1. Introduction
Proximity biotinylation is a promising and increasingly important method to identify protein–protein interactions [
1]. In the original version of this approach, the protein of interest was fused to BioID, a mutant biotin ligase from
Escherichia coli. This mutant enzyme activates biotin, which is then released from the active site rather than being transferred specifically to an acceptor peptide. The resulting cloud of activated biotin quickly reacts with proteins in the vicinity of the protein of interest. The biotinylated proteins are then purified and identified by mass spectrometry. A second-generation version, BioID2, was developed based on a biotin ligase from the thermophilic bacterium
Aquifex aeolicus, which is smaller, more stable and more effective at identifying protein–protein interactions [
2].
Proximity biotinylation has several advantages over other approaches for identifying protein–protein interactions. BioID is performed in the cells of interest, which should result in the identification of more physiologically relevant interactions and interactions with proteins that are not properly folded or appropriately modified when expressed in yeast or bacteria. Since interacting proteins are labeled with biotin in intact, living cells, there is less chance for non-specific interactions that may occur after lysis and subsequent mixing of proteins from distinct compartments. Since BioID covalently tags proteins with biotin, weak or transient interactions that would be missed by approaches requiring complexes to be maintained through purification and washing steps can be identified. The covalent linkage also allows harsher wash conditions to be employed, which reduces non-specific binding.
Although powerful, there are several issues that can present challenges to generating high-quality proximity biotinylation data. The promiscuous nature of the labelling often results in a large number of biotinylated proteins. Rigorous approaches and closely matched negative control proteins are needed to distinguish true