Development of Targeted Mass Spectrometry-Based Approaches for Quantitation of Proteins Enriched in the Postsynaptic Density (PSD)

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
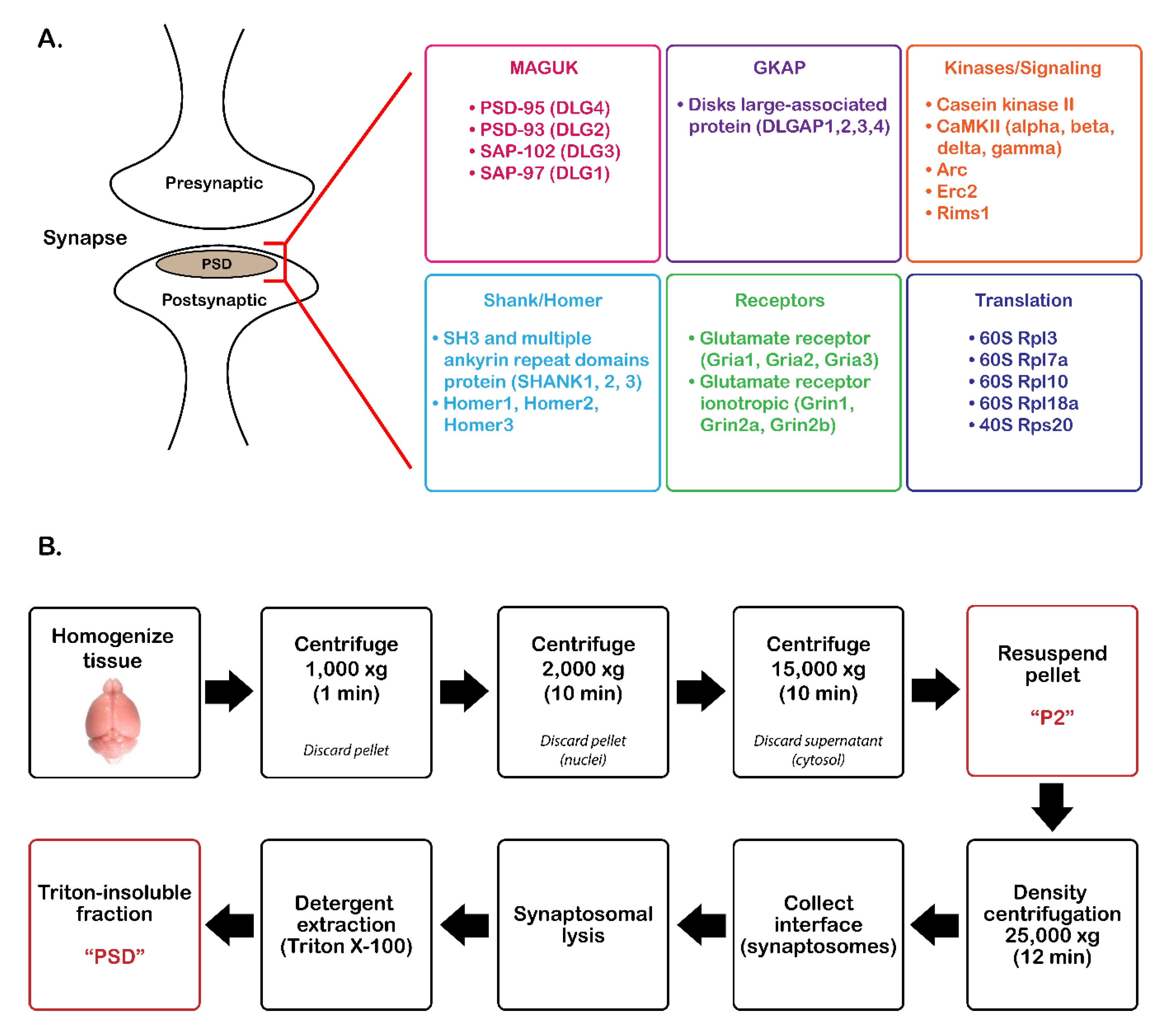
2.1. Tissue Collection
2.2. PSD Enrichment
2.3. Immunoblot Analysis
2.4. Sample Preparation for LC–MS/MS
2.5. Parallel Reaction Monitoring (PRM) Method Development
2.5.1. Peptide Design and Synthesis
2.5.2. SIL Peptide Dilution Series (Neat)
2.5.3. SIL Peptide Dilution Series in Fixed Biological Peptide Matrix
2.6. LC–MS/MS
2.6.1. Data-Independent Acquisition (DIA)
2.6.2. Parallel Reaction Monitoring (PRM)
2.7. Data Analysis
2.7.1. Data-Independent Acquisition (DIA)
2.7.2. Parallel Reaction Monitoring (PRM)
3. Results
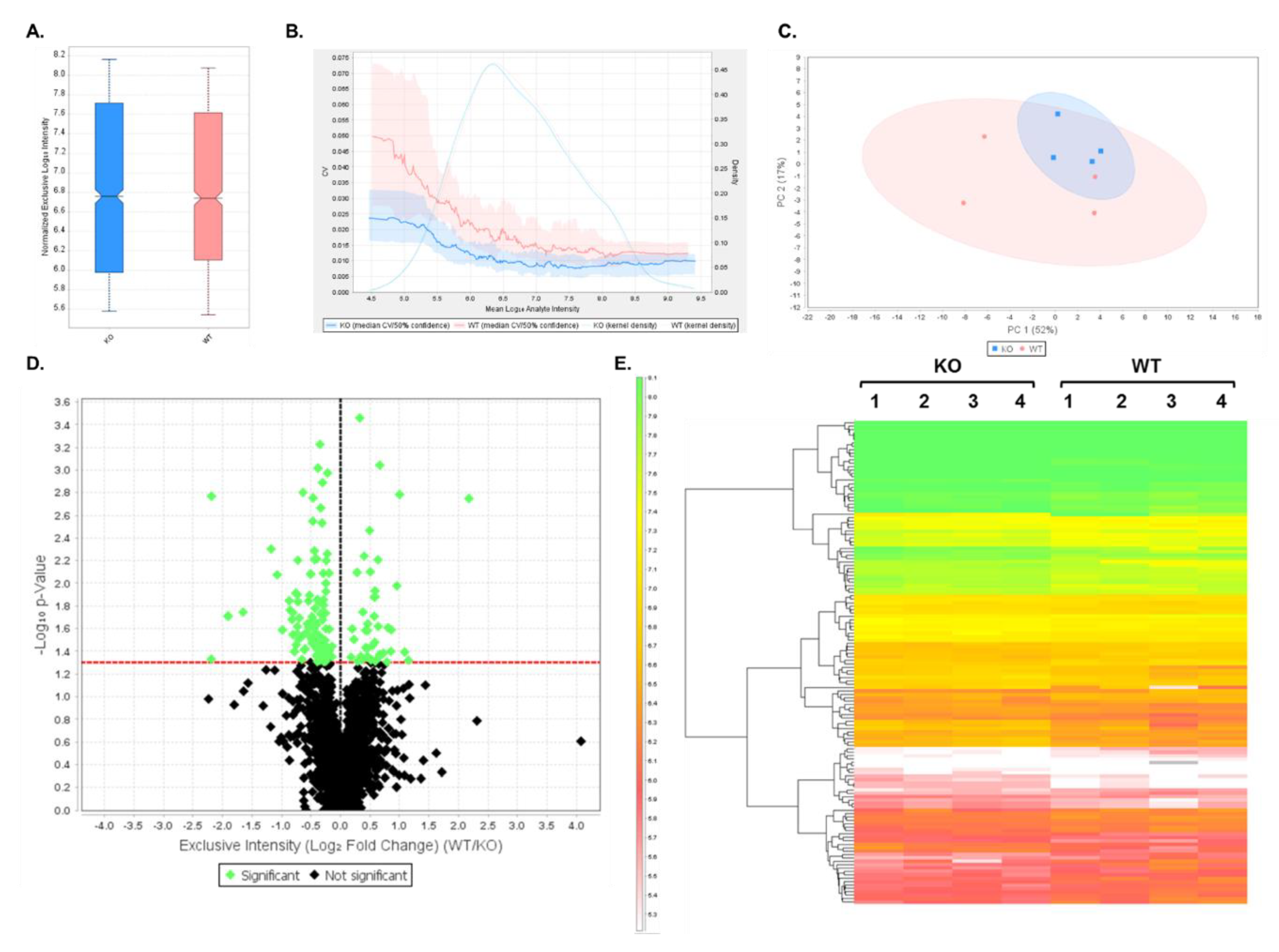
3.1. Validation of PSD Enrichment
3.2. DIA Results Indicated Minor Differences Between WT and Shank3B KO PSD-Enriched Proteins
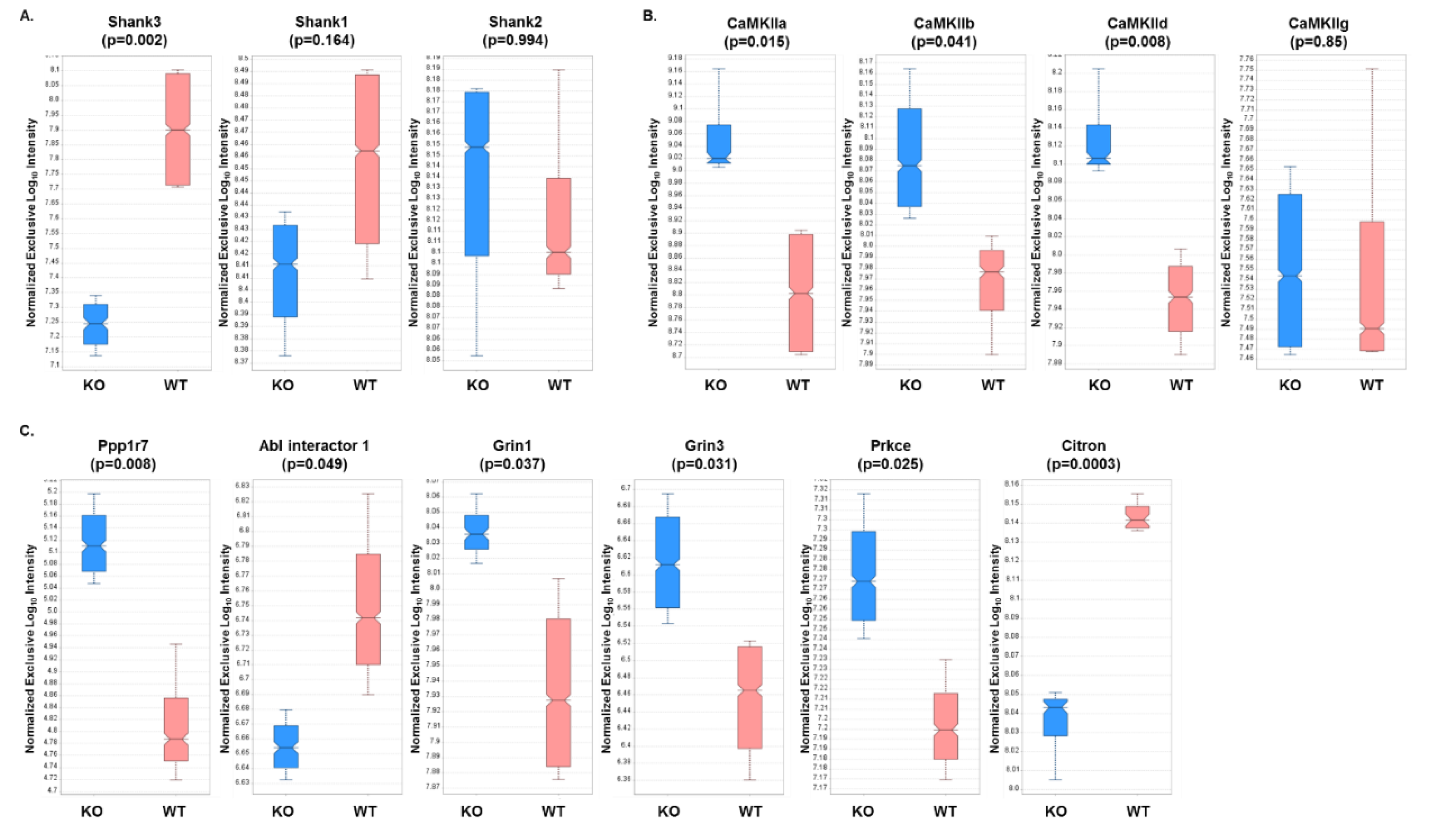
3.3. Expression Profiles from DIA Analysis of Wild-Type and Shank3B KO PSD Fractions Revealed Shank3-Associated Patterns
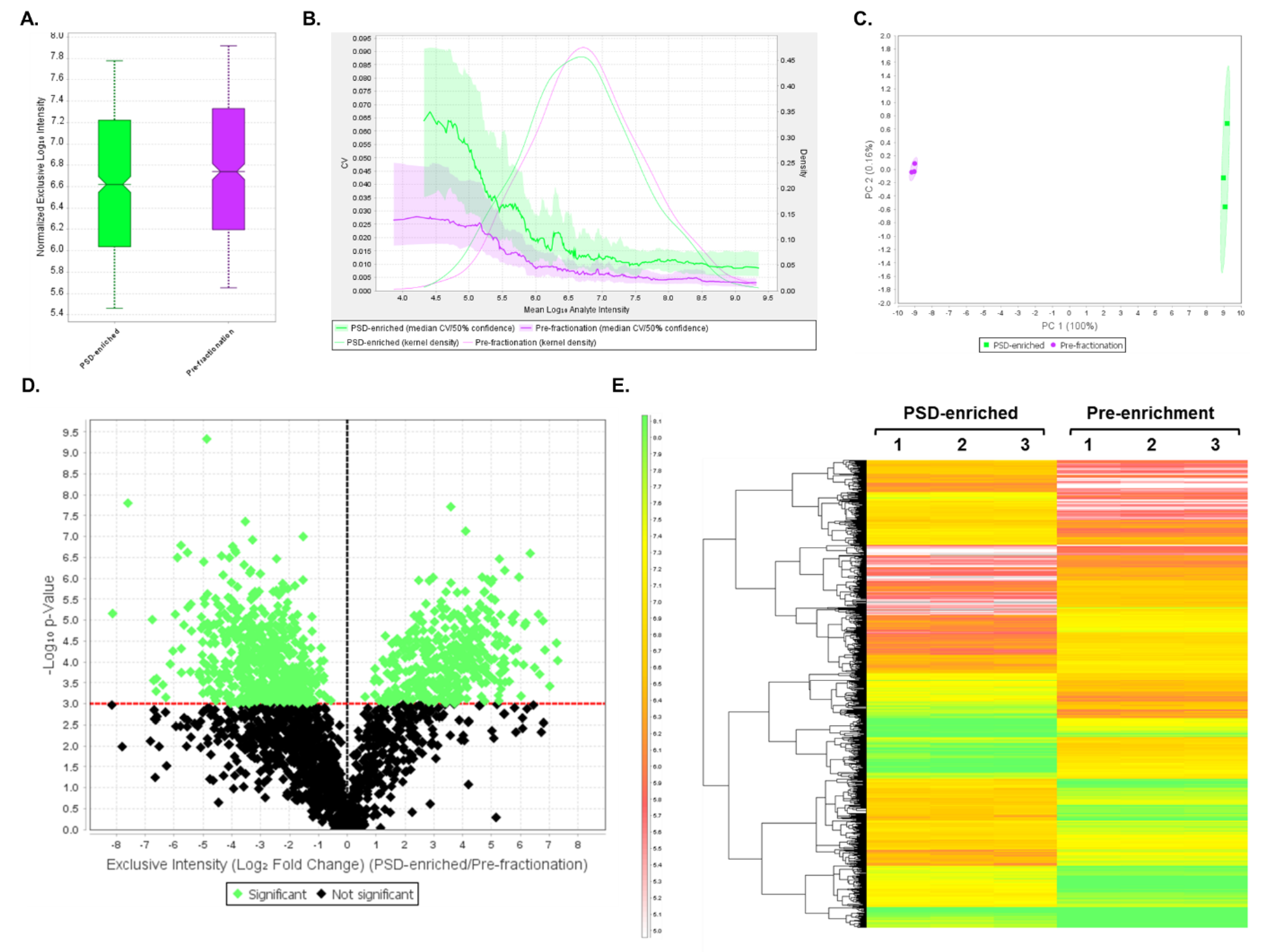
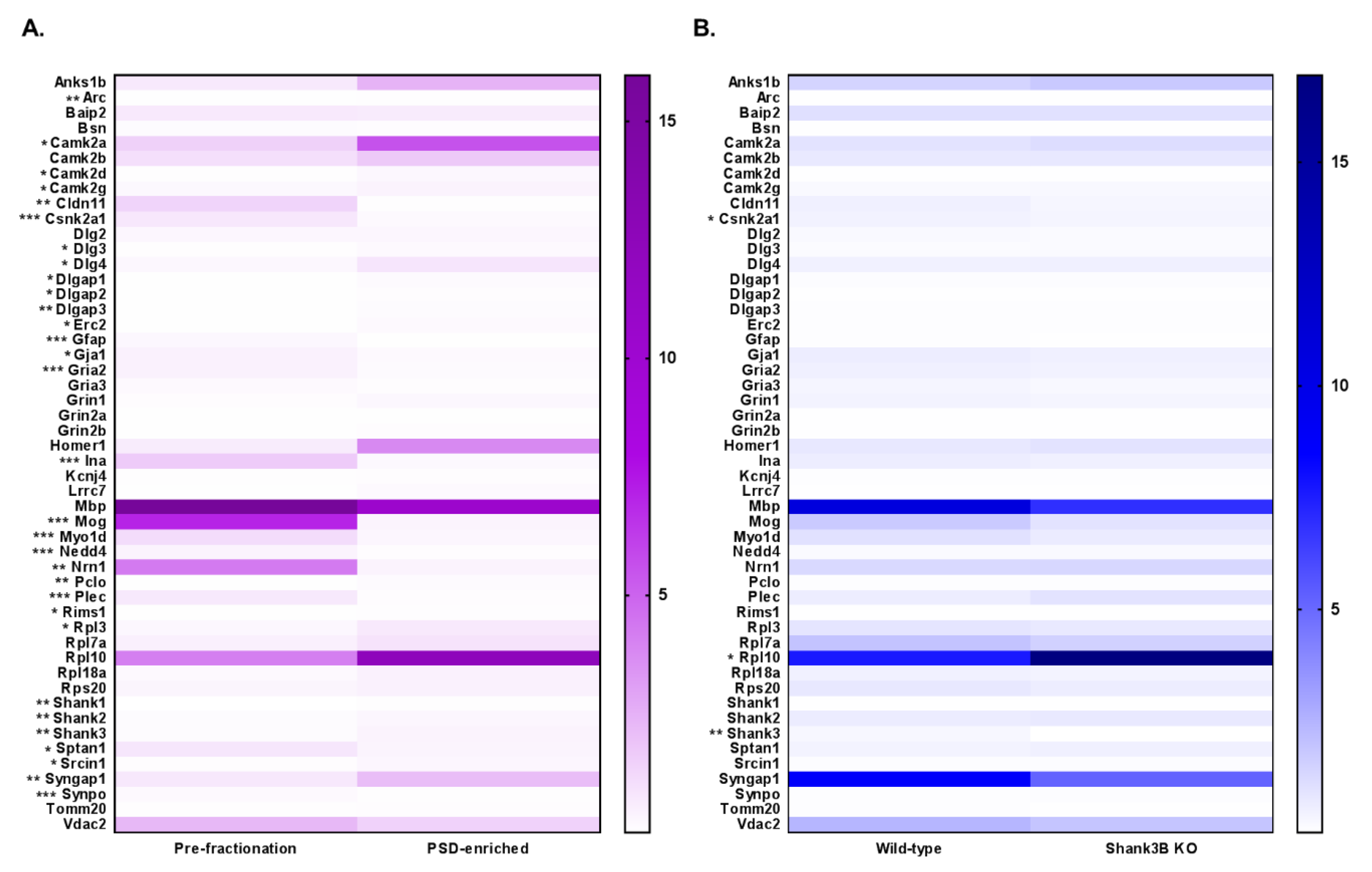
3.4. DIA Analyses Indicated Significant Differences in Protein Expression between Pre-Fractionation and PSD-Enriched Samples
3.5. DIA Expression Profiles Displayed Enrichment of PSD Proteins and Depletion of Contaminants in PSD-Enriched Fractions Comparerd to Pre-Fractionation Samples
3.6. Peptide Design for PRM Analysis
3.7. PRM Analysis of PSD Target Proteins Revealed Quantitative Differences in Protein Expression in WT Versus Shank3B KO Mouse Brain Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Palay, S.L. Synapses in the Central Nervous System. J. Cell Biol. 1956, 2, 193–201. [Google Scholar] [CrossRef]
- Cohen, R.S.; Blomberg, F.; Berzins, K.; Siekevitz, P. The structure of postsynaptic densities isolated from dog cerebral cortex. J. Cell Biol. 1977, 74, 181–203. [Google Scholar] [CrossRef]
- Harris, K.M.; Weinberg, R.J. Ultrastructure of Synapses in the Mammalian Brain. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Dosemeci, A.; Weinberg, R.J.; Reese, T.S.; Tao-Cheng, J.H. The postsynaptic density: There is more than meets the eye. Front. Synaptic Neurosci. 2016, 8. [Google Scholar] [CrossRef]
- Cho, K.-O.; Hunt, C.A.; Kennedy, M.B. The Rat Brain Postsynaptic Density Fraction Contains a Homolog of the Drosophila Discs-Large Tumor Suppressor Protein. Neuron 1992, 9, 929–942. [Google Scholar] [CrossRef]
- Kistner, U.; Wenzel, B.M.; Vehs, R.W.; Cases-langhoff, C.; Garner, A.M.; Appeltauer, U.; Voss, B.; Gundelfinger, E.D.; Garner, C.C. SAP90, a Rat Presynaptic Protein Related to the Product of the Drosophila Tumor Suppressor Gene dlg-A. J. Biol. Chem. 1993, 268, 4580–4583. [Google Scholar] [PubMed]
- Chen, X.; Vinade, L.; Leapman, R.D.; Petersen, J.D.; Nakagawa, T.; Phillips, T.M.; Sheng, M.; Reese, T.S. Mass of the postsynaptic density and enumeration of three key molecules. Proc. Natl. Acad. Sci. USA 2005, 102, 11551–11556. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Kawabata, I.; Sobue, K.; Okabe, S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat. Methods 2005, 2, 677–684. [Google Scholar] [CrossRef]
