What is Normalization? The Strategies Employed in Top-Down and Bottom-Up Proteome Analysis Workflows



Abstract
1. Introduction
2. Cellular Normalization
3. Normalization During and after Lysis and Protein Extraction
4. Pre-Digestion/Digestion Normalization
4.1. Normalization Using Total Protein Quantification
4.2. Digestion Optimization to Reduce Bias
4.3. Monitoring Digestion Efficiency
4.4. Normalization Issues Unique to Top Down Methodologies
5. Post-digestion Normalization
5.1. Peptide Assays for as a Means of Post-Digestion Normalization
5.2. Normalization using Internal Standards
5.3. Normalization Using Endogenous Molecules
6. Post-Analysis Normalization
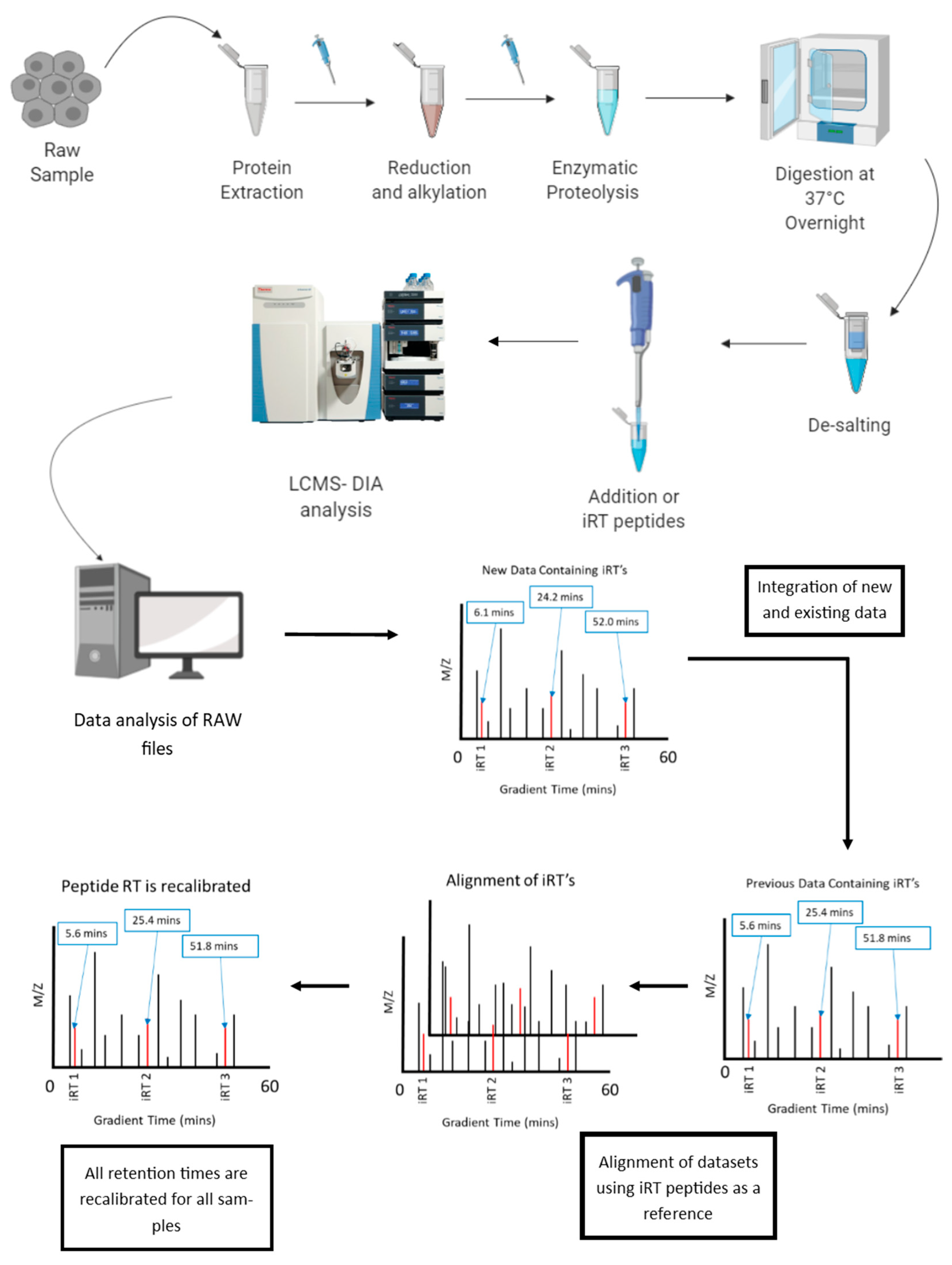
6.1. Normalization of Retention Time in LC-MS
6.2. Approaches of Normalization of MS-Derived Data
6.3. Limitations of Normalization: When an Outlier Stays an Outlier
6.4. Cross Run Normalization, Quality Control and the Removal of “Batch Effects”
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Valikangas, T.; Suomi, T.; Elo, L.L. A comprehensive evaluation of popular proteomics software workflows for label-free proteome quantification and imputation. Brief. Bioinform. 2018, 19, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Kingsmore, S.F. Multiplexed protein measurement: Technologies and applications of protein and antibody arrays. Nat. Rev. Drug Discov. 2006, 5, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteom. MCP 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Schmidt, A.; Malmstroem, J.; Claassen, M.; Ori, A.; Szymborska, A.; Herzog, F.; Rinner, O.; Ellenberg, J.; Aebersold, R. The quantitative proteome of a human cell line. Mol. Syst. Biol. 2011, 7, 549. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Kratchmarova, I.; Mann, M. Properties of 13c-substituted arginine in stable isotope labeling by amino acids in cell culture (silac). J. Proteome Res. 2003, 2, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.J.; Thalassinos, K.; Slade, S.E.; Connolly, J.B.; Crombie, A.; Murrell, J.C.; Scrivens, J.H. A comparison of labeling and label-free mass spectrometry-based proteomics approaches. J. Proteome Res. 2009, 8, 3752–3759. [Google Scholar] [CrossRef] [PubMed]
- Hecht, E.S.; McCord, J.P.; Muddiman, D.C. A quantitative glycomics and proteomics combined purification strategy. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.A.; Curtis, B.S.; Curtis, W.R. Improving accuracy of cell and chromophore concentration measurements using optical density. BMC Biophys. 2013, 6, 4. [Google Scholar] [CrossRef]
- Stenz, L.; Francois, P.; Fischer, A.; Huyghe, A.; Tangomo, M.; Hernandez, D.; Cassat, J.; Linder, P.; Schrenzel, J. Impact of oleic acid (cis-9-octadecenoic acid) on bacterial viability and biofilm production in staphylococcus aureus. FEMS Microbiol. Lett. 2008, 287, 149–155. [Google Scholar] [CrossRef]
- Cadena-Herrera, D.; Esparza-De Lara, J.E.; Ramirez-Ibanez, N.D.; Lopez-Morales, C.A.; Perez, N.O.; Flores-Ortiz, L.F.; Medina-Rivero, E. Validation of three viable-cell counting methods: Manual, semi-automated, and automated. Biotechnol. Rep. (Amst) 2015, 7, 9–16. [Google Scholar] [CrossRef]