interactors from background or non-specifically labeled proteins. In the current version of BioID, it is not possible to monitor expression or localization of the target protein in real time. Finally, the addition of the full-length BioID or BioID2 may not be tolerated by some viruses because large insertions disrupt packaging of the virus genome.
In this study, we sought to address these issues by recruiting BioID to target proteins via a small tag. We considered two small, self-associating peptide tags: GFP11 [
3] and HiBiT [
4,
5]. GFP11 corresponds to the 16 amino acids that comprise the 11th β-sheet of GFP. It spontaneously associates with GFP1–10 (the first ten β strands of super-folder GFP) to generate a green fluorescent complex [
3]. This tag has been used to monitor solubility and localization of proteins in living cells [
3,
6,
7]. Similarly, HiBiT is an engineered 13 amino acid peptide that binds to and complements the catalytically inactive LgBiT fragment of nanoluciferase (amino acids 1–156) [
4,
5]. Both GFP11 and HiBiT bind with similar affinities (~80 pM) to their respective partners [
5,
8]. Although both systems offer the potential to monitor recruitment of BioID with its target protein via complementation of the self-assembling proteins, we chose to pursue the GFP11 system because it enables easy monitoring of the expression and localization of the protein of interest in the cell population being assayed.
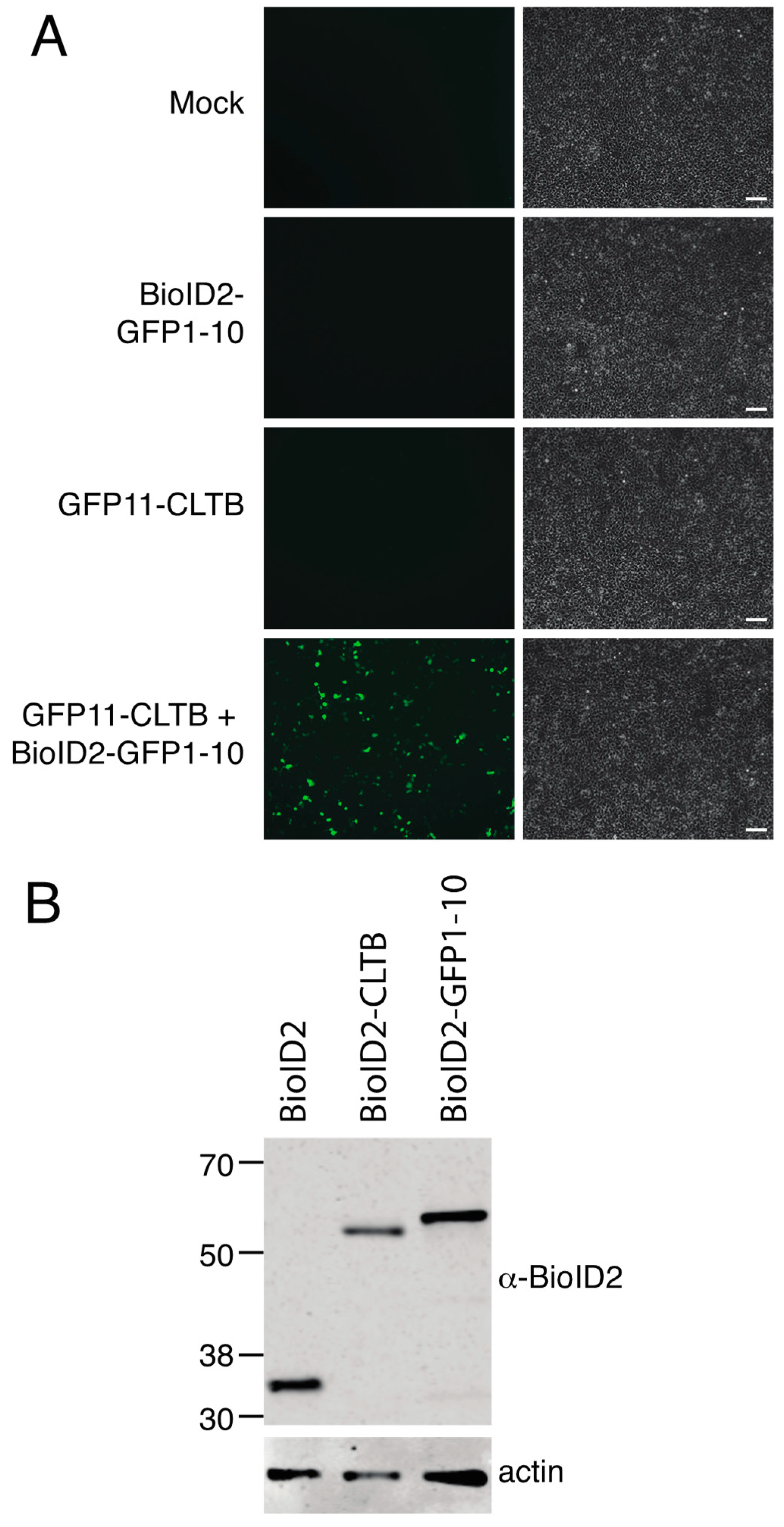
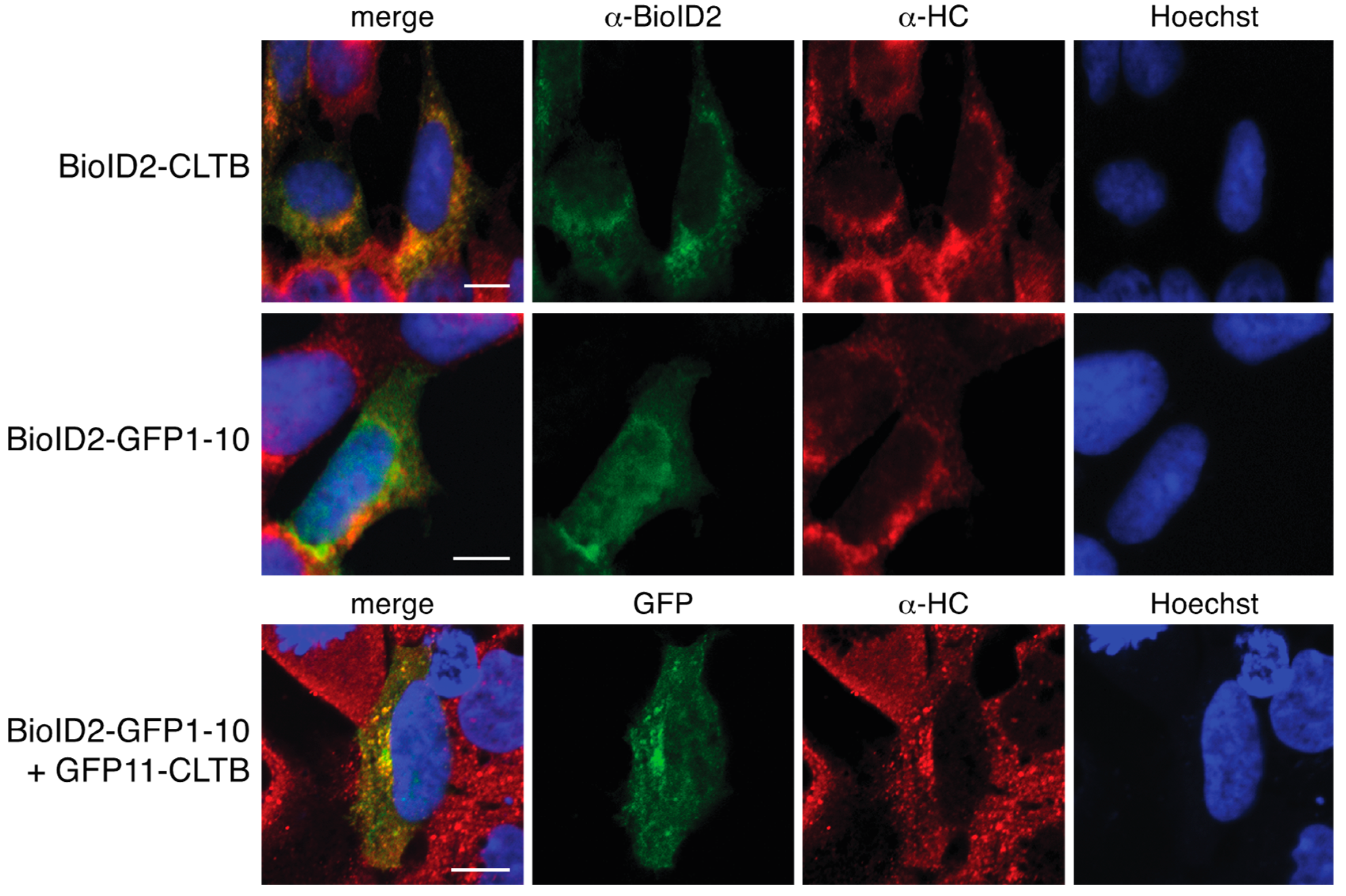
We report here a novel proximity biotinylation system based on the self-assembling split-GFP pair GFP11 and GFP1–10. BioID2 was fused to GFP1–10 and co-expressed with the protein of interest tagged with GFP11. Since localization of clathrin light chain was previously characterized in this split-GFP system and since some of the interacting partners of clathrin light chain are relatively well studied, we tested this system with clathrin light chain tagged with GFP11 [