- Takeuchi, M.; Hata, Y.; Hirao, K.; Toyoda, A.; Irie, M.; Takai, Y. SAPAPs: A family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J. Biol. Chem. 1997, 272, 11943–11951. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, I.; Malinow, R. Postsynaptic Density 95 controls AMPA Receptor Incorporation during Long-Term Potentiation and Experience-Driven Synaptic Plasticity. J. Neurosci. 2004, 24, 916–927. [Google Scholar] [CrossRef]
- Garner, C.C.; Nash, J.; Huganir, R.L. PDZ domains in synapse assembly and signalling. Trends Cell Biol. 2000, 10, 274–280. [Google Scholar] [CrossRef]
- Sheng, M.; Sala, C. PDZ Domains and the Organization of Supramolecular Complexes. Annu. Rev. Neurosci. 2001, 24, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Naisbitt, S.; Hsueh, Y.-P.; Rao, A.; Rothschild, A.; Craig, A.M.; Sheng, M. GKAP, a Novel Synaptic Protein That Interacts with the Guanylate Kinase-like Domain of the PSD-95. J. Cell Biol. 1997, 136, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Naisbitt, S.; Kim, E.; Tu, J.C.; Xiao, B.; Sala, C.; Valtschanoff, J.; Weinberg, R.J.; Worley, P.F.; Sheng, M. Shank, a Novel Family of Postsynaptic Density Proteins that Binds to the NMDA Receptor/PSD-95/GKAP Complex and Cortactin. Neuron 1999, 23, 569–582. [Google Scholar] [CrossRef]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef]
- Hung, A.Y.; Futai, K.; Sala, C.; Valtschanoff, J.G.; Ryu, J.; Woodworth, M.A.; Kidd, F.L.; Sung, C.C.; Miyakawa, T.; Bear, M.F.; et al. Smaller Dendritic Spines, Weaker Synaptic Transmission, but Enhanced Spatial Learning in Mice Lacking Shank1. Neurosci. Res. 2009, 28, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Inokuchi, K.; Ozawa, F.; Fukazawa, Y.; Saitoh, Y.; Sugiyama, H. Novel Members of the Vesl/Homer Family of PDZ Proteins That Bind Metabotropic Glutamate Receptors. J. Biol. Chem. 1998, 273, 23969–23975. [Google Scholar] [CrossRef] [PubMed]
- Brakeman, P.R.; Lanahan, A.A.; O’Brien, R.; Roche, K.; Barnes, C.A.; Huganir, R.L.; Worley, P.F. Homer: A protein that selectively binds metabotropic glutamate receptors. Nature 1997, 386, 284–288. [Google Scholar] [CrossRef]
- Xiao, B.; Tu, J.C.; Petralia, R.S.; Yuan, J.P.; Doan, A.; Breder, C.D.; Ruggiero, A.; Lanahan, A.A.; Wenthold, R.J.; Worley, P.F. Homer regulates the association of group 1 metabotropic glutamate receptors with multivalent complexes of Homer-related, synaptic proteins. Neuron 1998, 21, 707–716. [Google Scholar] [CrossRef]
- Barzik, M.; Carl, U.D.; Schubert, W.D.; Frank, R.; Wehland, J.; Heinz, D.W. The N-terminal domain of Homer/Vesl is a new class II EVH1 domain. J. Mol. Biol. 2001, 309, 155–169. [Google Scholar] [CrossRef]
- Sun, J.; Tadokoro, S.; Imanaka, T.; Murakami, S.D.; Nakamura, M.; Kashiwada, K.; Ko, J.; Nishida, W.; Sobue, K. Isolation of PSD-Zip45, a novel Homer/vesl family protein containing leucine zipper motifs, from rat brain 1. FEBS Lett. 1998, 437, 304–308. [Google Scholar] [CrossRef]
- Tadokoro, S.; Tachibana, T.; Imanaka, T.; Nishida, W.; Sobue, K. Involvement of unique leucine-zipper motif of PSD-Zip45 (Homer 1c/vesl-1L) in group 1 metabotropic glutamate receptor clustering. Proc. Natl. Acad. Sci. USA 1999, 96, 13801–13806. [Google Scholar] [CrossRef]
