Lê Cao, K.-A. A Generic Multivariate Framework for the Integration of Microbiome Longitudinal Studies with Other Data Types. Front. Genet. 2019, 10, 963. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Jing, G.; Zhang, Y.; Wu, S. Method Development for Cross-Study Microbiome Data Mining: Challenges and Opportunities. Comput. Struct. Biotechnol. J. 2020, 18, 2075–2080. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Costello, E.K.; Knight, R. Supervised Classification of Human Microbiota. FEMS Microbiol. Rev. 2011, 35, 343–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, P.E.; Dai, Y. Metabolome of Human Gut Microbiome Is Predictive of Host Dysbiosis. Gigascience 2015, 4, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moitinho-Silva, L.; Steinert, G.; Nielsen, S.; Hardoim, C.C.P.; Wu, Y.-C.; McCormack, G.P.; López-Legentil, S.; Marchant, R.; Webster, N.; Thomas, T.; et al. Predicting the HMA-LMA Status in Marine Sponges by Machine Learning. Front. Microbiol. 2017, 8, 752. [Google Scholar] [CrossRef] [Green Version]
- Fukui, H.; Nishida, A.; Matsuda, S.; Kira, F.; Watanabe, S.; Kuriyama, M.; Kawakami, K.; Aikawa, Y.; Oda, N.; Arai, K.; et al. Usefulness of Machine Learning-Based Gut Microbiome Analysis for Identifying Patients with Irritable Bowels Syndrome. J. Clin. Med. 2020, 9, 2403. [Google Scholar] [CrossRef]
- Hacilar, H.; Nalbantoglu, O.U.; Aran, O.; Bakir-Gungor, B. Inflammatory Bowel Disease Biomarkers of Human Gut Microbiota Selected via Ensemble Feature Selection Methods. arXiv 2020, arXiv:2001.03019. [Google Scholar]
- McGeachie, M.J.; Sordillo, J.E.; Gibson, T.; Weinstock, G.M.; Liu, Y.-Y.; Gold, D.R.; Weiss, S.T.; Litonjua, A. Longitudinal Prediction of the Infant Gut Microbiome with Dynamic Bayesian Networks. Sci. Rep. 2016, 6, 20359. [Google Scholar] [CrossRef] [Green Version]
- Noyes, N.; Cho, K.-C.; Ravel, J.; Forney, L.J.; Abdo, Z. Associations between Sexual Habits, Menstrual Hygiene Practices, Demographics and the Vaginal Microbiome as Revealed by Bayesian Network Analysis. PLoS ONE 2018, 13, e0191625. [Google Scholar] [CrossRef] [Green Version]
- Lugo-Martinez, J.; Ruiz-Perez, D.; Narasimhan, G.; Bar-Joseph, Z. Dynamic Interaction Network Inference from Longitudinal Microbiome Data. Microbiome 2019, 7, 54. [Google Scholar] [CrossRef] [Green Version]
- Howey, R.; Shin, S.-Y.; Relton, C.; Smith, G.D.; Cordell, H.J. Bayesian Network Analysis Incorporating Genetic Anchors Complements Conventional Mendelian Randomization Approaches for Exploratory Analysis of Causal Relationships in Complex Data. PLoS Genet. 2020, 16, e1008198. [Google Scholar] [CrossRef] [Green Version]
- Jang, B.-S.; Chang, J.H.; Chie, E.K.; Kim, K.; Park, J.W.; Kim, M.J.; Song, E.-J.; Nam, Y.-D.; Kang, S.W.; Jeong, S.-Y.; et al. Gut Microbiome Composition Is Associated with a Pathologic Response After Preoperative Chemoradiation in Patients with Rectal Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2020, 107, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Kharrat, N.; Assidi, M.; Abu-Elmagd, M.; Pushparaj, P.N.; Alkhaldy, A.; Arfaoui, L.; Naseer, M.I.; El Omri, A.; Messaoudi, S.; Buhmeida, A.; et al. Data Mining Analysis of Human Gut Microbiota Links Fusobacterium spp. with Colorectal Cancer Onset. Bioinformation 2019, 15, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Sazal, M.; Mathee, K.; Ruiz-Perez, D.; Cickovski, T.; Narasimhan, G. Inferring Directional Relationships in Microbial Communities Using Signed Bayesian Networks. BMC Genom. 2020, 21, 663. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Perez, D.; Lugo-Martinez, J.; Bourguignon, N.; Mathee, K.; Lerner, B.; Bar-Joseph, Z.; Narasimhan, G. Dynamic Bayesian Networks for Integrating Multi-Omics Time Series Microbiome Data. Msystems 2021, 6, e01105-20. [Google Scholar] [CrossRef]