7]. To further improve the removal of non-specifically biotinylated proteins, we established a stable cell line that inducibly expressed BioID2-GFP1–10. We show that complementation of BioID2-GFP1–10 with GFP11 clathrin light chain produced a green fluorescent complex that colocalized with clathrin heavy chain. Proximity biotinylation using this approach yielded a higher quality set of interacting proteins than direct fusion to BioID2.
2. Materials and Methods
2.1. Plasmids
Plasmids pcDNA3.1-GFP1–10 [
7], pEGFP-GFP11-clathrin light chain [
7], pMyc-BioID2-MCS [
2], and pCAG-FLPo [
9] were gifts from Bo Huang (
http://n2t.net/addgene:70219; RRID:Addgene_70219;
http://n2t.net/addgene:70217; RRID:Addgene_70217), Kyle Roux (Addgene plasmid # 74223;
http://n2t.net/addgene:74223; RRID:Addgene_74223), and Philippe Soriano (Addgene plasmid # 13,792;
http://n2t.net/addgene:13792; RRID:Addgene_13792), respectively. Plasmid pcDNA3.1 Myc-BioID2-GFP1–10 was constructed by digesting pcDNA3.1-GFP1–10 with
NheI and
EcoRI and inserting a DNA fragment encoding the Myc tag and BioID2, which was obtained by digesting plasmid Myc-BioID2-MCS. To generate pEGFP-Myc-BioID2-clathrin light chain, Myc-BioID2 was amplified from plasmid Myc-BioID2-MCS using primers (5′-CCAAGCTGGCTAGCCACCATG-3′) and (5′-CGACTGCAGAATTCTCGCTTCTTCTCAGGCTGAACTCG-3′), digested with
NheI and
EcoRI, and ligated into pEGFP-GFP11-clathrin light chain cut using the same enzymes. To construct pHiBiT-clathrin light chain, oligonucleotides (5′-CTAGCATGGTGAGCGGCTGGCGGCTGTTCAAG AAGATTAGCGGGAGTTCTGGCGGCTCGAGCGGTGGAGCT-3′) and (5′-CCACCGCTCGAGCCG CCAGAACTCCCGCTAATCTTCTTGAACAGCCGCCAGCCGCTCACCATG-3’) were phosphorylated, annealed, and ligated into
NheI- and
SacI-digested pEGFP-GFP11-clathrin light chain. pcDNA5/FRT/TO-BioID2-GFP1–10 was constructed by amplifying GFP1–10 from pcDNA3.1-GFP1–10 with primers (5′-CAGCGGCAGTTCTGGCGGTGGATCCATGTCCAAAGGAGAAGAACT GTTTAC-3′) and (5′-CGGGCCCTCTAGACTCGAGCGGCCGCTTATGTTCCTTTTTCATTTGGAT CT-3′), digesting with
BamHI and
NotI, and inserting the purified fragment into plasmid pcDNA5/FRT/TO-BioID2 cut with the same enzymes.