- Hayashi, M.K.; Ames, H.M.; Hayashi, Y. Tetrameric Hub Structure of Postsynaptic Scaffolding Protein Homer. J. Neurosci. 2006, 26, 8492–8501. [Google Scholar] [CrossRef]
- Hayashi, M.K.; Tang, C.; Verpelli, C.; Narayanan, R.; Stearns, M.H.; Xu, R.-M.; Li, H.; Sala, C.; Hayashi, Y. The Postsynaptic Density Proteins Homer and Shank Form a Polymeric Network Structure. Cell 2009, 137, 159–171. [Google Scholar] [CrossRef]
- Shiraishi-Yamaguchi, Y.; Sato, Y.; Sakai, R.; Mizutani, A.; Knöpfel, T.; Mori, N.; Mikoshiba, K.; Furuichi, T. Interaction of Cupidin/Homer2 with two actin cytoskeletal regulators, Cdc42 small GTPase and Drebrin, in dendritic spines. BMC Neurosci. 2009, 10, 1–14. [Google Scholar] [CrossRef]
- Baron, M.K.; Boeckers, T.M.; Vaida, B.; Faham, S.; Gingery, M.; Sawaya, D.; Gundelfinger, E.D.; Bowie, J.U. An architectural framework that may lie at the core of the postsynaptic density. Science 2006, 311, 531–535. [Google Scholar] [CrossRef]
- Goulding, S.P.; Szumlinski, K.K.; Contet, C.; MacCoss, M.J.; Wu, C.C. A mass spectrometry-based proteomic analysis of Homer2-interacting proteins in the mouse brain. J. Proteomics 2017, 166, 127–137. [Google Scholar] [CrossRef]
- Erondu, N.E.; Kennedy, M.B. Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J. Neurosci. 1985, 5, 3270–3277. [Google Scholar] [CrossRef]
- Dosemeci, A.; Tao-Cheng, J.H.; Vinade, L.; Winters, C.A.; Pozzo-Miller, L.; Reese, T.S. Glutamate-induced transient modification of the postsynaptic density. Proc. Natl. Acad. Sci. USA 2001, 98, 10428–10432. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.R.; Park, M.; Martone, M.E.; Fischer, W.H.; Ellisman, M.H.; Zivin, J.A. Assembly of proteins to postsynaptic densities after transient cerebral ischemia. J. Neurosci. 1998, 18, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Okumura-Noji, K.; Tanaka, R.; Tada, T. Rapid Translocation of Cytosolic Ca2+/Calmodulin-Dependent Protein Kinase II into Postsynaptic Density After Decapitation. J. Neurochem. 1994, 63, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Martone, M.E.; Jones, Y.Z.; Young, S.J.; Ellisman, M.H.; Zivin, J.A.; Hu, B.-R. Modification of Postsynaptic Densities after Transient Cerebral Ischemia: A Quantitative and Three-Dimensional Ultrastructural Study. J. Neurosci. 1999, 19, 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef]
- Shonesy, B.C.; Jalan-Sakrikar, N.; Cavener, V.S.; Colbran, R.J. CaMKII: A molecular substrate for synaptic plasticity and memory. Prog. Mol. Biol. Transl. Sci. 2014, 122, 61–87. [Google Scholar] [CrossRef] [PubMed]
- Dhamne, S.C.; Silverman, J.L.; Super, C.E.; Lammers, S.H.T.; Hameed, M.Q.; Modi, M.E.; Copping, N.A.; Pride, M.C.; Smith, D.G.; Rotenberg, A.; et al. Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol. Autism 2017, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.T.; Wang, W.; Croney, D.M.; Kozorovitskiy, Y.; Sabatini, B.L. Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B-/- mice. Nat. Neurosci. 2016, 19, 716–724. [Google Scholar] [CrossRef]