- Montes, M.; Jaensson, E.A.; Orozco, A.F.; Lewis, D.E.; Corry, D.B. A general method for bead-enhanced quantitation by flow cytometry. J. Immunol. Methods 2006, 317, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Davey, H.M.; Kell, D.B. Flow cytometry and cell sorting of heterogeneous microbial populations: The importance of single-cell analyses. Microbiol. Rev. 1996, 60, 641–696. [Google Scholar] [PubMed]
- Goranov, A.I.; Cook, M.; Ricicova, M.; Ben-Ari, G.; Gonzalez, C.; Hansen, C.; Tyers, M.; Amon, A. The rate of cell growth is governed by cell cycle stage. Genes Dev. 2009, 23, 1408–1422. [Google Scholar] [CrossRef] [PubMed]
- Wilfinger, W.W.; Mackey, K.; Chomczynski, P. Effect of ph and ionic strength on the spectrophotometric assessment of nucleic acid purity. BioTechniques 1997, 22, 474–476, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Bokes, P.; Singh, A. Protein copy number distributions for a self-regulating gene in the presence of decoy binding sites. PLoS ONE 2015, 10, e0120555. [Google Scholar] [CrossRef] [PubMed]
- Marzluff, W.F.; Duronio, R.J. Histone mrna expression: Multiple levels of cell cycle regulation and important developmental consequences. Curr. Opin. Cell Biol. 2002, 14, 692–699. [Google Scholar] [CrossRef]
- Langan, T.J.; Chou, R.C. Synchronization of mammalian cell cultures by serum deprivation. Methods Mol. Biol. 2011, 761, 75–83. [Google Scholar] [PubMed]
- Shimada, H.; Obayashi, T.; Takahashi, N.; Matsui, M.; Sakamoto, A. Normalization using ploidy and genomic DNA copy number allows absolute quantification of transcripts, proteins and metabolites in cells. Plant Methods 2010, 6, 29. [Google Scholar] [CrossRef]
- Soppa, J. Polyploidy in archaea and bacteria: About desiccation resistance, giant cell size, long-term survival, enforcement by a eukaryotic host and additional aspects. J. Mol. Microbiol. Biotechnol. 2014, 24, 409–419. [Google Scholar] [CrossRef]
- Thompson, J.N.; Nuismer, S.L.; Merg, K. Plant polyploidy and the evolutionary ecology of plant/animal interactions. Biological. J. Linn. Soc. 2004, 82, 511–519. [Google Scholar] [CrossRef][Green Version]
- Suda, J.; Krahulcova, A.; Travnicek, P.; Rosenbaumova, R.; Peckert, T.; Krahulec, F. Genome size variation and species relationships in hieracium sub-genus pilosella (asteraceae) as inferred by flow cytometry. Ann. Bot. 2007, 100, 1323–1335. [Google Scholar] [CrossRef] [PubMed]
- Reznik, E.; Miller, M.L.; Senbabaoglu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Blagoev, B.; Mann, M. Quantitative proteomics to study mitogen-activated protein kinases. Methods 2006, 40, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Mann, M. A practical recipe for stable isotope labeling by amino acids in cell culture (silac). Nat. Protoc. 2006, 1, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Wu, W.W.; Zhou, Y.; Shen, R.F.; Martin, B.; Maudsley, S. Effective correction of experimental errors in quantitative proteomics using stable isotope labeling by amino acids in cell culture (silac). J. Proteom. 2012, 75, 3720–3732. [Google Scholar] [CrossRef] [PubMed]