2.2. Cell Lines
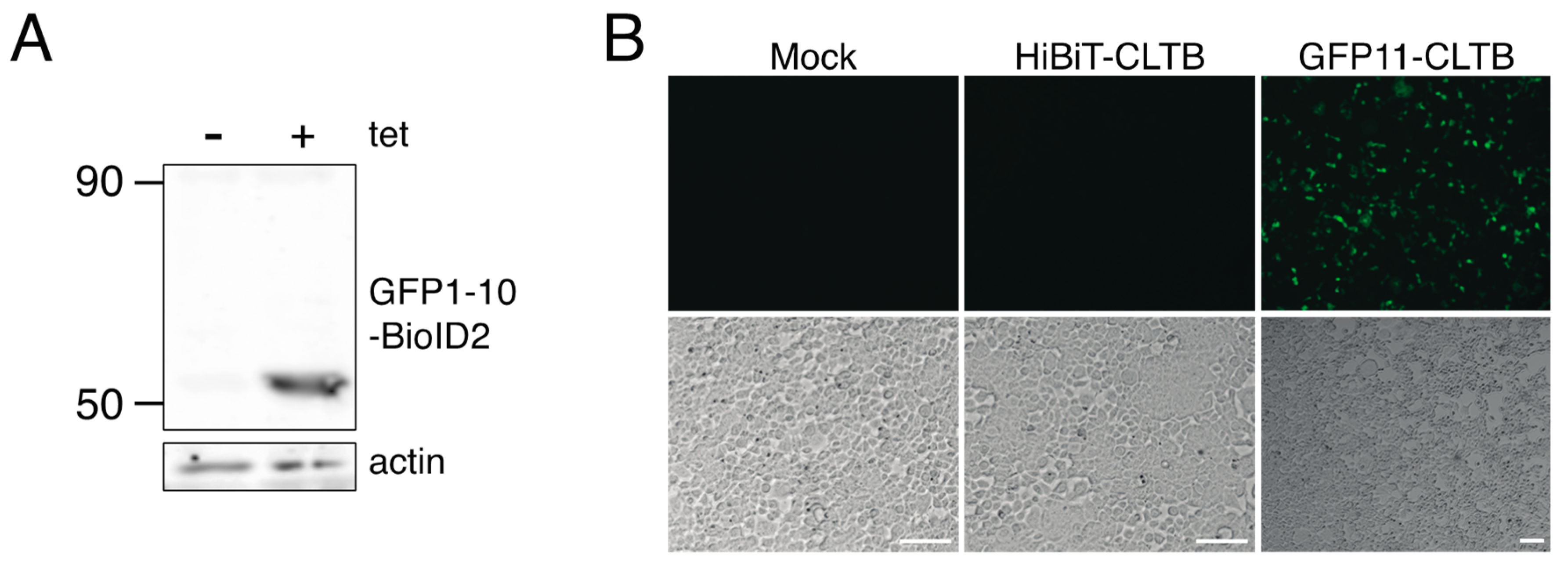
Human embryonic kidney 293 [HEK293] (ATCC® CRL1573™) cells were purchased from ATCC and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco™, ThermoFisher Scientific, Waltham, MA, USA) + 10% FBS (R & D Systems, formerly known as Atlanta Biologicals, Minneapolis, MN, USA) at 37 °C in 5% CO2. Flp-In™ T-REx™ 293 cells (R78007) were purchased from ThermoFisher Scientific (Waltham, MA, USA) and maintained in DMEM + 10% FBS + 100 μg/mL Zeocin (Gibco™, Waltham, MA, USA). Flp-In™ T-REx™ 293-BioID2-GFP1–10 stable cells were maintained in DMEM + 10% FBS supplemented with 200 μg/mL hygromycin (Gibco™) at 37 °C in 5% CO2.
Cells stably expressing BioID2-GFP1–10 were generated as per the manufacturer’s protocol (Invitrogen™, ThermoFisher Scientific, Waltham, MA, USA) with minor changes. Flp-In™ T-REx™ 293 cells were grown in a 6-well plate overnight, then transfected with pcDNA5/FRT/TO-BioID2-GFP1–10 (100 ng) and pFLPo (1 μg); transfection medium was replaced with fresh culture medium after 4 h. On the following day, the cells were transferred into a 10 cm plate and hygromycin B (200 μg/mL) was added 24 h later. The cells were maintained in medium containing hygromycin B for at least two weeks. Medium was changed regularly to remove dead cells. Once colonies were visible, individual colonies were picked, expanded in medium containing hygromycin B and analyzed for protein expression.