- Berryer, M.H.; Hamdan, F.F.; Klitten, L.L.; Møller, R.S.; Carmant, L.; Schwartzentruber, J.; Patry, L.; Dobrzeniecka, S.; Rochefort, D.; Neugnot-Cerioli, M.; et al. Mutations in SYNGAP1 Cause Intellectual Disability, Autism, and a Specific Form of Epilepsy by Inducing Haploinsufficiency. Hum. Mutat. 2013, 34, 385–394. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Gauthier, J.; Spiegelman, D.; Noreau, A.; Yang, Y.; Pellerin, S.; Dobrzeniecka, S.; Cote, M.; Perreau-Linck, E.; Carmant, L.; et al. Mutations in SYNGAP1 in Autosomal Nonsyndromic Mental Retardation. N. Engl. J. Med. 2009, 360, 599–605. [Google Scholar] [CrossRef]
- Parker, M.J.; Fryer, A.E.; Shears, D.J.; Lachlan, K.L.; Mckee, S.A.; Magee, A.C.; Mohammed, S.; Vasudevan, P.C.; Park, S.M.; Benoit, V.; et al. De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am. J. Med. Genet. Part A 2015, 167, 2231–2237. [Google Scholar] [CrossRef]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.M.; Hooli, B.; DiVito, J.; Ionita, I.; et al. Genome-wide Association Analysis Reveals Putative Alzheimer’s Disease Susceptibility Loci in Addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef]
- Stephenson, J.R.; Wang, X.; Perfitt, T.L.; Parrish, W.P.; Shonesy, B.C.; Marks, C.R.; Mortlock, D.P.; Nakagawa, T.; Sutcliffe, J.S.; Colbran, R.J. A Novel Human CAMK2A Mutation Disrupts Dendritic Morphology and Synaptic Transmission, and Causes ASD-Related Behaviors. J. Neurosci. 2017, 37, 2216–2233. [Google Scholar] [CrossRef]
- Fernández, E.; Collins, M.O.; Uren, R.T.; Kopanitsa, M.V.; Komiyama, N.H.; Croning, M.D.R.; Zografos, L.; Armstrong, J.D.; Choudhary, J.S.; Grant, S.G.N. Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol. Syst. Biol. 2009, 5, 1–17. [Google Scholar] [CrossRef]
- Carlin, R.K.; Grab, D.J.; Cohen, R.S.; Siekevitz, P. Isolation and characterization of postsynaptic densities from various brain regions: Enrichment of different types of postsynaptic densities. J. Cell Biol. 1980, 86. [Google Scholar] [CrossRef]
- De Robertis, E.; Azcurra, J.M.; Fiszer, S. Ultrastructure and cholinergic binding capacity of junctional complexes isolated from rat brain. Brain Res. 1967, 5, 45–56. [Google Scholar] [CrossRef]
- Davis, G.A.; Bloom, F.E. Isolation of synaptic junctional complexes from rat brain. Brain Res. 1973, 62, 135–153. [Google Scholar] [CrossRef]
- Zeng, M.; Shang, Y.; Araki, Y.; Guo, T.; Huganir, R.L.; Zhang, M. Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell 2016, 166, 1163–1175.e12. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Chen, X.; Guan, D.; Xu, J.; Wu, H.; Tong, P.; Zhang, M. Reconstituted Postsynaptic Density as a Molecular Platform for Understanding Synapse Formation and Plasticity. Cell 2018, 174, 1172–1187.e16. [Google Scholar] [CrossRef] [PubMed]
- Bartol, T.M.; Bromer, C.; Kinney, J.; Chirillo, M.A.; Bourne, J.N.; Harris, K.M.; Sejnowski, T.J. Nanoconnectomic upper bound on the variability of synaptic plasticity. Elife 2015, 4, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, M.; Honkura, N.; Ellis-Davies, G.C.R.; Kasai, H. Structural basis of long-term potentiation in single dendritic spines. Nature 2004, 429, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, J.; Yasuda, R. Biochemical Computation for Spine Structural Plasticity. Neuron 2015, 87, 63–75. [Google Scholar] [CrossRef]