- La Rosa, P.S.; Warner, B.B.; Zhou, Y.; Weinstock, G.M.; Sodergren, E.; Hall-Moore, C.M.; Stevens, H.J.; Bennett, W.E.; Shaikh, N.; Linneman, L.A.; et al. Patterned Progression of Bacterial Populations in the Premature Infant Gut. Proc. Natl. Acad. Sci. USA 2014, 111, 12522–12527. [Google Scholar] [CrossRef] [Green Version]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.K.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal Microbiome of Reproductive-Age Women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef] [Green Version]
- Moayyeri, A.; Hammond, C.J.; Hart, D.J.; Spector, T.D. The UK Adult Twin Registry (TwinsUK Resource). Twin Res. Hum. Genet. 2013, 16, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Marchesi, J.R.; Dutilh, B.E.; Hall, N.; Peters, W.H.M.; Roelofs, R.; Boleij, A.; Tjalsma, H. Towards the Human Colorectal Cancer Microbiome. PLoS ONE 2011, 6, e20447. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-Omics of the Gut Microbial Ecosystem in Inflammatory Bowel Diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Castelvecchi, D. Can We Open the Black Box of AI? Nat. News 2016, 538, 20. [Google Scholar] [CrossRef] [Green Version]
- Holzinger, A.; Biemann, C.; Pattichis, C.S.; Kell, D.B. What Do We Need to Build Explainable AI Systems for the Medical Domain? arXiv 2017, arXiv:1712.09923. [Google Scholar]
- Barredo Arrieta, A.; Díaz-Rodríguez, N.; Del Ser, J.; Bennetot, A.; Tabik, S.; Barbado, A.; Garcia, S.; Gil-Lopez, S.; Molina, D.; Benjamins, R.; et al. Explainable Artificial Intelligence (XAI): Concepts, Taxonomies, Opportunities and Challenges toward Responsible AI. Inf. Fusion 2020, 58, 82–115. [Google Scholar] [CrossRef] [Green Version]
- Prifti, E.; Chevaleyre, Y.; Hanczar, B.; Belda, E.; Danchin, A.; Clément, K.; Zucker, J.-D. Interpretable and Accurate Prediction Models for Metagenomics Data. GigaScience 2020, 9, giaa010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrieri, A.P.; Haiminen, N.; Maudsley-Barton, S.; Gardiner, L.-J.; Murphy, B.; Mayes, A.E.; Paterson, S.; Grimshaw, S.; Winn, M.; Shand, C.; et al. Explainable AI Reveals Changes in Skin Microbiome Composition Linked to Phenotypic Differences. Sci. Rep. 2021, 11, 4565. [Google Scholar] [CrossRef]
- Wong, C.W.; Yost, S.E.; Lee, J.S.; Gillece, J.D.; Folkerts, M.; Reining, L.; Highlander, S.K.; Eftekhari, Z.; Mortimer, J.; Yuan, Y. Analysis of Gut Microbiome Using Explainable Machine Learning Predicts Risk of Diarrhea Associated with Tyrosine Kinase Inhibitor Neratinib: A Pilot Study. Front. Oncol. 2021, 11, 283. [Google Scholar] [CrossRef]
- Pan, A.Y. Statistical Analysis of Microbiome Data: The Challenge of Sparsity. Curr. Opin. Endocr. Metab. Res. 2021, 19, 35–40. [Google Scholar] [CrossRef]
- Wright, E.K.; Kamm, M.A.; Teo, S.M.; Inouye, M.; Wagner, J.; Kirkwood, C.D. Recent Advances in Characterizing the Gastrointestinal Microbiome in Crohn’s Disease: A Systematic Review. Inflamm. Bowel Dis. 2015, 21, 1219–1228. [Google Scholar] [CrossRef] [Green Version]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Robust Methods for Differential Abundance Analysis in Marker Gene Surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [Green Version]
- Badri, M.; Kurtz, Z.D.; Müller, C.L.; Bonneau, R. Normalization Methods for Microbial Abundance Data Strongly Affect Correlation Estimates. BioRxiv 2018, 406264. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [Green Version]
- Mars, R.A.T.; Yang, Y.; Ward, T.; Houtti, M.; Priya, S.; Lekatz, H.R.; Tang, X.; Sun, Z.; Kalari, K.R.; Korem, T.; et al. Longitudinal Multi-Omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome. Cell 2020, 182, 1460–1473.e17. [Google Scholar] [CrossRef] [PubMed]
- Aitchison, J. The Statistical Analysis of Compositional Data. J. R. Stat. Soc. Ser. B 1982, 44, 139–177. [Google Scholar] [CrossRef]