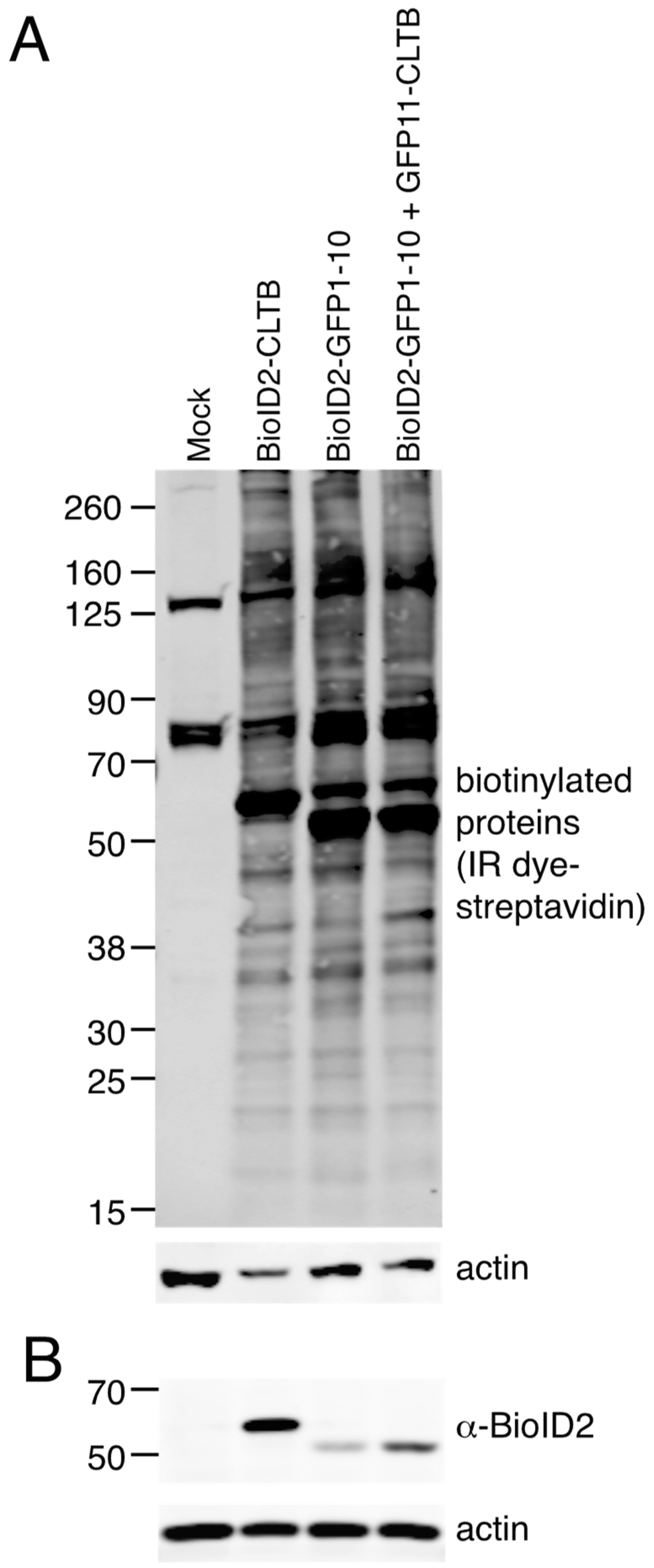
2.3. Proximity Biotinylation
HEK293 and Flp-In™ T-REx™ 293-BioID2-GFP1–10 stable cells were mock transfected or transfected with plasmids as described in the results section and figure legends using Lipofectamine 2000. For stable GFP1–10 cells, transfection medium was replaced with fresh medium containing 1 μg/mL tetracycline at 3–4 h after transfection. Uninduced, mock-transfected cells were maintained in parallel. At 24 h post-transfection, biotin (50 μM) was added to the culture medium and cells were incubated for 24 h.