- Roy, M.; Sorokina, O.; Skene, N.; Simonnet, C.; Mazzo, F.; Zwart, R.; Sher, E.; Smith, C.; Armstrong, J.D.; Grant, S.G.N. Proteomic analysis of postsynaptic proteins in regions of the human neocortex. Nat. Neurosci. 2018, 21, 130–141. [Google Scholar] [CrossRef]
- Bayés, Á.; Van De Lagemaat, L.N.; Collins, M.O.; Croning, M.D.R.; Whittle, I.R.; Choudhary, J.S.; Grant, S.G.N. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 2011, 14, 19–21. [Google Scholar] [CrossRef]
- Bayés, À; Collins, M.O.; Croning, M.D.R.; van de Lagemaat, L.N.; Choudhary, J.S.; Grant, S.G.N. Comparative Study of Human and Mouse Postsynaptic Proteomes Finds High Compositional Conservation and Abundance Differences for Key Synaptic Proteins. PLoS ONE 2012, 7, 1–13. [Google Scholar] [CrossRef]
- Bayés, À.; Collins, M.O.; Galtrey, C.M.; Simonnet, C.; Roy, M.; Croning, M.D.R.; Gou, G.; Van De Lagemaat, L.N.; Milward, D.; Whittle, I.R.; Smith, C.; et al. Human post-mortem synapse proteome integrity screening for proteomic studies of postsynaptic complexes. Mol. Brain 2014, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Kim, M.J.; Cheng, D.; Duong, D.M.; Gygi, S.P.; Sheng, M. Semi-quantitative Proteomic Analysis of Rat Forebrain Postsynaptic Density Fractions by Mass Spectrometry. J. Biol. Chem. 2004, 179, 21003–21011. [Google Scholar] [CrossRef]
- Li, J.; Wilkinson, B.; Clementel, V.A.; Hou, J.; O’Dell, T.J.; Coba, M.P. Long-term potentiation modulates synaptic phosphorylation networks and reshapes the structure of the postsynaptic interactome. Sci. Signal. 2016, 9, rs8. [Google Scholar] [CrossRef]
- Li, J.; Zhang, W.; Yang, H.; Howrigan, D.P.; Wilkinson, B.; Souaiaia, T.; Evgrafov, O.V.; Genovese, G.; Clementel, V.A.; Tudor, J.C.; et al. Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nat. Neurosci. 2017, 20, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Colangelo, C.M.; Ivosev, G.; Chung, L.; Abbott, T.; Shifman, M.; Sakaue, F.; Cox, D.; Kitchen, R.R.; Burton, L.; Tate, S.A.; et al. Development of a highly automated and multiplexed targeted proteome pipeline and assay for 112 rat brain synaptic proteins. Proteomics 2015, 15, 1202–1214. [Google Scholar] [CrossRef]
- Gillette, M.A.; Carr, S.A. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat. Methods 2013, 10, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Venable, J.D.; Dong, M.Q.; Wohlschlegel, J.; Dillin, A.; Yates, J.R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 2004, 1, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics 2012, 11, O111.016717. [Google Scholar] [CrossRef]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Miladinović, S.M.; Cheng, L.-Y.; Messner, S.; Ehrenberger, T.; Zanotelli, V.; Butscheid, Y.; Escher, C.; et al. Extending the Limits of Quantitative Proteome Profiling with Data-Independent Acquisition and Application to Acetaminophen-Treated Three-Dimensional Liver Microtissues. Mol. Cell. Proteomics 2015, 14. [Google Scholar] [CrossRef]
- Geiger, T.; Cox, J.; Mann, M. Proteomics on an Orbitrap Benchtop Mass Spectrometer Using All-ion Fragmentation. Mol. Cell. Proteomics 2010, 9, 2252–2261. [Google Scholar] [CrossRef]
- Egertson, J.D.; Kuehn, A.; Merrihew, G.E.; Bateman, N.W.; MacLean, B.X.; Ting, Y.S.; Canterbury, J.D.; Marsh, D.M.; Kellmann, M.; Zabrouskov, V.; et al. Multiplexed MS/MS for improved data-independent acquisition. Nat. Methods 2013, 10, 744–746. [Google Scholar] [CrossRef] [PubMed]