- Saeys, Y.; Inza, I.; Larrañaga, P. A Review of Feature Selection Techniques in Bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-W.; Kuo, C.-H.; Kuo, F.-C.; Wang, Y.-K.; Hsu, W.-H.; Yu, F.-J.; Hu, H.-M.; Hsu, P.-I.; Wang, J.-Y.; Wu, D.-C. Fecal Microbiota Transplantation: Review and Update. J. Formos Med. Assoc. 2019, 118 (Suppl. S1), S23–S31. [Google Scholar] [CrossRef]
- Mihaljević, B.; Bielza, C.; Larrañaga, P. Bayesian Networks for Interpretable Machine Learning and Optimization. Neurocomputing 2021, 456, 648–665. [Google Scholar] [CrossRef]
- Needham, C.J.; Bradford, J.R.; Bulpitt, A.J.; Westhead, D.R. A Primer on Learning in Bayesian Networks for Computational Biology. PLoS Comput. Biol. 2007, 3, e129. [Google Scholar] [CrossRef]
- Pearl, J. Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference; Morgan Kaufmann: Burlington, MA, USA, 1988. [Google Scholar]
- Chickering, D.M. Learning Bayesian Networks Is NP-Complete. In Learning from Data: Artificial Intelligence and Statistics V.; Fisher, D., Lenz, H.-J., Eds.; Lecture Notes in Statistics; Springer: New York, NY, USA, 1996; pp. 121–130. ISBN 978-1-4612-2404-4. [Google Scholar]
- Verma, T.; Pearl, J. Equivalence and Synthesis of Causal Models. In Proceedings of the Sixth Annual Conference on Uncertainty in Artificial Intelligence, Virtual Event, 27–29 July 1990; Elsevier Science Inc.: New York, NY, USA, 1990; pp. 255–270. [Google Scholar]
- Spirtes, P.; Glymour, C.; Scheines, R. Causation, Prediction, and Search, 2nd ed.; Adaptive Computation and Machine Learning Series; A Bradford Book: Cambridge, MA, USA, 2001; ISBN 978-0-262-19440-2. [Google Scholar]
- Borchani, H.; Bielza, C.; Martı´nez-Martı´n, P.; Larrañaga, P. Markov Blanket-Based Approach for Learning Multi-Dimensional Bayesian Network Classifiers: An Application to Predict the European Quality of Life-5 Dimensions (EQ-5D) from the 39-Item Parkinson’s Disease Questionnaire (PDQ-39). J. Biomed. Inform. 2012, 45, 1175–1184. [Google Scholar] [CrossRef]
- Margaritis, D. Learning Bayesian Network Model Structure from Data; Carnegie-Mellon Univ Pittsburgh Pa School of Computer Science: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Tsamardinos, I.; Aliferis, C.F.; Statnikov, A. Algorithms for Large Scale Markov Blanket Discovery. FLAIRS Conf. 2003, 2, 376–380. [Google Scholar]
- Henrion, M. An Introduction to Algorithms for Inference in Belief Nets. In Machine Intelligence and Pattern Recognition; Henrion, M., Shachter, R.D., Kanal, L.N., Lemmer, J.F., Eds.; Uncertainty in Artificial Intelligence; Elsevier: Amsterdam, The Netherlands, 1990; Volume 10, pp. 129–138. [Google Scholar]
- Shachter, R.D.; Peot, M.A. Simulation Approaches to General Probabilistic Inference on Belief Networks. In Machine Intelligence and Pattern Recognition; Henrion, M., Shachter, R.D., Kanal, L.N., Lemmer, J.F., Eds.; Uncertainty in Artificial Intelligence; Elsevier: Amsterdam, The Netherlands, 1990; Volume 10, pp. 221–231. [Google Scholar]
- Golightly, A.; Wilkinson, D.J. Bayesian Parameter Inference for Stochastic Biochemical Network Models Using Particle Markov Chain Monte Carlo. Interface Focus 2011, 1, 807–820. [Google Scholar] [CrossRef] [Green Version]
- Dagum, P.; Luby, M. Approximating Probabilistic Inference in Bayesian Belief Networks Is NP-Hard. Artif. Intell. 1993, 60, 141–153. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, D. Gaussian Mixture Models. In Encyclopedia of Biometrics; Li, S.Z., Jain, A., Eds.; Springer US: Boston, MA, USA, 2009; pp. 659–663. ISBN 978-0-387-73003-5. [Google Scholar]
- Madsen, A.L. Belief Update in CLG Bayesian Networks with Lazy Propagation. Int. J. Approx. Reason. 2008, 49, 503–521. [Google Scholar] [CrossRef] [Green Version]
- Dean, T.; Kanazawa, K. A Model for Reasoning about Persistence and Causation. Comput. Intell. 1989, 5, 142–150. [Google Scholar] [CrossRef]
- Quesada, D. DbnR: Dynamic Bayesian Network Learning and Inference. Available online: https://github.com/dkesada/dbnR (accessed on 10 January 2022).
