
Combining Mass Spectrometry-Based Phosphoproteomics with a Network-Based Approach to Reveal FLT3-Dependent Mechanisms of Chemoresistance

 , , and
, , and
Abstract
1. Introduction
2. FLT3
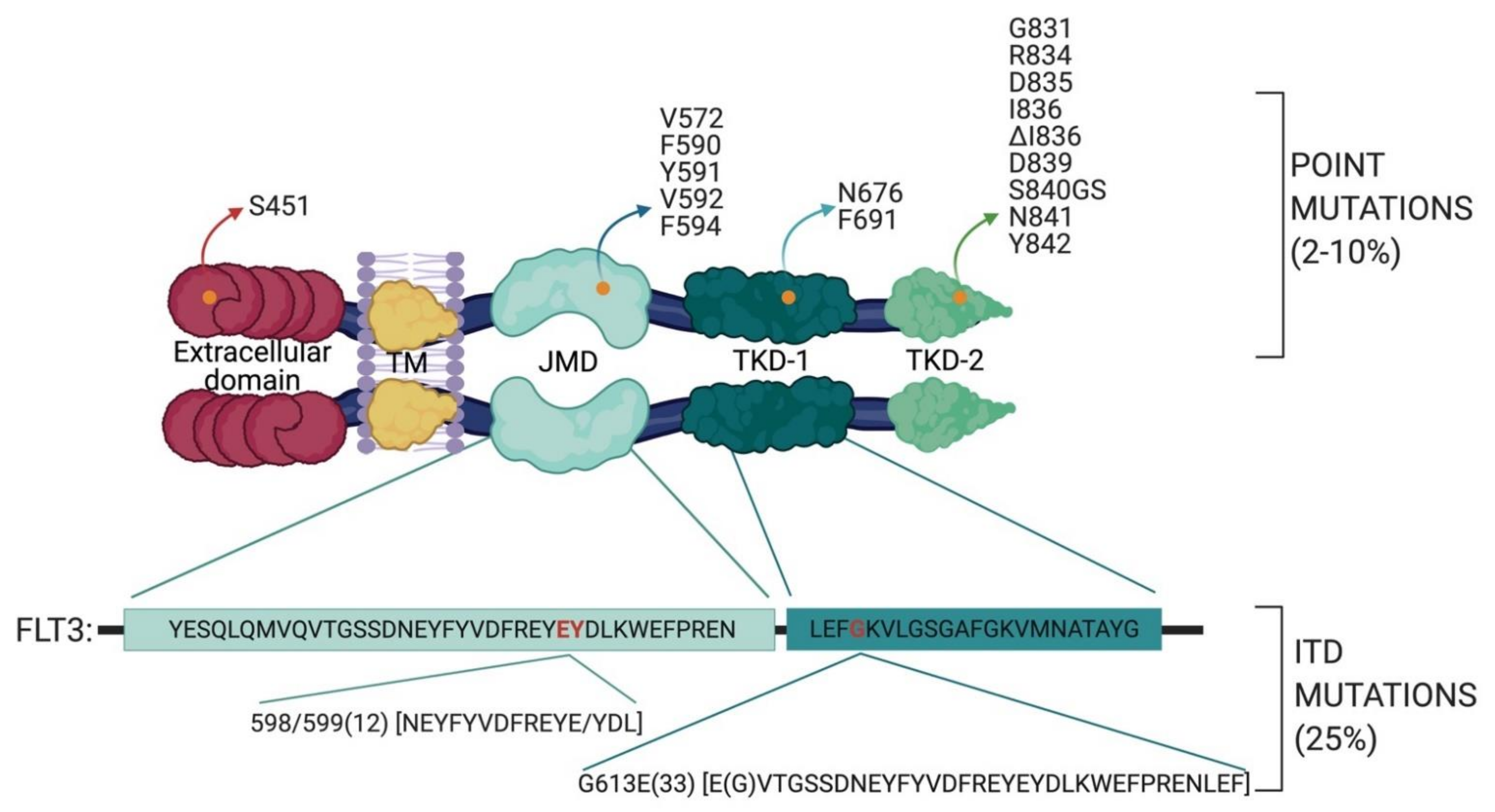
2.1. FLT3 Mutations in AML
- the TKD1 beta-sheet1 (amino acids 610 to 615) in 24.6% of FLT3-ITD positive AML patients
- in the nucleotide binding loop (NBL) (amino acids 616 to 623) in 2% of FLT3-ITD positive AML patients
- in the TKD2 beta-sheet2 (amino acids 624 to 630) in 1.3 % of FLT3-ITD positive AML patients.