- Selevsek, N.; Chang, C.-Y.; Gillet, L.C.; Navarro, P.; Bernhardt, O.M.; Reiter, L.; Cheng, L.-Y.; Vitek, O.; Aebersold, R. Reproducible and Consistent Quantification of the Saccharomyces cerevisiae Proteome by SWATH-mass spectrometry. Mol. Cell. Proteomics 2015, 14, 739–749. [Google Scholar] [CrossRef]
- Distler, U.; Schmeisser, M.J.; Pelosi, A.; Reim, D.; Kuharev, J.; Weiczner, R.; Baumgart, J.; Boeckers, T.M.; Nitsch, R.; Vogt, J.; et al. In-depth protein profiling of the postsynaptic density from mouse hippocampus using data-independent acquisition proteomics. Proteomics 2014, 14, 2607–2613. [Google Scholar] [CrossRef]
- Peterson, A.C.; Russell, J.D.; Bailey, D.J.; Westphall, M.S.; Coon, J.J. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol. Cell. Proteomics 2012, 11, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Gallien, S.; Duriez, E.; Crone, C.; Kellmann, M.; Moehring, T.; Domon, B. Targeted Proteomic Quantification on Quadrupole-Orbitrap Mass Spectrometer. Mol. Cell. Proteomics 2012, 11, 1709–1723. [Google Scholar] [CrossRef] [PubMed]
- Ankney, J.A.; Muneer, A.; Chen, X. Relative and Absolute Quantitation in Mass Spectrometry–Based Proteomics. Annu. Rev. Anal. Chem. 2018, 11, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. Sistema séptico domiciliario | Rotomoldeo en Colombia Tanques Plasticos En Colombia Rotoplast. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Searle, B.C.; Pino, L.K.; Egertson, J.D.; Ting, Y.S.; Lawrence, R.T.; Villen, J.; MacCoss, M.J. Comprehensive peptide quantification for data independent acquisition mass spectrometry using chromatogram libraries. bioRxiv 2018, 277822. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Baucum, A.J.; Shonesy, B.C.; Rose, K.L.; Colbran, R.J. Quantitative Proteomics Analysis of CaMKII Phosphorylation and the CaMKII Interactome in the Mouse Forebrain. ACS Chem. Neurosci. 2015, 6, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Reim, D.; Distler, U.; Halbedl, S.; Verpelli, C.; Sala, C.; Bockmann, J.; Tenzer, S.; Boeckers, T.M.; Schmeisser, M.J. Proteomic Analysis of Post-synaptic Density Fractions from Shank3 Mutant Mice Reveals Brain Region Specific Changes Relevant to Autism Spectrum Disorder. Front. Mol. Neurosci. 2017, 10, 26. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein#. | Gene Name | Protein Description | Peptide # | Peptide Sequence |
|---|---|---|---|---|
| 1 | Anks1b | Ankyrin repeat & sterile alpha motif domain-containing protein 1B | 1 | TLANLPWIVEPGQEAK |
| 2 | LIFQSCDYK | |||
| 3 | ILQAIQLLPK | |||
| 2 | Arc | Activity-regulated cytoskeleton-associated protein | 4 | GGPAAKPNVILQIGK |
| 5 | TLEQLIQR | |||
| 3 | Baiap2 | Brain-specific angiogenesis inhibitor 1-associated protein 2 | 6 | EGDLITLLVPEAR |
| 7 | AFHNELLTQLEQK | |||
| 8 | AIFSHAAGDNSTLLSFK | |||
| 4 | Bsn | Protein bassoon | 9 | ATAEFSTQTPSLTPSSDIPR |
| 10 | HGGGSGGPDLVPYQPQHGPGLNAPQGLASLR | |||
| 11 | ATSVPGPTQATAPPEVGR | |||
| 5 | Camk2a | Calcium/calmodulin-dependent protein kinase type II subunit alpha | 12 | FTEEYQLFEELGK |
| 13 | VLAGQEYAAK | |||
| 14 | ITQYLDAGGIPR | |||