2.4. Immuno- and Streptavidin Blots
Cells were lysed with 50 mM Tris pH 8, 5 mM EDTA, 150 mM NaCl and 1% Triton X. After collecting cell debris by centrifugation, supernatant was removed and SDS-PAGE sample buffer containing 2% β-mercaptoethanol was added. Electrophoresis was carried out on an SDS-PAGE gel and proteins were transferred to nitrocellulose membranes. BioID2 fusion proteins were detected with chicken anti-BioID2 primary antibody (BioFront Technologies, Tallahassee, FL, USA, product number BID2-CP-100) and IRDye
® 800CW donkey anti-chicken secondary antibodies (LI-COR Biosciences, Lincoln, NE, USA, product number 926-32218). Actin was detected with mouse monoclonal anti-actin antibody AC-15 (Sigma-Aldrich, St. Louis, MO, USA, product number A1978) and IRDye
® 680RD goat anti-mouse secondary antibodies (LI-COR Biosciences, product number 925-68070). Blots were imaged with an Odyssey
® Imaging System (LI-COR Biosciences, Lincoln, NE, USA). Biotinylated proteins were detected with IRDye
® 800CW streptavidin (LI-COR Biosciences, product number 925-32230). The original unmodified blots along with the quantitation of the bands are included in
Supplementary File 1.
2.5. Fluorescent Microscopy
HEK 293 and Flp-In™ T-REx™ 293-BioID2-GFP1–10 stable cells were transfected with GFP expression plasmids as indicated in the text. Flp-In™ T-REx™ 293-BioID2-GFP1–10 stable cells were incubated in the presence of tetracycline to induce BioID2-GFP1–10 expression. Green fluorescence in live cells was imaged using a Zeiss Axio Observer inverted fluorescence microscope equipped with an Axiocam 506 mono camera.
To localize clathrin and BioID2, HEK 293 cells were transfected as described above. On the following day, cells were fixed with 4% paraformaldehyde and permeablilized with 1% Triton X-100. Clathrin was detected by sequentially probing with rabbit anti-clathrin heavy Chain (Abcam, Cambridge, UK, product number ab21679) primary antibodies and Alexa Fluor 594 goat anti-rabbit IgG secondary antibodies (Invitrogen, ThermoFisher Scientific, Waltham, MA, USA, product number A11012). BioID2 was detected with chicken anti-BioID2 primary antibody and Alexa Fluor 488 goat anti-chicken IgG secondary antibodies (Invitrogen, ThermoFisher Scientific, Waltham, MA, USA, product number A11039). Cells imaging was performed using a Nikon A1 Confocal System on Nikon Eclipse Ti Microscope.
2.6. Purification of Biotinylated Proteins
HEK 293 cells were plated in T75 flasks and mock transfected or transfected with pcDNA3.1 Myc-BioID2-GFP1–10, pEGFP-Myc-BioID2-clathrin light chain or pEGFP-GFP11-clathrin light chain plus pcDNA3.1 Myc-BioID2-GFP1–10 plasmids in triplicate. At 24 h after the transfection, biotin (50 μM) was added to the cells. After 24 h, cells were washed gently with PBS, flushed from the surface of the flask with fresh PBS, and collected by centrifugation at 500× g. Cells pellets were frozen on dry ice and stored at −80 °C.
Triplicate cultures of T-REx™ 293-BioID2-GFP1–10 cells were mock transfected or transfected with pEGFP-GFP11-clathrin light chain or pEGFP-HiBiT-clathrin light chain followed by induction with tetracycline. At 24 h after the transfection, 50 μM biotin was added and cells were processed as described above at 48 h.
Biotinylated proteins were purified according to the protocol of Hesketh et al. with slight modifications [
10]. Cell pellets were weighed, thawed on ice and resuspended at a 1:4 wt:vol ratio in BioID Lysis buffer (50 mM Tris pH7.5, 150 mM NaCl, 0.4% SDS, 1% IGEPAL, 1.5 mM MgCl
2, 1 mM EGTA) supplemented with 1X Protease Inhibitor mix (Sigma-Aldrich, St. Louis, MO, USA, product number S8830) and 250 U Benzonase (MilliporeSigma, St. Louis, MO, USA, product number 712053) per mL of Lysis Buffer. Cells were frozen on dry ice and immediately thawed at 37 °C. As soon as the cells started thawing, cells were placed on ice and transferred to an end-over-end mixer for 30 min at 4 °C. Cell debris was removed by centrifugation at 21,130×
g for 20 min. The supernatant was transferred to a fresh tube, to which was added 35 μL of streptavidin beads that were washed three times with BioID Lysis Buffer. The mixture was incubated overnight at 4 °C with rotation using an end-over-end mixer. Beads were collected by centrifugation at 500×
g for 2 min and washed once with BioID Lysis Buffer, once with BioID Wash Buffer (2% SDS, 50 mM Tris, pH 7.5), twice with BioID Lysis Buffer and three times with 50 mM ammonium bicarbonate solution, pH 8.0. The beads were suspended in ammonium bicarbonate solution and stored at −80 °C.