- Scutari, M. Learning Bayesian Networks with the Bnlearn R Package. J. Stat. Softw. 2010, 35, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Wilczyński, B.; Dojer, N. BNFinder: Exact and Efficient Method for Learning Bayesian Networks. Bioinformatics 2009, 25, 286–287. [Google Scholar] [CrossRef]
- McGeachie, M.J.; Chang, H.-H.; Weiss, S.T. CGBayesNets: Conditional Gaussian Bayesian Network Learning and Inference with Mixed Discrete and Continuous Data. PLoS Comput. Biol. 2014, 10, e1003676. [Google Scholar] [CrossRef] [Green Version]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Favera, R.D.; Califano, A. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinform. 2006, 7, S7. [Google Scholar] [CrossRef] [Green Version]
- Tsamardinos, I.; Brown, L.E.; Aliferis, C.F. The Max-Min Hill-Climbing Bayesian Network Structure Learning Algorithm. Mach. Learn. 2006, 65, 31–78. [Google Scholar] [CrossRef] [Green Version]
- Gasse, M.; Aussem, A.; Elghazel, H. An Experimental Comparison of Hybrid Algorithms for Bayesian Network Structure Learning. In Machine Learning and Knowledge Discovery in Databases; Flach, P.A., De Bie, T., Cristianini, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 58–73. [Google Scholar]
- Heckerman, D.; Geiger, D.; Chickering, D.M. Learning Bayesian Networks: The Combination of Knowledge and Statistical Data. Mach. Learn. 1995, 20, 197–243. [Google Scholar] [CrossRef] [Green Version]
- Rissanen, J. Modeling by Shortest Data Description. Automatica 1978, 14, 465–471. [Google Scholar] [CrossRef]
- Grünwald, P.D. The Minimum Description Length Principle; Adaptive Computation and Machine Learning Series; MIT Press: Cambridge, MA, USA, 2007; ISBN 978-0-262-07281-6. [Google Scholar]
- Cooper, G.F.; Herskovits, E. A Bayesian Method for the Induction of Probabilistic Networks from Data. Mach. Learn. 1992, 9, 309–347. [Google Scholar] [CrossRef]
- Chang, H.-H.; McGeachie, M. Phenotype Prediction by Integrative Network Analysis of SNP and Gene Expression Microarrays. In Proceedings of the Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Boston, MA, USA, 30 August—3 September 2011; pp. 6849–6852. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by Simulated Annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Cowell, R.G. Local Propagation in Conditional Gaussian Bayesian Networks. J. Mach. Learn. Res. 2005, 6, 1517–1550. [Google Scholar]