- Cairns, D.A.; Barrett, J.H.; Billingham, L.J.; Stanley, A.J.; Xinarianos, G.; Field, J.K.; Johnson, P.J.; Selby, P.J.; Banks, R.E. Sample size determination in clinical proteomic profiling experiments using mass spectrometry for class comparison. Proteomics 2009, 9, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Zybailov, B.; Mosley, A.L.; Sardiu, M.E.; Coleman, M.K.; Florens, L.; Washburn, M.P. Statistical analysis of membrane proteome expression changes in saccharomyces cerevisiae. J. Proteome Res. 2006, 5, 2339–2347. [Google Scholar] [CrossRef]
- McCarthy, J.; Hopwood, F.; Oxley, D.; Laver, M.; Castagna, A.; Righetti, P.G.; Williams, K.; Herbert, B. Carbamylation of proteins in 2-d electrophoresis--myth or reality? J. Proteome Res. 2003, 2, 239–242. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Yergey, A.L. Proteomics is analytical chemistry: Fitness-for-purpose in the application of top-down and bottom-up analyses. Proteomes 2015, 3, 440–453. [Google Scholar] [CrossRef]
- Goodwin, R.J. Sample preparation for mass spectrometry imaging: Small mistakes can lead to big consequences. J. Proteom. 2012, 75, 4893–4911. [Google Scholar] [CrossRef]
- Russell, B.; Suwanarusk, R.; Malleret, B.; Costa, F.T.; Snounou, G.; Kevin Baird, J.; Nosten, F.; Renia, L. Human ex vivo studies on asexual plasmodium vivax: The best way forward. Int. J. Parasitol. 2012, 42, 1063–1070. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis chemical analysis working group (cawg) metabolomics standards initiative (msi). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.L.; Goodman, S.R.; Branton, D. The effect of endogenous proteases on the spectrin binding proteins of human erythrocytes. Biochim. Biophys. Acta 1980, 598, 517–527. [Google Scholar] [CrossRef]
- Havanapan, P.O.; Thongboonkerd, V. Are protease inhibitors required for gel-based proteomics of kidney and urine? J. Proteome Res. 2009, 8, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Livesey, J.H.; Ellis, M.J.; Yandle, T.G. Effect of anticoagulants and storage temperatures on stability of plasma and serum hormones. Clin. Biochem. 2001, 34, 107–112. [Google Scholar] [CrossRef]
- Tran, J.C.; Zamdborg, L.; Ahlf, D.R.; Lee, J.E.; Catherman, A.D.; Durbin, K.R.; Tipton, J.D.; Vellaichamy, A.; Kellie, J.F.; Li, M.; et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480, 254–258. [Google Scholar] [CrossRef]
- Sapan, C.V.; Lundblad, R.L.; Price, N.C. Colorimetric protein assay techniques. Biotechnol. Appl. Biochem. 1999, 29, 99–108. [Google Scholar]
- Kruger, N.J. The bradford method for protein quantitation. Methods Mol. Biol. 1994, 32, 9–15. [Google Scholar]
- Noble, J.E.; Bailey, M.J. Quantitation of protein. Methods Enzymol. 2009, 463, 73–95. [Google Scholar]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Wisniewski, J.R.; Gaugaz, F.Z. Fast and sensitive total protein and peptide assays for proteomic analysis. Anal. Chem. 2015, 87, 4110–4116. [Google Scholar] [CrossRef] [PubMed]