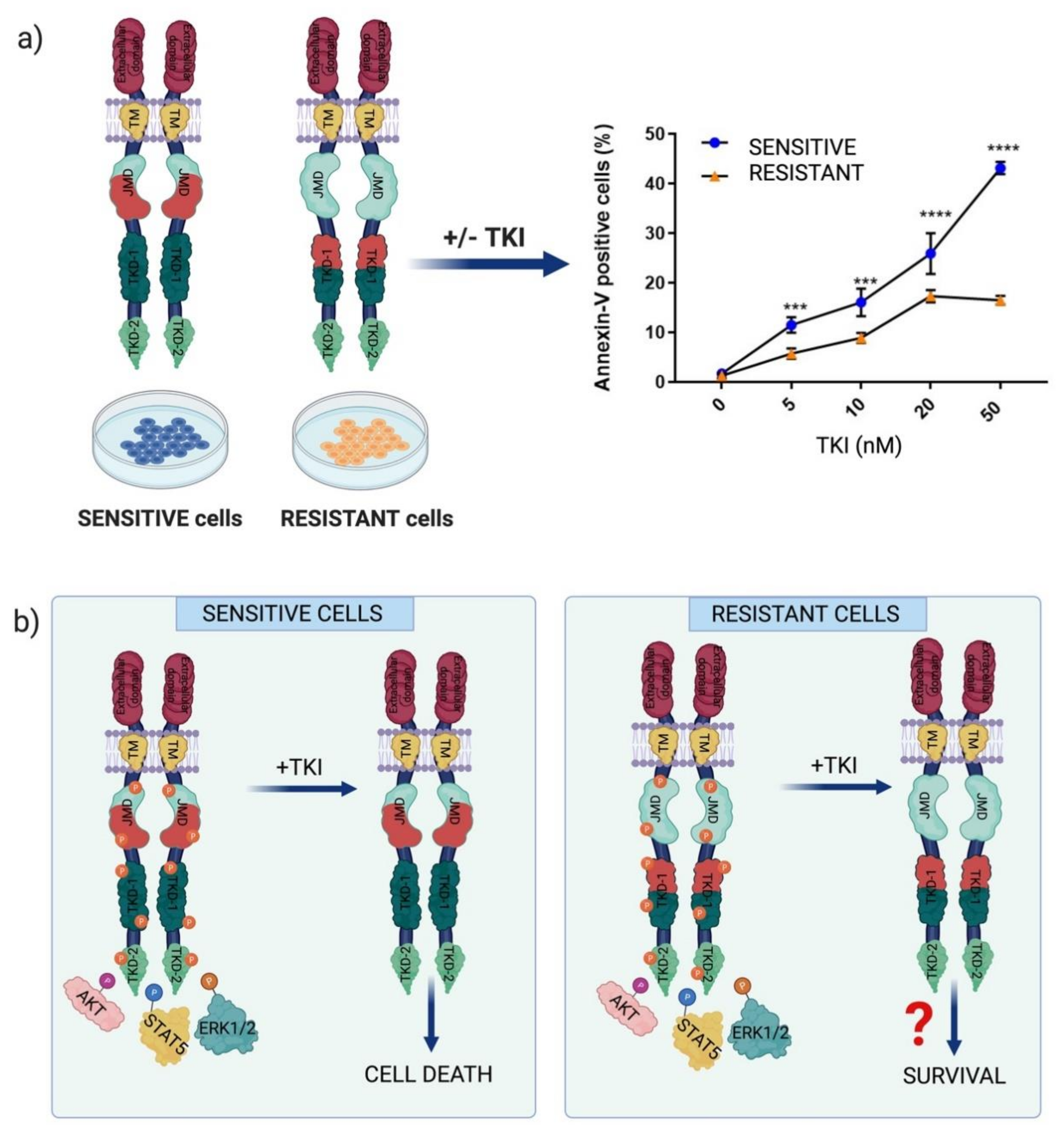
2.2. Chemotherapeutic Resistance Mechanisms in FLT3-Dependent AML
3. A Network-Based Strategy to Revert Chemotherapeutic Resistance in AML
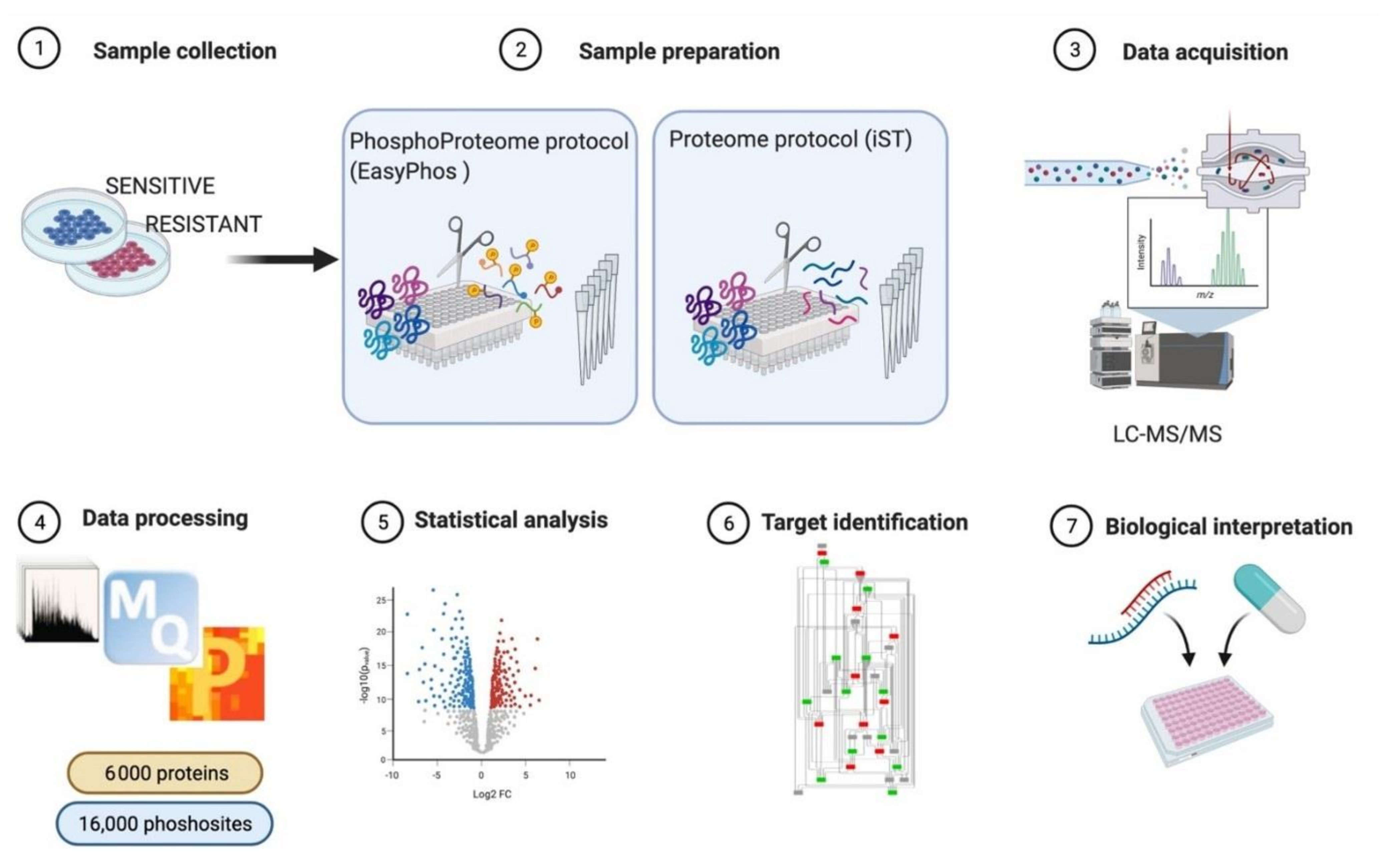
3.1. MS-Based Phosphoproteomics
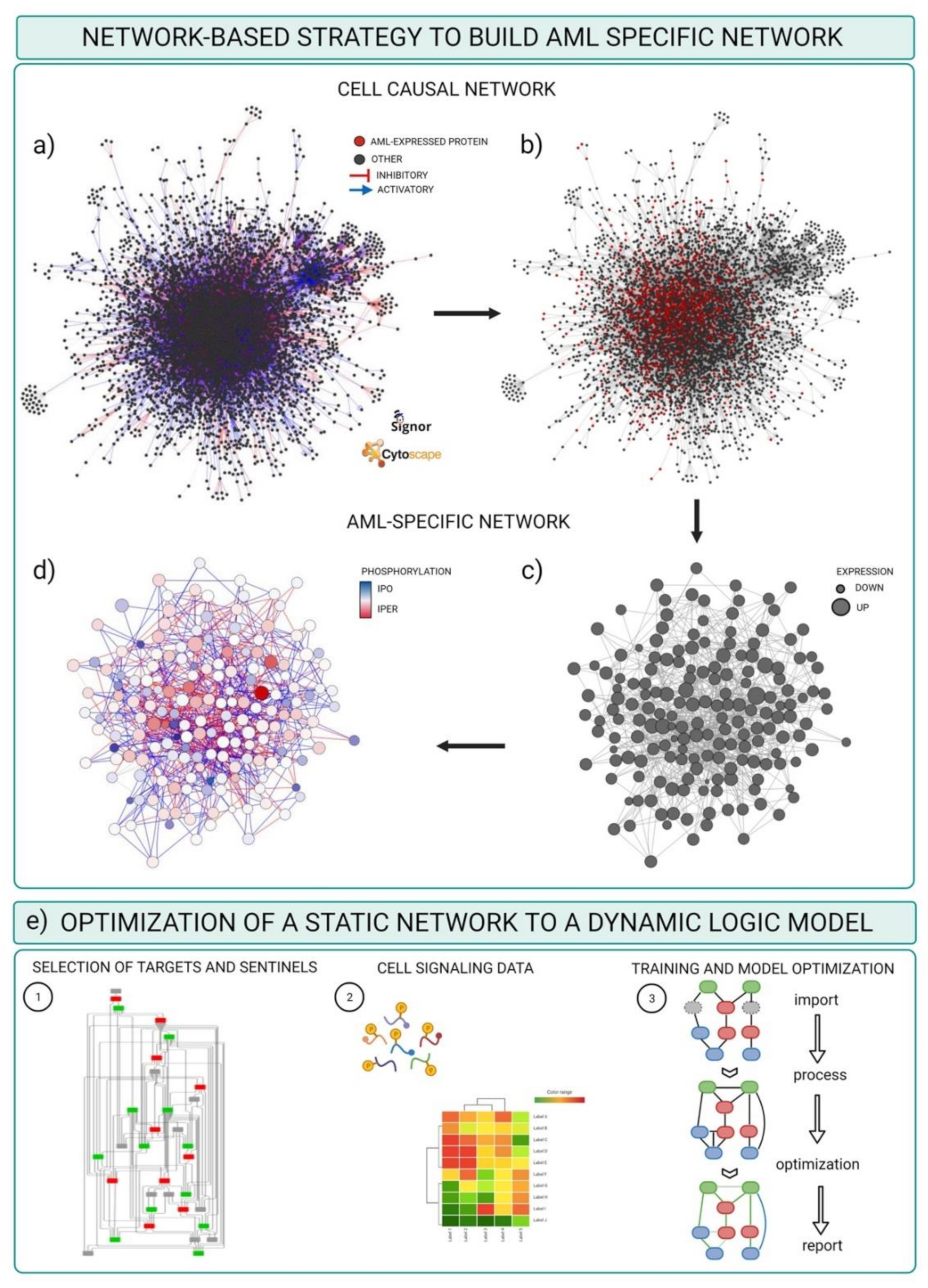
3.2. Integrating Mass-Spectrometry Based Proteomics with Literature-Derived Signaling Networks
- (a)
- Network template: the file should contain a table with at least five columns listing the source nodes, the target nodes, the causal effects (up- or downregulation) and the information about the amino acid position of the phosphorylated site as well as the amino acid sequence context of the phosphosite (sequence window). In our case, this file is the complete list of causal interactions available from the SIGNOR database.