2.7. On Bead Digestion
The bead suspension was thawed on ice and the beads were collected by centrifugation. After removing the supernatant, the beads were resuspended in 10 μL of 8 M urea, 10 mM dithiothreitol (DTT) and incubated for 1 h at 37 °C for reduction. Alkylation was performed by adding 2% (v/v) of alkylating solution (97.5% acetonitrile, 0.5% triethylphosphine, and 2% iodoethanol) and incubating for 1 h at 37 °C. After drying by vacuum centrifugation, the proteins were digested using 80 μL (0.05 μg/μL) of sequence grade Lys-C/Trypsin (Promega, Madison, WI, USA) in a Barocycler NEP2320 (Pressure Biosciences, Inc., Boston, MA, USA) at 50 °C under 20,000 psi for 1 h. The resulting peptides were cleaned and recovered from the beads using C18 spin columns (Nest Group), dried by vacuum centrifugation, and resuspended in 97% purified water/3% acetonitrile (ACN)/0.1% formic acid (FA).
2.8. LC–MS/MS Data Collection and Data Analysis
Mass spectrometry was performed using a Dionex UltiMate 3000 RSLC Nano System coupled to a Q Exactive™ HF Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific, Waltham, MA, USA). Peptides were loaded onto a 300 µm x 5mm C18 PepMap™ 100 trap column and washed for 5 min with 98% purified water/2% ACN/0.1% FA at a flow rate of 5 µl/minute. After washing, the trap column was switched in line with a 75 µm × 50 cm reverse-phase Acclaim™ PepMap™ RSLC C18 analytical column heated to 50 °C. Peptides were separated using a 120 min linear gradient at a flow rate of 0.3 µL/min. Mobile phase A consisted of 0.1% FA in purified water while mobile phase B was 0.1% FA in 80% ACN. The method began at 2% B and reached 10% B in 5 min, 30% B in 80 min, 45% B in 93 min, and 100% B in 93 min. The column was held at 100% B for 5 min before being brought back to 2% B and equilibrated for 20 min. Samples were injected into the QE-HF through the Nanospray Flex™ Ion Source using an emitter tip from New Objective (Littleton, MA, USA). MS data were collected between 400 and 1600 m/z using 120,000 resolution at 200 m/z, 100 ms maximum injection time, and 15 s dynamic exclusion. The top 20 precursor ions were fragmented by higher energy C-trap dissociation (HCD) at a normalized collision energy of 27%. MS/MS spectra were acquired using the Orbitrap at a resolution of 15,000 at 200 m/z and a maximum injection time of 20 ms.
LC–MS/MS RAW data files were converted into MGF in Mascot Daemon (ver 2.5.1.) using ProteoWizard RAW data import filter. MS/MS spectra were searched against Uniprot human protein database downloaded on 18 November 2018 in Mascot Daemon. To control the FDR (False Discovery Rate), spectra were searched against the corresponding reverse sequence database by selecting a decoy option. The search parameters were as follows: (1) precursor ion (MS1) mass tolerance of 0.05 Da, and product ion (MS/MS) mass tolerance of 0.2 Da, respectively; (2) ethanolyl of cysteine as a fixed modification and oxidation of methionine (M) and acetyl (N-term) as variable modifications, and 1 missed cleavage allowed. Peptide matches were accepted if the significance scores of their match had a p value < 0.05. All the matched peptides were filtered to accept peptides with rank 1. The FDR was adjusted to 1% prior to exporting results files. Protein identification generally required a minimum of two peptides and a minimum of one unique peptide if other matched peptides were shared among multiple protein/protein isoforms. Relative protein abundances across samples were determined by spectral counts. The LC-MS/MS RAW data files are available in the MassIVE data repository (massive.ucsd.edu) under ID MSV000086141.