- Waddell, W.J. A simple ultraviolet spectrophotometric method for the determination of protein. J. Lab. Clin. Med. 1956, 48, 311–314. [Google Scholar] [PubMed]
- Noaman, N.; Abbineni, P.S.; Withers, M.; Coorssen, J.R. Coomassie staining provides routine (sub)femtomole in-gel detection of intact proteoforms: Expanding opportunities for genuine top-down proteomics. Electrophoresis 2017, 38, 3086–3099. [Google Scholar] [CrossRef] [PubMed]
- Thiede, B.; Lamer, S.; Mattow, J.; Siejak, F.; Dimmler, C.; Rudel, T.; Jungblut, P.R. Analysis of missed cleavage sites, tryptophan oxidation and n-terminal pyroglutamylation after in-gel tryptic digestion. Rapid Commun. Mass Spectrom. RCM 2000, 14, 496–502. [Google Scholar] [CrossRef]
- Sun, W.; Gao, S.; Wang, L.; Chen, Y.; Wu, S.; Wang, X.; Zheng, D.; Gao, Y. Microwave-assisted protein preparation and enzymatic digestion in proteomics. Mol. Cell. Proteom. 2006, 5, 769–776. [Google Scholar] [CrossRef]
- Glatter, T.; Ludwig, C.; Ahrne, E.; Aebersold, R.; Heck, A.J.; Schmidt, A. Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem lys-c/trypsin proteolysis over trypsin digestion. J. Proteome Res. 2012, 11, 5145–5156. [Google Scholar] [CrossRef]
- Tsiatsiani, L.; Heck, A.J. Proteomics beyond trypsin. FEBS J. 2015, 282, 2612–2626. [Google Scholar] [CrossRef]
- Peng, C.; Lu, Z.; Xie, Z.; Cheng, Z.; Chen, Y.; Tan, M.; Luo, H.; Zhang, Y.; He, W.; Yang, K.; et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteom. MCP 2011, 10, M111. [Google Scholar] [CrossRef]
- Mukherjee, S.; Kapp, E.A.; Lothian, A.; Roberts, A.M.; Vasil’ev, Y.V.; Boughton, B.A.; Barnham, K.J.; Kok, W.M.; Hutton, C.A.; Masters, C.L.; et al. Characterization and identification of dityrosine cross-linked peptides using tandem mass spectrometry. Anal. Chem. 2017, 89, 6136–6145. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Teng, Y.; O’Connell, J.T.; Charytan, D.; Muller, G.A.; Muller, C.A.; Sugimoto, H.; Kalluri, R. Identification of human epididymis protein-4 as a fibroblast-derived mediator of fibrosis. Nat. Med. 2013, 19, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Somiari, R.I.; Renganathan, K.; Russell, S.; Wolfe, S.; Mayko, F.; Somiari, S.B. A colorimetric method for monitoring tryptic digestion prior to shotgun proteomics. Int. J. Proteom. 2014, 2014, 125482. [Google Scholar] [CrossRef] [PubMed]
- Karp, N.A.; Kreil, D.P.; Lilley, K.S. Determining a significant change in protein expression with decyder during a pair-wise comparison using two-dimensional difference gel electrophoresis. Proteomics 2004, 4, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Tonge, R.; Shaw, J.; Middleton, B.; Rowlinson, R.; Rayner, S.; Young, J.; Pognan, F.; Hawkins, E.; Currie, I.; Davison, M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics 2001, 1, 377–396. [Google Scholar] [CrossRef]
- Karp, N.A.; Lilley, K.S. Maximising sensitivity for detecting changes in protein expression: Experimental design using minimal cydyes. Proteomics 2005, 5, 3105–3115. [Google Scholar] [CrossRef] [PubMed]