- (b)
- Node experimental attributes: a table containing the protein expression levels in specific experimental conditions, as revealed by MS-based proteomic experiments.
- (c)
- Edge attributes: a table listing the phosphorylation level of the regulatory phosphopeptides involved in each activation/inactivation reaction, as revealed by the MS-based phosphoproteomic experiment.
- (d)
- Cytoscape software installed. For the scope of this example, we used default options. Alternatively, a plethora of adds on applications developed to visualize and analyze networks and omics data are made available at the Cytoscape App store (Table 3).
- Download the complete list of interactions from the Download all data section of the SIGNOR database (https://signor.uniroma2.it/downloads.php accessed on 24 April 2021).
- Upload the complete dataset on the Cytoscape software by setting the columns as follows: ENTITYA>Source Node; ENTITYB>Target Node; TYPEA/B, IDA/B>Source/Target Attribute; EFFECT>Interaction Type; MECHANISM, SEQUENCE, RESIDUE, DIRECT>Interaction Attribute (Figure 4a).
- Import proteomic data as node attributes.
- Use the “filter” tab in Cytoscape to select nodes identified in specific experimental conditions as revealed by MS-based proteomic experiments (Figure 4b).
- Create a subnetwork containing only the selected nodes to obtain a network of proteins expressed in the reference system.
- Use the “filter” tab in Cytoscape to select nodes with degree (number of connections) ≥ 0 to remove unconnected nodes.
- Use the “style” tab to modify the layout of the network, e.g., the size of the nodes to reflect protein expression level (Figure 4c).
- Import phosphoproteomic data as attribute of the edges, using the 15mer sequence in SIGNOR as key (see field SEQUENCE).
- Use the “style” tab to modify the visual properties of the edges. Use arrow style to show effect and directionality and modify color according to phosphoproteomic data (Figure 4d).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description |
|---|---|
| Omnipath App | It allows access to the large collection of network resources of the Omnipath web server. From the 61 web resources the user can import any combination of networks and their respective annotations. The purpose of the app is to link the access to this kind of data to the Cytoscape functionalities [65]. |
| Omics Visualizer | It is a data visualization app; it is ideal for omics data in which each node of the network is associated to multiple values. Indeed, the app allows the user to import files with multiple rows of data for a single node and offers different ways to visualize these data [66]. |
| BiNGO | It is a Cytoscape plug-in of the Biological Networks Gene Ontology resource. It analyzes GO term enrichments and it maps them onto a given network, it uses either the full GO ontologies annotation or the GOSlim ontologies. The annotated graphs generated by BiNGO are flexible and customizable by the standard Cytoscape functionalities [67]. |
| CytoCopteR | It is the graphical interface of CellNOptR. With this App the user can combine literature-derived network with experimental data to build and optimize cell specific and predictive logic networks. It uses different kind of logic formalisms (Boolean steady-state, Boolean multiple steady-state, Boolean time courses through synchronous update, steady-state constrained fuzzy logic and continuous logic-based ODEs) and the user can choose between them depending on the kind and the amount of data to analyze [68]. |
3.3. Optimizing and Building Dynamic Network trough Cell Signaling Experimental Data
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palma, A.; Iannuccelli, M.; Rozzo, I.; Licata, L.; Perfetto, L.; Massacci, G.; Castagnoli, L.; Cesareni, G.; Sacco, F. Integrating Patient-Specific Information into Logic Models of Complex Diseases: Application to Acute Myeloid Leukemia. J. Pers. Med. 2021, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Cusick, M.E.; Barabási, A.-L. Interactome Networks and Human Disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Cremers, C.G.; Nguyen, L.K. Network rewiring, adaptive resistance and combating strategies in breast cancer. Cancer Drug Resist. 2019, 2, 1106–1126. [Google Scholar] [CrossRef]