2.9. Analysis of BioID Data
Proteins identified by LC–MS/MS analysis were analyzed using the Contaminant Repository for Affinity Purification (CRAPome) at
www.crapome.org [
11]. Spectral counts from biotinylated proteins identified in clathrin light chain samples were compared to samples from controls cells expressing Myc-BioID2-GFP1–10 using default SAINTexpress parameters (Incorporate Known Data = none, Number of Replicates Per Bait = all) to calculate fold change, SAINT score and Bayesian false discovery rate (BFDR) [
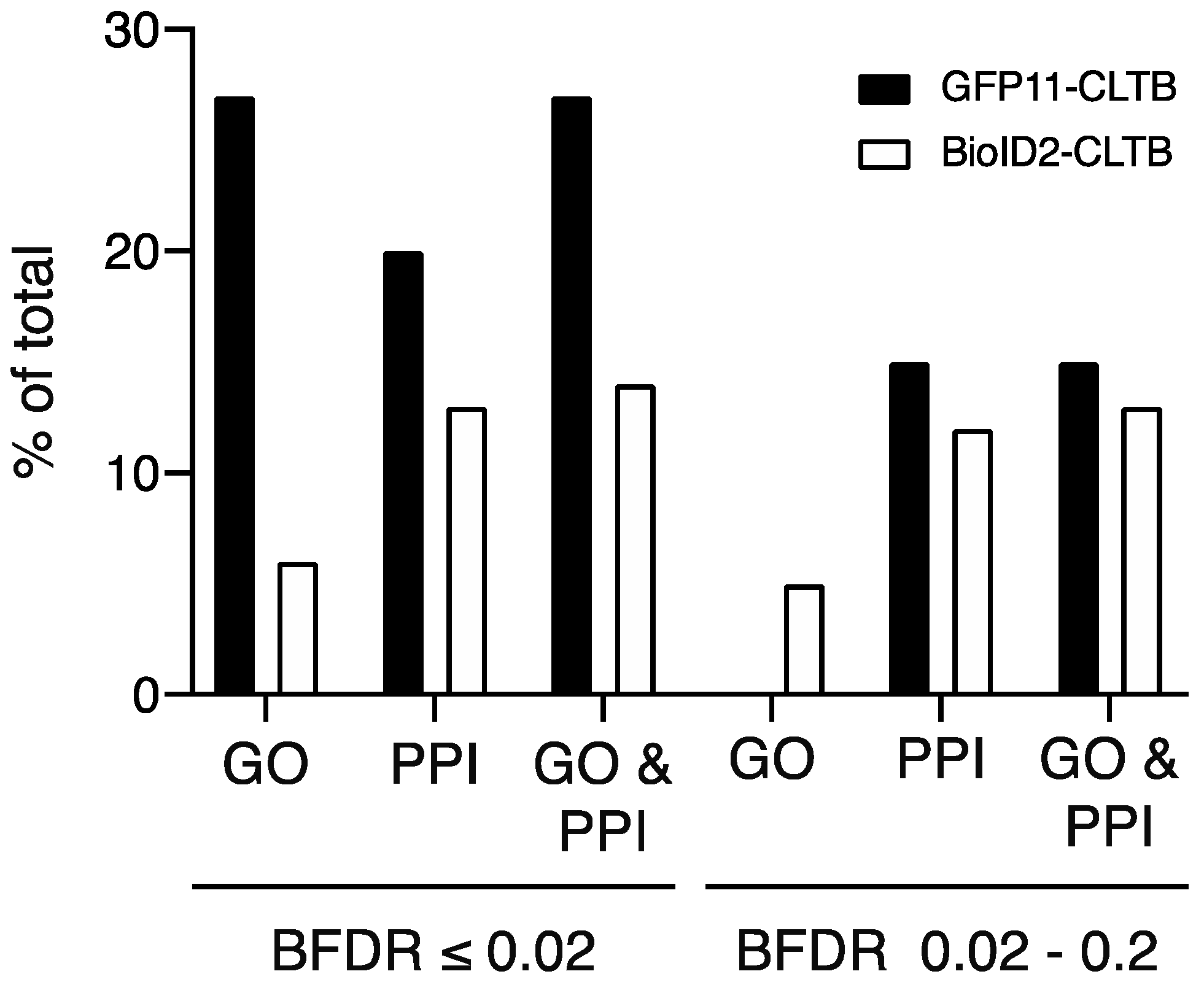
12,
13]. For purposes of comparison, we report all interactions with a BFDR ≤ 0.02 (high confidence) and BFDR ≤ 0.2 (low confidence). Proteins with BFDR > 0.2 were excluded.
Human genes annotated with clathrin-related GO terms were downloaded from Amigo (
http://amigo.geneontology.org) on 25 August 2020 [
14,
15]. The set of human proteins that bind to human clathrin light or heavy chain were downloaded from BioGrid version 3.5.188 [
16,
17]. High and low confidence interactors from clathrin light chain BioID experiments were queried against the GO and clathrin-interacting protein lists.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