- McNamara, L.E.; Kantawong, F.A.; Dalby, M.J.; Riehle, M.O.; Burchmore, R. Preventing and troubleshooting artefacts in saturation labelled fluorescence 2-d difference gel electrophoresis (saturation dige). Proteomics 2011, 11, 4610–4621. [Google Scholar] [CrossRef] [PubMed]
- Thiede, B.; Koehler, C.J.; Strozynski, M.; Treumann, A.; Stein, R.; Zimny-Arndt, U.; Schmid, M.; Jungblut, P.R. High resolution quantitative proteomics of hela cells protein species using stable isotope labeling with amino acids in cell culture(silac), two-dimensional gel electrophoresis(2de) and nano-liquid chromatograpohy coupled to an ltq-orbitrapmass spectrometer. Mol. Cell. Proteom. 2013, 12, 529–538. [Google Scholar]
- Zhan, X.; Yang, H.; Peng, F.; Li, J.; Mu, Y.; Long, Y.; Cheng, T.; Huang, Y.; Li, Z.; Lu, M.; et al. How many proteins can be identified in a 2de gel spot within an analysis of a complex human cancer tissue proteome? Electrophoresis 2018, 39, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Takemori, N.; Takemori, A.; Wongkongkathep, P.; Nshanian, M.; Loo, R.R.O.; Lermyte, F.; Loo, J.A. Top-down/bottom-up mass spectrometry workflow using dissolvable polyacrylamide gels. Anal. Chem. 2017, 89, 8244–8250. [Google Scholar] [CrossRef] [PubMed]
- Weist, S.; Eravci, M.; Broedel, O.; Fuxius, S.; Eravci, S.; Baumgartner, A. Results and reliability of protein quantification for two-dimensional gel electrophoresis strongly depend on the type of protein sample and the method employed. Proteomics 2008, 8, 3389–3396. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.E.; Pawar, V.; Smith, S.J.; Graumann, K. Protein interactions at the higher plant nuclear envelope: Evidence for a linker of nucleoskeleton and cytoskeleton complex. Front. Plant Sci. 2014, 5, 183. [Google Scholar] [CrossRef] [PubMed]
- Achour, B.; Dantonio, A.; Niosi, M.; Novak, J.J.; Al-Majdoub, Z.M.; Goosen, T.C.; Rostami-Hodjegan, A.; Barber, J. Data generated by quantitative liquid chromatography-mass spectrometry proteomics are only the start and not the endpoint: Optimization of quantitative concatemer-based measurement of hepatic uridine-5’-diphosphate-glucuronosyltransferase enzymes with reference to catalytic activity. Drug Metab. Dispos. 2018, 46, 805–812. [Google Scholar] [PubMed]
- Unwin, R.D.; Griffiths, J.R.; Whetton, A.D. Simultaneous analysis of relative protein expression levels across multiple samples using itraq isobaric tags with 2d nano lc-ms/ms. Nat. Protoc. 2010, 5, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Warwood, S.; Mohammed, S.; Cristea, I.M.; Evans, C.; Whetton, A.D.; Gaskell, S.J. Guanidination chemistry for qualitative and quantitative proteomics. Rapid Commun. Mass Spectrom. RCM 2006, 20, 3245–3256. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, P.; Hansen, J.B.; Allen, M. Microvolume protein concentration determination using the nanodrop 2000c spectrophotometer. J. Vis. Exp. 2009. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Harvey, A.; Yen, T.Y.; Aizman, I.; Tate, C.; Case, C. Proteomic analysis of the extracellular matrix produced by mesenchymal stromal cells: Implications for cell therapy mechanism. PLoS ONE 2013, 8, e79283. [Google Scholar] [CrossRef]