| 6 | Camk2b | Calcium/calmodulin-dependent protein kinase type II subunit beta | 15 | TTEQLIEAVNNGDFEAYAK |
| 16 | GSLPPAALEPQTTVIHNPVDGIK | |||
| 17 | ESSDSTNTTIEDEDAK | |||
| 7 | Camk2d | Calcium/calmodulin-dependent protein kinase type II subunit delta | 18 | FTDEYQLFEELGK |
| 19 | IPTGQEYAAK | |||
| 8 | Camk2g | Calcium/calmodulin-dependent protein kinase type II subunit gamma | 20 | FYFENLLSK |
| 21 | ITEQLIEAINNGDFEAYTK | |||
| 22 | FTDDYQLFEELGK | |||
| 9 | Cldn11 | Claudin-11 | 23 | FYYSSGSSSPTHAK |
| 10 | Csnk2a1 | CK2 | 24 | GGPNIITLADIVKDPVSR |
| 25 | TPALVFEHVNNTDFK | |||
| 26 | LIDWGLAEFYHPGQEYNVR | |||
| 11 | Dlg2 | Disks large homolog 2 | 27 | DSGLPSQGLSFK |
| 28 | GQEDLILSYEPVTR | |||
| 29 | FIEAGQYNDNLYGTSVQSVR | |||
| 12 | Dlg3 | Disks large homolog 3 | 30 | VNEVDVSEVVHSR |
| 31 | ILSVNGVNLR | |||
| 32 | LLAVNNTNLQDVR | |||
| 13 | Dlg4 | PSD-95 | 33 | NAGQTVTIIAQYKPEEYSR |
| 34 | EVTHSAAVEALK | |||
| 35 | IIPGGAAAQDGR | |||
| 14 | Dlgap1 | Disks large-associated protein 1 | 36 | AVSEVSINR |
| 37 | FQSVGVQVEEEK | |||
| 38 | SLDSLDPAGLLTSPK | |||
| 15 | Dlgap2 | Disks large-associated protein 2 | 39 | TQGLFSYR |
| 40 | CSSIGVQDSEFPDHQPYPR | |||
| 41 | TSPTVALRPEPLLK | |||
| 16 | Dlgap3 | Disks large-associated protein 3 | 42 | EAEDYELPEEILEK |
| 43 | FLELQQLK | |||
| 44 | GPAGPGPGPGSGAAPEAR | |||
| 17 | Erc2 | ERC protein 2 | 45 | DLNHLLQQESGNR |
| 46 | VNALQAELTEK | |||
| 47 | IAELESLTLR | |||
| 18 | Gfap | Glial fibrillary acidic protein | 48 | ALAAELNQLR |
| 49 | ITIPVQTFSNLQIR | |||
| 50 | LADVYQAELR | |||
| 19 | Gja1 | Gap junction alpha-1 protein | 51 | SDPYHATTGPLSPSK |
| 20 | Gria2 | Glutamate receptor 2 | 52 | LTIVGDGK |
| 53 | ADIAIAPLTITLVR | |||
| 54 | GADQEYSAFR | |||
| 21 | Gria3 | Glutamate receptor 3 | 55 | GSALGNAVNLAVLK |
| 56 | NTQNFKPAPATNTQNYATYR | |||
| 57 | ADIAVAPLTITLVR | |||
| 22 | Grin1 | Glutamate receptor ionotropic, NMDA 1 | 58 | VIILSASEDDAATVYR |
| 59 | HNYESAAEAIQAVR | |||
| 60 | IPVLGLTTR | |||
| 23 | Grin2a | Glutamate receptor ionotropic, NMDA 2A | 61 | FSYIPEAK |
| 62 | GVEDALVSLK | |||
| 63 | YLPEEVAHSDISETSSR | |||
| 24 | Grin2b | Glutamate receptor ionotropic, NMDA 2B | 64 | FQRPNDFSPPFR |
| 65 | SDVSDISTHTVTYGNIEGNAAK | |||
| 25 | Homer1 | Homer1 | 66 | LTAALLESTANVK |
| 67 | HAVTVSYFYDSTR | |||
| 68 | ANTVYGLGFSSEHHLSK | |||
| 26 | Ina | Alpha-internexin | 69 | ALEAELAALR |
| 70 | FANLNEQAAR | |||
| 71 | HSAEVAGYQDSIGQLESDLR | |||
| 27 | Kcnj4 | Inward rectifier potassium channel 4 | 72 | FEPVVFEEK |
| 73 | SSYLASEILWGHR | |||
| 74 | TYEVAGTPCCSAR | |||
| 28 | Lrrc7 | Leucine-rich repeat-containing protein 7 | 75 | VLNLSDNR |
| 76 | ALIPLQTEAHPETK | |||
| 77 | IVGVPLELEQSTHR | |||
| 29 | Mbp | Myelin basic protein | 78 | DTGILDSIGR |
| 79 | TPPPSQGK | |||
| 80 | TQDENPVVHFFK | |||
| 30 | Mog | Myelin-oligodendrocyte glycoprotein | 81 | ALVGDEAELPCR |
| 82 | DQDAEQAPEYR | |||
| 83 | FSDEGGYTCFFR | |||
| 31 | Myo1d | Unconventional myosin-1d | 84 | VVSVIAELLSTK |
| 85 | HQVEYLGLLENVR | |||
| 86 | IGELVGVLVNHFK | |||
| 32 | Nedd4 | E3 ubiquitin-protein ligase NEDD4 | 87 | EWFFLISK |
| 88 | LLDGFFIRPFYK | |||
| 89 | LLQFVTGTSR | |||
| 33 | Nrn1 | Neuritin | 90 | FSTFSGSITGPLYTHR |
| 91 | GFSDCLLK | |||
| 34 | Pclo | Protein piccolo | 92 | NYVLIDDIGDITK |