- Sacco, F.; Perfetto, L.; Cesareni, G. Combining Phosphoproteomics Datasets and Literature Information to Reveal the Functional Connections in a Cell Phosphorylation Network. Proteomics 2018, 18, e1700311. [Google Scholar] [CrossRef]
- Humphrey, S.J.; Karayel, O.; James, D.E.; Mann, M. High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nat. Protoc. 2018, 13, 1897–1916. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nat. Cell Biol. 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.D.; Thielert, M.; Vasilopoulou, C.; Ammar, C.; Coscia, F.; Mund, A.; Horning, O.B.; Bache, N.; Apalategui, A.; Lubeck, M.; et al. Ultra-High Sensitivity Mass Spectrometry Quantifies Single-Cell Proteome Changes upon Perturbation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Grove, C.S.; Vassiliou, G.S. Acute myeloid leukaemia: A paradigm for the clonal evolution of cancer? Dis. Model. Mech. 2014, 7, 941–951. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Mrózek, K.; Marcucci, G.; Nicolet, D.; Maharry, K.S.; Becker, H.; Whitman, S.P.; Metzeler, K.H.; Schwind, S.; Wu, Y.-Z.; Kohlschmidt, J.; et al. Prognostic Significance of the European LeukemiaNet Standardized System for Reporting Cytogenetic and Molecular Alterations in Adults with Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 4515–4523. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef]
- Lam, S.S.; Leung, A.Y. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. Int. J. Mol. Sci. 2020, 21, 1537. [Google Scholar] [CrossRef]
- Kazi, J.U.; Rönnstrand, L. FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The Structural Basis for Autoinhibition of FLT3 by the Juxtamembrane Domain. Mol. Cell 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M. The importance ofFLT3mutational analysis in acute myeloid leukemia. Leuk. Lymphoma 2018, 59, 2273–2286. [Google Scholar] [CrossRef]
- Reindl, C.; Bagrintseva, K.; Vempati, S.; Schnittger, S.; Ellwart, J.W.; Wenig, K.; Hopfner, K.-P.; Hiddemann, W.; Spiekermann, K. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood 2006, 107, 3700–3707. [Google Scholar] [CrossRef]
- Bacher, U.; Haferlach, C.; Kern, W.; Haferlach, T.; Schnittger, S. Prognostic relevance of FLT3-TKD mutations in AML: The combination matters—An analysis of 3082 patients. Blood 2008, 111, 2527–2537. [Google Scholar] [CrossRef]
- Zorn, J.A.; Wang, Q.; Fujimura, E.; Barros, T.; Kuriyan, J. Crystal Structure of the FLT3 Kinase Domain Bound to the Inhibitor Quizartinib (AC220). PLoS ONE 2015, 10, e0121177. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Ohno, R.; Ueda, R.; Saito, H.; Naoe, T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene 2002, 21, 2555–2563. [Google Scholar] [CrossRef]
- Kayser, S.; Schlenk, R.F.; Londono, M.C.; Breitenbuecher, F.; Wittke, K.; Du, J.; Groner, S.; Späth, D.; Krauter, J.; Ganser, A.; et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood 2009, 114, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Stirewalt, D.L.; Kopecky, K.J.; Meshinchi, S.; Engel, J.H.; Pogosova-Agadjanyan, E.L.; Linsley, J.; Slovak, M.L.; Willman, C.L.; Radich, J.P. Size of FLT3 internal tandem duplication has prognostic significance in patients with acute myeloid leukemia. Blood 2006, 107, 3724–3726. [Google Scholar] [CrossRef] [PubMed]
- Breitenbuecher, F.; Schnittger, S.; Grundler, R.; Markova, B.; Carius, B.; Brecht, A.; Duyster, J.; Haferlach, T.; Huber, C.; Fischer, T. Identification of a novel type of ITD mutations located in nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor. Blood 2009, 113, 4074–4077. [Google Scholar] [CrossRef] [PubMed]
- Arreba-Tutusaus, P.; Mack, T.S.; Bullinger, L.; Schnöder, T.M.; Polanetzki, A.; Weinert, S.; Ballaschk, A.; Wang, Z.; Deshpande, A.J.; Armstrong, S.A.; et al. Impact of FLT3-ITD location on sensitivity to TKI-therapy in vitro and in vivo. Leukemia 2015, 30, 1220–1225. [Google Scholar] [CrossRef]
- Kiyoi, H. Flt3 Inhibitors: Recent Advances and Problems for Clinical Application. Nagoya J. Med. Sci. 2015, 77, 7–17. [Google Scholar]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of drug resistance in acute myeloid leukemia. OncoTargets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef]
- Traer, E.; Martinez, J.; Javidi-Sharifi, N.; Agarwal, A.; Dunlap, J.; English, I.; Kovacsovics, T.; Tyner, J.W.; Wong, M.; Druker, B.J. FGF2 from Marrow Microenvironment Promotes Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Cancer Res. 2016, 76, 6471–6482. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-T.; Hernandez, D.; Alonso, S.; Gao, M.; Su, M.; Ghiaur, G.; Levis, M.J.; Jones, R.J. Role of CYP3A4 in bone marrow microenvironment–mediated protection of FLT3/ITD AML from tyrosine kinase inhibitors. Blood Adv. 2019, 3, 908–916. [Google Scholar] [CrossRef]