- Fonslow, B.R.; Stein, B.D.; Webb, K.J.; Xu, T.; Choi, J.; Park, S.K.; Yates, J.R., 3rd. Digestion and depletion of abundant proteins improves proteomic coverage. Nat. Methods 2013, 10, 54–56. [Google Scholar] [CrossRef]
- Zhou, Q.; Andersson, R.; Hu, D.; Bauden, M.; Kristl, T.; Sasor, A.; Pawlowski, K.; Pla, I.; Hilmersson, K.S.; Zhou, M.; et al. Quantitative proteomics identifies brain acid soluble protein 1 (basp1) as a prognostic biomarker candidate in pancreatic cancer tissue. EBioMedicine 2019, 43, 282–294. [Google Scholar] [CrossRef]
- Escher, C.; Reiter, L.; MacLean, B.; Ossola, R.; Herzog, F.; Chilton, J.; MacCoss, M.J.; Rinner, O. Using irt, a normalized retention time for more targeted measurement of peptides. Proteomics 2012, 12, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.R.; Lilley, K.S. Pipeline to assess the greatest source of technical variance in quantitative proteomics using metabolic labelling. J. Proteom. 2012, 77, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Mann, M. A proteomics approach to the protein normalization problem: Selection of unvarying proteins for ms-based proteomics and western blotting. J. Proteome Res. 2016, 15, 2321–2326. [Google Scholar] [CrossRef] [PubMed]
- Eltoweissy, M.; Dihazi, G.H.; Muller, G.A.; Asif, A.R.; Dihazi, H. Protein dj-1 and its anti-oxidative stress function play an important role in renal cell mediated response to profibrotic agents. Mol. Biosyst. 2016, 12, 1842–1859. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallstrom, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.H.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.W.B.; Wong, L. Advanced bioinformatics methods for practical applications in proteomics. Brief. Bioinform. 2019, 20, 347–355. [Google Scholar] [CrossRef]
- Bruderer, R.; Muntel, J.; Muller, S.; Bernhardt, O.M.; Gandhi, T.; Cominetti, O.; Macron, C.; Carayol, J.; Rinner, O.; Astrup, A.; et al. Analysis of 1508 plasma samples by capillary-flow data-independent acquisition profiles proteomics of weight loss and maintenance. Mol. Cell. Proteom. MCP 2019, 18, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Lecchi, P.; Zhao, J.; Wiggins, W.S.; Chen, T.H.; Bertenshaw, G.P.; Yip, P.F.; Mansfield, B.C.; Peltier, J.M. A method for assessing and maintaining the reproducibility of mass spectrometric analyses of complex samples. Rapid Commun. Mass Spectrom. RCM 2009, 23, 1817–1824. [Google Scholar] [CrossRef]
- Nesatyy, V.J.; Groh, K.; Nestler, H.; Suter, M.J. On the acquisition of +1 charge states during high-throughput proteomics: Implications on reproducibility, number and confidence of protein identifications. J. Proteom. 2009, 72, 761–770. [Google Scholar] [CrossRef]
- Rudnick, P.A.; Wang, X.; Yan, X.; Sedransk, N.; Stein, S.E. Improved normalization of systematic biases affecting ion current measurements in label-free proteomics data. Mol. Cell. Proteom. MCP 2014, 13, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- LeDuc, R.D.; Taylor, G.K.; Kim, Y.B.; Januszyk, T.E.; Bynum, L.H.; Sola, J.V.; Garavelli, J.S.; Kelleher, N.L. Prosight ptm: An integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res. 2004, 32, W340–W345. [Google Scholar] [CrossRef] [PubMed]