- Koller, D.; Friedman, N. Probabilistic Graphical Models: Principles and Techniques; Adaptive Computation and Machine Learning Series; MIT Press: Cambridge, MA, USA, 2009; ISBN 978-0-262-01319-2. [Google Scholar]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The Genus Alistipes: Gut Bacteria With Emerging Implications to Inflammation, Cancer, and Mental Health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhang, X.; Hu, Z. Application of Artificial Intelligence Modeling Technology Based on Multi-Omics in Noninvasive Diagnosis of Inflammatory Bowel Disease. J. Inflamm. Res. 2021, 14, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, P.; Abad, M.; Ramoni, M.F. Bayesian Networks for Genomic Analysis. Genom. Signal Process. Stat. 2005, 2, 281–320. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Study | Method | Dataset | Longitudinal Data | Meta-Omics | Goal |
|---|---|---|---|---|---|---|
| 1 | [28] | DBNs | Premature infant gut [36] | ☒ | ☐ | Build a DBN model to identify important relationships between microbiome taxa and predict future changes in microbiome composition |
| 2 | [29] | BNs | Vaginal microbiome [37] | ☐ | ☐ | Demonstrate associations between women’s sexual and menstrual habits, demographics, vaginal microbiome composition, and symptoms and diagnostics of bacterial vaginosis (BV) |
| 3 | [30] | DBNs | Infant gut [36] | ☒ | ☐ | Obtaining inferences from time-series data |
| 4 | [31] | BNs | Twins UK [38] | ☐ | ☐ | Possible causal relationships between metabolites and body mass index (BMI) |
| 5 | [32] | BNs | Rectal cancer [32] | ☐ | ☒ | Reveal differential microbial communities and functions in terms of therapeutic responses |
| 6 | [33] | BNs with the incremental dynamic analysis (IDA) method | Colorectal cancer [14,39] | ☐ | ☐ | Identify key species that are likely to be causal agents of colorectal cancer (CRC) |
| 7 | [34] | BNs with co-occurrence networks (CoNs) | Infant gut [36], vaginal [37], oral data [5] | ☐ | ☐ | Make an inference about colonisation order |
| 8 | [35] | DBNs | IBDMD (inflammatory bowel disease multi-omics database) [40] | ☒ | ☒ | Infer temporal relationships between entities in a microbial community and extend (Lugo-Martinez et al., 2019) to other omics |
| # | Contributions |
|---|---|
| 1 | Statistical analysis of longitudinal, multi-omic human microbiome data |
| 2 | State-of-the-art review of interpretable artificial intelligence approaches (models and tools) for human microbiome data |