- Yu, L.J.; Matias, J.; Scudiero, D.A.; Hite, K.M.; Monks, A.; Sausville, E.A.; Waxman, D.J. P450 enzyme expression patterns in the NCI human tumor cell line panel. Drug Metab. Dispos. 2001, 29, 304–312. [Google Scholar] [PubMed]
- Moore, A.S.; Faisal, A.; De Castro, D.G.; Bavetsias, V.; Sun, C.; Atrash, B.; Valenti, M.; Brandon, A.D.H.; Avery, S.; Mair, D.; et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: A model for emerging clinical resistance patterns. Leukemia 2012, 26, 1462–1470. [Google Scholar] [CrossRef]
- Marhäll, A.; Heidel, F.; Fischer, T.; Rönnstrand, L. Internal tandem duplication mutations in the tyrosine kinase domain of FLT3 display a higher oncogenic potential than the activation loop D835Y mutation. Ann. Hematol. 2018, 97, 773–780. [Google Scholar] [CrossRef]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wiśniewski, J.R.; Cox, J.; Mann, M. Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Xie, Z.; Zhu, H.; Qian, J. Understanding protein phosphorylation on a systems level. Briefings Funct. Genom. 2010, 9, 32–42. [Google Scholar] [CrossRef]
- Scheltema, R.A.; Hauschild, J.-P.; Lange, O.; Hornburg, D.; Denisov, E.; Damoc, E.; Kuehn, A.; Makarov, A.; Mann, M. The Q Exactive HF, a Benchtop Mass Spectrometer with a Pre-filter, High-performance Quadrupole and an Ultra-high-field Orbitrap Analyzer. Mol. Cell. Proteom. 2014, 13, 3698–3708. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, E.; Berven, F.S.; Bartaula-Brevik, S.; Stokowy, T.; Hovland, R.; Vaudel, M.; Døskeland, S.O.; McCormack, E.; Batth, T.S.; Olsen, J.V.; et al. Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia. Cancers 2020, 12, 709. [Google Scholar] [CrossRef]
- Walters, D.K.; Goss, V.L.; Stoffregen, E.P.; Gu, T.-L.; Lee, K.; Nardone, J.; McGreevey, L.; Heinrich, M.C.; Deininger, M.W.; Polakiewicz, R.; et al. Phosphoproteomic analysis of AML cell lines identifies leukemic oncogenes. Leuk. Res. 2006, 30, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Alcolea, M.P.; Casado, P.; Rodríguez-Prados, J.-C.; Vanhaesebroeck, B.; Cutillas, P.R. Phosphoproteomic Analysis of Leukemia Cells under Basal and Drug-treated Conditions Identifies Markers of Kinase Pathway Activation and Mechanisms of Resistance*. Mol. Cell. Proteom. 2012, 11, 453–466. [Google Scholar] [CrossRef]
- Savage, S.R.; Zhang, B. Using phosphoproteomics data to understand cellular signaling: A comprehensive guide to bioinformatics resources. Clin. Proteom. 2020, 17, 1–18. [Google Scholar] [CrossRef]
- Luo, Y.; Jiang, Q.; Zhu, Z.; Sattar, H.; Wu, J.; Huang, W.; Su, S.; Liang, Y.; Wang, P.; Meng, X. Phosphoproteomics and Proteomics Reveal Metabolism as a Key Node in LPS-Induced Acute Inflammation in RAW264.7. Inflammation 2020, 43, 1667–1679. [Google Scholar] [CrossRef]
- Jünger, M.A.; Aebersold, R. Mass spectrometry-driven phosphoproteomics: Patterning the systems biology mosaic. Wiley Interdiscip. Rev. Dev. Biol. 2014, 3, 83–112. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chance, M.R. Integrating phosphoproteomics in systems biology. Comput. Struct. Biotechnol. J. 2014, 10, 90–97. [Google Scholar] [CrossRef]
- Oppermann, F.S.; Grundner-Culemann, K.; Kumar, C.; Gruss, O.J.; Jallepalli, P.V.; Daub, H. Combination of Chemical Genetics and Phosphoproteomics for Kinase Signaling Analysis Enables Confident Identification of Cellular Downstream Targets. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef]
- Casado, P.; Rodriguez-Prados, J.-C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinase-Substrate Enrichment Analysis Provides Insights into the Heterogeneity of Signaling Pathway Activation in Leukemia Cells. Sci. Signal. 2013, 6, rs6. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Xue, Y. Phosphoproteomics-based network medicine. FEBS J. 2013, 280, 5696–5704. [Google Scholar] [CrossRef]