- Horn, D.M.; Zubarev, R.A.; McLafferty, F.W. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J. Am. Soc. Mass Spectrom. 2000, 11, 320–332. [Google Scholar] [CrossRef]
- Kultima, K.; Nilsson, A.; Scholz, B.; Rossbach, U.L.; Falth, M.; Andren, P.E. Development and evaluation of normalization methods for label-free relative quantification of endogenous peptides. Mol. Cell. Proteom. MCP 2009, 8, 2285–2295. [Google Scholar] [CrossRef]
- Stratton, K.G.; Webb-Robertson, B.M.; McCue, L.A.; Stanfill, B.; Claborne, D.; Godinez, I.; Johansen, T.; Thompson, A.M.; Burnum-Johnson, K.E.; Waters, K.M.; et al. Pmartr: Quality control and statistics for mass spectrometry-based biological data. J. Proteome Res. 2019, 18, 1418–1425. [Google Scholar] [CrossRef]
- Willforss, J.; Chawade, A.; Levander, F. Normalyzerde: Online tool for improved normalization of omics expression data and high-sensitivity differential expression analysis. J. Proteome Res. 2019, 18, 732–740. [Google Scholar] [CrossRef]
- Murie, C.; Sandri, B.; Sandberg, A.S.; Griffin, T.J.; Lehtio, J.; Wendt, C.; Larsson, O. Normalization of mass spectrometry data (nomad). Adv. Biol. Regul. 2018, 67, 128–133. [Google Scholar] [CrossRef]
- Ting, L.; Cowley, M.J.; Hoon, S.L.; Guilhaus, M.; Raftery, M.J.; Cavicchioli, R. Normalization and statistical analysis of quantitative proteomics data generated by metabolic labeling. Mol. Cell. Proteom. MCP 2009, 8, 2227–2242. [Google Scholar] [CrossRef]
- Berg, P.; McConnell, E.W.; Hicks, L.M.; Popescu, S.C.; Popescu, G.V. Evaluation of linear models and missing value imputation for the analysis of peptide-centric proteomics. BMC Bioinform. 2019, 20, 102. [Google Scholar] [CrossRef]
- Van Houtven, J.; Agten, A.; Boonen, K.; Baggerman, G.; Hooyberghs, J.; Laukens, K.; Valkenborg, D. Qcquan: A web tool for the automated assessment of protein expression and data quality of labeled mass spectrometry experiments. J. Proteome Res. 2019, 18, 2221–2227. [Google Scholar] [CrossRef]
- Navarro, P.; Kuharev, J.; Gillet, L.C.; Bernhardt, O.M.; MacLean, B.; Rost, H.L.; Tate, S.A.; Tsou, C.C.; Reiter, L.; Distler, U.; et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat. Biotechnol. 2016, 34, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.M.; Hu, Y.; Xia, Y.; Ouyang, Z. Power normalization for mass spectrometry data analysis and analytical method assessment. Anal. Chem. 2016, 88, 3156–3163. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, R.E.; Carroll, H.P.; Harris, A.; Maher, E.R.; Selby, P.J.; Banks, R.E. Housekeeping proteins: A preliminary study illustrating some limitations as useful references in protein expression studies. Proteomics 2005, 5, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Herbrich, S.M.; Cole, R.N.; West, K.P., Jr.; Schulze, K.; Yager, J.D.; Groopman, J.D.; Christian, P.; Wu, L.; O’Meally, R.N.; May, D.H.; et al. Statistical inference from multiple itraq experiments without using common reference standards. J. Proteome Res. 2013, 12, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, S.; Huh, W.K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Padula, M.P.; Davey, P.; Pernice, M.; Jiang, Z.; Sablok, G.; Contreras-Porcia, L.; Ralph, P.J. Proteome analysis reveals extensive light stress-response reprogramming in the seagrass zostera muelleri (alismatales, zosteraceae) metabolism. Front. Plant Sci. 2016, 7, 2023. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Category | Method | Positives | Negatives |
|---|---|---|---|
| Fluorometric | UV absorption (Tryptophan) | Rapid Low cost Sample recoverable for proteomic preparations | Quantitates only the amino acids tyrosine, tryptophan and phenylalanine Sensitive to detergents |
| Qubit Protein Assay | Sensitive at low protein concentrations Small sample volumes (≤10 µL) | Sensitive to temperature fluctuations Easily saturated | |
| Colorimetric | BCA | Compatible with detergents at low concentrations | Quantitates only the amino acids tyrosine, tryptophan and cysteine Sample not recoverable Sensitive to detergents |
| Bradford/Coomassie | Compatible with reducing agents Reagent binds to protein rather than to individual amino acids | Sample not recoverable | |
| Lowry | Sensitive | Timely and laborious procedure Sample not recoverable Sensitive to detergents and other common reagents | |
| Densitometry | SDS-Page (In-gel) | Highly accurate Sample recoverable for proteomic preparations however, laborious process | Analysis susceptible to bias depending on gating of bands |
| Western-Blot and ELISA (also considered Flourometric or Colorimetric depending on tag or application) | Target-protein specific | Analysis susceptible to bias depending on gating of bands |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Rourke, M.B.; Town, S.E.L.; Dalla, P.V.; Bicknell, F.; Koh Belic, N.; Violi, J.P.; Steele, J.R.; Padula, M.P. What is Normalization? The Strategies Employed in Top-Down and Bottom-Up Proteome Analysis Workflows. Proteomes 2019, 7, 29. https://doi.org/10.3390/proteomes7030029
O’Rourke MB, Town SEL, Dalla PV, Bicknell F, Koh Belic N, Violi JP, Steele JR, Padula MP. What is Normalization? The Strategies Employed in Top-Down and Bottom-Up Proteome Analysis Workflows. Proteomes. 2019; 7(3):29. https://doi.org/10.3390/proteomes7030029
Chicago/Turabian StyleO’Rourke, Matthew B., Stephanie E. L. Town, Penelope V. Dalla, Fiona Bicknell, Naomi Koh Belic, Jake P. Violi, Joel R. Steele, and Matthew P. Padula. 2019. "What is Normalization? The Strategies Employed in Top-Down and Bottom-Up Proteome Analysis Workflows" Proteomes 7, no. 3: 29. https://doi.org/10.3390/proteomes7030029
APA StyleO’Rourke, M. B., Town, S. E. L., Dalla, P. V., Bicknell, F., Koh Belic, N., Violi, J. P., Steele, J. R., & Padula, M. P. (2019). What is Normalization? The Strategies Employed in Top-Down and Bottom-Up Proteome Analysis Workflows. Proteomes, 7(3), 29. https://doi.org/10.3390/proteomes7030029