| 3 | Identification of temporal interactions and connections between the biological entities: microbial taxa, microbial metabolic pathways, metabolites |
| 4 | Address both taxonomic composition and functional profile |
| 5 | Network model for each specific disease state (UC, CD) |
| 6 | Novel proposed preprocessing framework for the IBD Human Microbiome Project data to serve as an analysis tool for non-ML experts |
| Data Type | File Name | File Description | File Dimension |
|---|---|---|---|
| Metadata | hmp2_metadata.csv | Full sample metadata table; samples as rows and metadata as columns | 178 × 490 |
| Metabolomics | iHMP_metabolomics.csv | Metabolomics profiles | 81,867 × 553 |
| Metagenomics | taxonomic_profiles.tsv | MetaPhlAn2 taxonomic profiles | 1479 × 1639 |
| Metatranscriptomics | pathabundance_relab.tsv | MTX pathway abundances with stratification | 6061 × 736 |
| Data Type | FSS Technique | Evaluation Metric (Accuracy) |
|---|---|---|
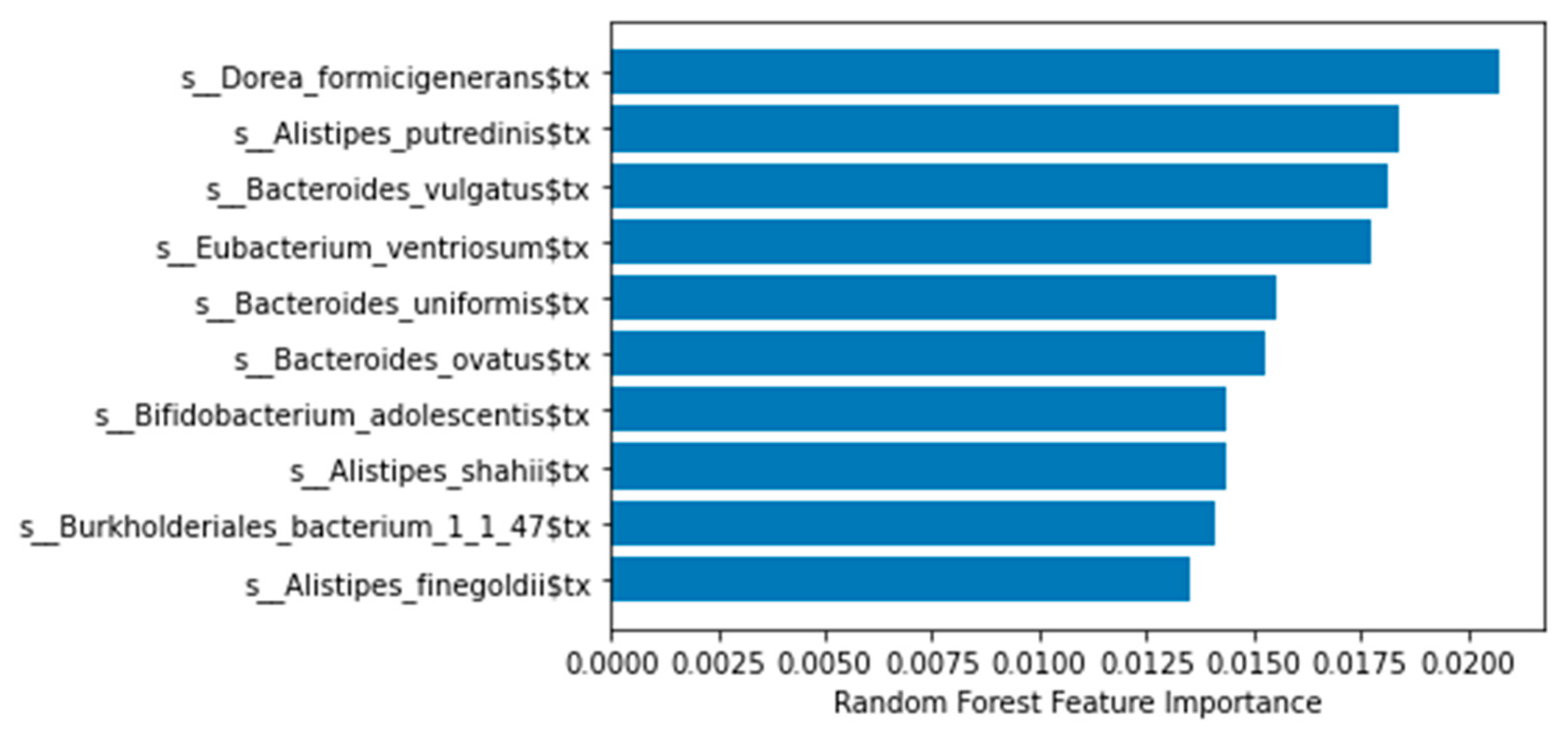
| Metagenomics | Univariate Chi2 | 0.82 |
| Univariate ANOVA | 0.78 | |
| Univariate MI | 0.73 | |
| Random forest | 0.92 | |
| Metabolomics | Univariate Chi2 | 0.67 |
| Univariate ANOVA | 0.69 | |
| Univariate MI | 0.69 | |
| Random forest | 0.78 | |
| Metatranscriptomics | Univariate Chi2 | 0.55 |
| Univariate ANOVA | 0.56 | |
| Univariate MI | 0.50 | |
| Random forest | 0.68 |
| Name | Language | Data Type | Learning | Inference |
|---|---|---|---|---|
| dbnR [72] | R | Continuous data | Yes | Yes |
| Bnlearn [73] | R | Discrete and continuous | Yes | No |
| Bnfinder [74] | Python | Discrete and continuous * | Yes | Yes |
| CGBayesNets [75] | Matlab | Discrete and continuous | Yes | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laccourreye, P.; Bielza, C.; Larrañaga, P. Explainable Machine Learning for Longitudinal Multi-Omic Microbiome. Mathematics 2022, 10, 1994. https://doi.org/10.3390/math10121994
Laccourreye P, Bielza C, Larrañaga P. Explainable Machine Learning for Longitudinal Multi-Omic Microbiome. Mathematics. 2022; 10(12):1994. https://doi.org/10.3390/math10121994
Chicago/Turabian StyleLaccourreye, Paula, Concha Bielza, and Pedro Larrañaga. 2022. "Explainable Machine Learning for Longitudinal Multi-Omic Microbiome" Mathematics 10, no. 12: 1994. https://doi.org/10.3390/math10121994
APA StyleLaccourreye, P., Bielza, C., & Larrañaga, P. (2022). Explainable Machine Learning for Longitudinal Multi-Omic Microbiome. Mathematics, 10(12), 1994. https://doi.org/10.3390/math10121994