- Satpathy, S.; Wagner, S.A.; Beli, P.; Gupta, R.; Kristiansen, T.A.; Malinova, D.; Francavilla, C.; Tolar, P.; Bishop, G.A.; Hostager, B.S.; et al. Systems-wide analysis of BCR signalosomes and downstream phosphorylation and ubiquitylation. Mol. Syst. Biol. 2015, 11, 810. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, M.; Smith, R.; Rajeeve, V.; Bessant, C.; Cutillas, P.R. Reconstructing kinase network topologies from phosphoproteomics data reveals cancer-associated rewiring. Nat. Biotechnol. 2020, 38, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Van Alphen, C.; Cloos, J.; Beekhof, R.; Cucchi, D.G.; Piersma, S.R.; Knol, J.C.; Henneman, A.A.; Pham, T.V.; van Meerloo, J.; Ossenkoppele, G.J.; et al. Phosphotyrosine-based Phosphoproteomics for Target Identification and Drug Response Prediction in AML Cell Lines. Mol. Cell. Proteom. 2020, 19, 884–899. [Google Scholar] [CrossRef]
- Sacco, F.; Humphrey, S.J.; Cox, J.; Mischnik, M.; Schulte, A.; Klabunde, T.; Schäfer, M.; Mann, M. Glucose-regulated and drug-perturbed phosphoproteome reveals molecular mechanisms controlling insulin secretion. Nat. Commun. 2016, 7, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Sacco, F.; Silvestri, A.; Posca, D.; Pirrò, S.; Gherardini, P.F.; Castagnoli, L.; Mann, M.; Cesareni, G. Deep Proteomics of Breast Cancer Cells Reveals that Metformin Rewires Signaling Networks Away from a Pro-growth State. Cell Syst. 2016, 2, 159–171. [Google Scholar] [CrossRef]
- Sacco, F.; Seelig, A.; Humphrey, S.J.; Krahmer, N.; Volta, F.; Reggio, A.; Marchetti, P.; Gerdes, J.; Mann, M. Phosphoproteomics Reveals the GSK3-PDX1 Axis as a Key Pathogenic Signaling Node in Diabetic Islets. Cell Metab. 2019, 29, 1422–1432. [Google Scholar] [CrossRef]
- Licata, L.; Surdo, P.L.; Iannuccelli, M.; Palma, A.; Micarelli, E.; Perfetto, L.; Peluso, D.; Calderone, A.; Castagnoli, L.; Cesareni, G. SIGNOR 2.0, the SIGnaling Network Open Resource 2.0: 2019 Update. Nucleic Acids Res. 2019, 48, D504–D510. [Google Scholar] [CrossRef]
- Perfetto, L.; Briganti, L.; Calderone, A.; Perpetuini, A.C.; Iannuccelli, M.; Langone, F.; Licata, L.; Marinkovic, M.; Mattioni, A.; Pavlidou, T.; et al. SIGNOR: A database of causal relationships between biological entities. Nucleic Acids Res. 2016, 44, D548–D554. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Robles, M.S.; Humphrey, S.J.; Mann, M. Phosphorylation Is a Central Mechanism for Circadian Control of Metabolism and Physiology. Cell Metab. 2017, 25, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Mischnik, M.; Sacco, F.; Cox, J.; Schneider, H.-C.; Schäfer, M.; Hendlich, M.; Crowther, D.; Mann, M.; Klabunde, T. IKAP: A heuristic framework for inference of kinase activities from Phosphoproteomics data. Bioinformatics 2015, 32, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Armenta, C.; Ochoa, D.; Gonçalves, E.; Saez-Rodriguez, J.; Beltrao, P. Benchmarking substrate-based kinase activity inference using phosphoproteomic data. Bioinformatics 2017, 33, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, D.; Jonikas, M.; Lawrence, R.T.; El Debs, B.; Selkrig, J.; Typas, A.; Villén, J.; Santos, S.D.; Beltrao, P. An atlas of human kinase regulation. Mol. Syst. Biol. 2016, 12, 888. [Google Scholar] [CrossRef]
- Ceccarelli, F.; Turei, D.; Gabor, A.; Saez-Rodriguez, J. Bringing data from curated pathway resources to Cytoscape with OmniPath. Bioinformatics 2019, 36, 2632–2633. [Google Scholar] [CrossRef]
- Legeay, M.; Doncheva, N.T.; Morris, J.H.; Jensen, L.J. Visualize omics data on networks with Omics Visualizer, a Cytoscape App. F1000Research 2020, 9, 157. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Terfve, C.; Cokelaer, T.; Henriques, D.; MacNamara, A.; Gonçalves, E.J.V.; Morris, M.K.; Van Iersel, M.P.; Lauffenburger, D.A.; Saez-Rodriguez, J. CellNOptR: A flexible toolkit to train protein signaling networks to data using multiple logic formalisms. BMC Syst. Biol. 2012, 6, 1–4. [Google Scholar] [CrossRef]
- Wooten, D.; Gebru, M.; Wang, H.-G.; Albert, R. Data-Driven Math Model of FLT3-ITD Acute Myeloid Leukemia Reveals Potential Therapeutic Targets. J. Pers. Med. 2021, 11, 193. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, S. Metabolic stability and epigenesis in randomly constructed genetic nets. J. Theor. Biol. 1969, 22, 437–467. [Google Scholar] [CrossRef]
- Huang, S.; Ingber, D.E. Shape-Dependent Control of Cell Growth, Differentiation, and Apoptosis: Switching between Attractors in Cell Regulatory Networks. Exp. Cell Res. 2000, 261, 91–103. [Google Scholar] [CrossRef]