| 93 | AQEAEALDVSFGHSSSSAR | |||
| 94 | AAAGPLPPISADTR | |||
| 35 | Plec | Plectin | 95 | DSQDAGGFGPEDR |
| 96 | IISLETYNLFR | |||
| 97 | LGFHLPLEVAYQR | |||
| 36 | Rims1 | Regulating synaptic membrane exocytosis protein 1 | 98 | ATTLTVPEQQR |
| 99 | ESGALLGLK | |||
| 100 | ETSPISSHPVTWQPSK | |||
| 37 | Rpl3 | 60S ribosomal protein L3 | 101 | VACIGAWHPAR |
| 102 | IGQGYLIKDGK | |||
| 103 | NNASTDYDLSDK | |||
| 38 | Rpl7a | 60S ribosomal protein L7a | 104 | NFGIGQDIQPK |
| 105 | LKVPPAINQFTQALDR | |||
| 106 | AGVNTVTTLVENK | |||
| 39 | Rpl10 | 60S ribosomal protein L10 | 107 | VHIGQVIMSIR |
| 40 | Rpl18a | 60S ribosomal protein L18a | 108 | IFAPNHVVAK |
| 109 | VKNFGIWLR | |||
| 110 | DLTTAGAVTQCYR | |||
| 41 | Rps20 | 40S ribosomal protein S20 | 111 | DTGKTPVEPEVAIHR |
| 112 | VCADLIR | |||
| 113 | LIDLHSPSEIVK | |||
| 42 | Shank1 | SH3 and multiple ankyrin repeat domains protein 1 | 114 | ALTASPPAAR |
| 115 | LESGGSSGGYGAYAAGSR | |||
| 116 | GSSTEDGPGVPPPSPR | |||
| 43 | Shank2 | SH3 and multiple ankyrin repeat domains protein 2 | 117 | AASVPALADLVK |
| 118 | LLDPSSPLALALSAR | |||
| 119 | IFLSGITEEER | |||
| 44 | Shank3 | SH3 and multiple ankyrin repeat domains protein 3 | 120 | AALAVGSPGPVGGSFAR |
| 121 | LDPTAPVWAAK | |||
| 122 | VLSIGEGGFWEGTVK | |||
| 45 | Sptan1 | Spectrin alpha chain, non-erythrocytic 1 | 123 | ELPTAFDYVEFTR |
| 124 | SSLSSAQADFNQLAELDR | |||
| 125 | HQAFEAELSANQSR | |||
| 46 | Srcin1 | SRC kinase signaling inhibitor 1 | 126 | GEGLYADPYGLLHEGR |
| 127 | AGAGGPLYGDGYGFR | |||
| 128 | LLEETQAELLK | |||
| 47 | Syngap1 | Ras GTPase-activating protein SynGAP | 129 | AGYVGLVTVPVATLAGR |
| 130 | GGEPPGDTFAPFHGYSK | |||
| 131 | SASGDTVFWGEHFEFNNLPAVR | |||
| 48 | Synpo | Synaptopodin | 132 | YVIESSGHAELAR |
| 133 | AASPAKPSSLDLVPNLPR | |||
| 134 | VASEEEEVPLVVYLK | |||
| 49 | Tomm20 | Mitochondrial import receptor subunit TOM20 | 135 | LPDLKDAEAVQK |
| 50 | Vdac2 | Voltage-dependent anion-selective channel protein 2 | 136 | GFGFGLVK |
| 137 | YQLDPTASISAK | |||
| 138 | WCEYGLTFTEK |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, R.S.; Rauniyar, N.; Sakaue, F.; Lam, T.T.; Williams, K.R.; Nairn, A.C. Development of Targeted Mass Spectrometry-Based Approaches for Quantitation of Proteins Enriched in the Postsynaptic Density (PSD). Proteomes 2019, 7, 12. https://doi.org/10.3390/proteomes7020012
Wilson RS, Rauniyar N, Sakaue F, Lam TT, Williams KR, Nairn AC. Development of Targeted Mass Spectrometry-Based Approaches for Quantitation of Proteins Enriched in the Postsynaptic Density (PSD). Proteomes. 2019; 7(2):12. https://doi.org/10.3390/proteomes7020012
Chicago/Turabian StyleWilson, Rashaun S., Navin Rauniyar, Fumika Sakaue, TuKiet T. Lam, Kenneth R. Williams, and Angus C. Nairn. 2019. "Development of Targeted Mass Spectrometry-Based Approaches for Quantitation of Proteins Enriched in the Postsynaptic Density (PSD)" Proteomes 7, no. 2: 12. https://doi.org/10.3390/proteomes7020012
APA StyleWilson, R. S., Rauniyar, N., Sakaue, F., Lam, T. T., Williams, K. R., & Nairn, A. C. (2019). Development of Targeted Mass Spectrometry-Based Approaches for Quantitation of Proteins Enriched in the Postsynaptic Density (PSD). Proteomes, 7(2), 12. https://doi.org/10.3390/proteomes7020012