- Saez-Rodriguez, J.; Alexopoulos, L.G.; Epperlein, J.; Samaga, R.; Lauffenburger, D.A.; Klamt, S.; Sorger, P.K. Discrete logic modelling as a means to link protein signalling networks with functional analysis of mammalian signal transduction. Mol. Syst. Biol. 2009, 5, 331. [Google Scholar] [CrossRef]
- Schoof, E.M.; Furtwängler, B.; Üresin, N.; Rapin, N.; Savickas, S.; Gentil, C.; Lechman, E.; auf dem Keller, U.; Dick, J.E.; Porse, B.T. Quantitative Single-Cell Proteomics as a Tool to Characterize Cellular Hierarchies. bioRxiv 2021, 745679. [Google Scholar]
- Han, L.; Qiu, P.; Zeng, Z.; Jorgensen, J.L.; Mak, D.H.; Burks, J.K.; Schober, W.; McQueen, T.J.; Cortes, J.; Tanner, S.D.; et al. Single-cell mass cytometry reveals intracellular survival/proliferative signaling in FLT3-ITD-mutated AML stem/progenitor cells. Cytom. Part A 2015, 87, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Lun, X.-K.; Bodenmiller, B. Profiling Cell Signaling Networks at Single-cell Resolution. Mol. Cell. Proteom. 2020, 19, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.H.; Nolan, G.P. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [Google Scholar] [CrossRef]



| Frequency of FLT3 Co-Occurrent Mutated Genes (N = 512) | Frequency of FLT3 Co-Occurrent Mutated Pairs (N = 512) | ||
|---|---|---|---|
| Gene | n of Patients (%) | Gene | n of Patients (%) |
| NPM1 | 242 (47.3) | NPM1:DNMT3A | 130 (25.4) |
| DNMT3A | 168 (32.8) | TET2:NPM1 | 35 (6.8) |
| TET2 | 59 (11.5) | NPM1:IDH1 | 24 (4.7) |
| NRAS | 51 (9,9) | NPM1:IDH2 | 24(4.7) |
| RUNX1 | 40 (7.8) | NRAS:NPM1 | 21 (4.1) |
| WT1 | 37 (7.2) | PTPN11:NPM1 | 21 (4.1) |
| CEBPA | 36 (7.0) | RAD21:NPM1 | 20 (3.9) |
| MLL | 35 (6.8) | TET2:DNMT3A | 19 (3.7) |
| IDH1 | 34 (6.6) | IDH1:DNMT3A | 16 (3.1) |
| IDH2 | 33 (6.4) | IDH2:DNMT3A | 16 (3.1) |
| PTPN11 | 29 (5.7) | MLL:DNMT3A | 15 (2.9) |
| RAD21 | 29 (5.7) | NRAS:DNMT3A | 15 (2.9) |
| SFRS2 | 15 (2.9) | RUNX1:DNMT3A | 13 (2.5) |
| MYC | 14 (2.7) | WT1:NPM1 | 13 (2.5) |
| ASXL1 | 12 (2.3) | DNMT3A:CEBPA | 11 (2.1) |
| CBL | 12 (2.3) | RUNX1:MLL | 11(2.1) |
| EZH2 | 12 (2.3) | NPM1:MYC | 8 (1.6) |
| KRAS | 12 (2.3) | SFRS2:RUNX1 | 8 (1.6) |
| PHF6 | 11 (2.1) | STAG2:NPM1 | 8 (1.6) |
| KIT | 10 (1.9) | NPM1:KRAS | 7 (1.4) |
| GATA2 | 9 (1.7) | RAD21: DNMT3A | 7 (1.4) |
| SF3B1 | 8 (1.6) | TET2:RUNX1 | 7 (1.4) |
| MLL2 | 7 (1.4) | KRAS:DNMT3A | 6 (1.2) |
| TP53 | 7 (1.4) | PHF6:NPM1 | 6 (1.2) |
| U2AF1 | 7 (1.4) | RUNX1:NRAS | 6 (1.2) |
| NF1 | 6 (1.2) | TET2:MLL | 6 (1.2) |
| ZRSR2 | 5 (1) | TET2:PTPN11 | 6 (1.2) |
| NPM1:NF1 | 5 (0.9) | ||
| NRAS:KRAS | 5 (0.9) | ||
| RUNX1:EZH2 | 5 (0.9) | ||
| STAG2:MLL | 5 (0.9) | ||
| TET2:RAD21 | 5 (0.9) | ||
| TET2:STAG2 | 5 (0.9) | ||
| 1° Generation | |||||
|---|---|---|---|---|---|
| Inhibitor | Sorafenib | Midostaurin | Sunitinib | Lestaurtinib | Tandutinib |
| Target | FLT3; c-KIT; VEGFR; PDGFR; RAF1 | FLT3; c-KIT; PDGFRB; VEGFR | FLT3; c-KIT; KDR; PDGFR | FLT3; JAK2; TRK A | FLT3; PDGFR; c-KIT |
| Trial phase | II/III | III | II | III | I |
| FDA approved | No | Yes | No | No | No |
| 2° Generation | |||||
| Inhibitor | Quizartinib | Gilteritinib | Crenolanib | Ponatinib | |
| Target | FLT3; c-KIT; PDGFRa | FLT3; AXL | FLT3; PDGFR | FLT3; BCR-ABL; c-KIT; FGFR1; PDGFRa | |
| Trial phase | III | III | III | I/II | |
| FDA approved | No | Yes | No | No | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pugliese, G.M.; Latini, S.; Massacci, G.; Perfetto, L.; Sacco, F. Combining Mass Spectrometry-Based Phosphoproteomics with a Network-Based Approach to Reveal FLT3-Dependent Mechanisms of Chemoresistance. Proteomes 2021, 9, 19. https://doi.org/10.3390/proteomes9020019
Pugliese GM, Latini S, Massacci G, Perfetto L, Sacco F. Combining Mass Spectrometry-Based Phosphoproteomics with a Network-Based Approach to Reveal FLT3-Dependent Mechanisms of Chemoresistance. Proteomes. 2021; 9(2):19. https://doi.org/10.3390/proteomes9020019
Chicago/Turabian StylePugliese, Giusj Monia, Sara Latini, Giorgia Massacci, Livia Perfetto, and Francesca Sacco. 2021. "Combining Mass Spectrometry-Based Phosphoproteomics with a Network-Based Approach to Reveal FLT3-Dependent Mechanisms of Chemoresistance" Proteomes 9, no. 2: 19. https://doi.org/10.3390/proteomes9020019
APA StylePugliese, G. M., Latini, S., Massacci, G., Perfetto, L., & Sacco, F. (2021). Combining Mass Spectrometry-Based Phosphoproteomics with a Network-Based Approach to Reveal FLT3-Dependent Mechanisms of Chemoresistance. Proteomes, 9(2), 19. https://doi.org/10.3390/proteomes9020019