Comparative Proteomic Analysis Unveils Critical Pathways Underlying the Role of Nitrogen Fertilizer Treatment in American Elderberry



Abstract
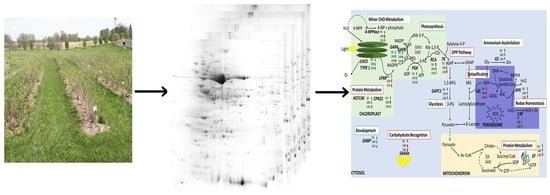
1. Introduction
2. Materials and Methods
3. Results
3.1. Comparative Proteome Analyses and Differentially Abundant Proteins in Elderberry Leaves
3.2. Protein Identification
3.3. Hierarchical Clustering of Differentially Abundant Proteins in the Three Treatment Groups
3.4. Principle Component Analysis (PCA) of Each Group
3.5. Functional Classification and Cellular Localization of Differentially Abundant Proteins
3.6. Expression Profiles of Unique Proteins in Response to N Across Three Genotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cernusca, M.M.; Gold, M.A.; Godsey, L.D. Using the Porter model to analyze the US elderberry industry. Agrofor. Syst. 2012, 86, 365–377. [Google Scholar] [CrossRef]
- Masclaux-Daubresse, C.; Daniel-Vedele, F.; Dechorgnat, J.; Chardon, F.; Gaufichon, L.; Suzuki, A. Nitrogen uptake, assimilation and remobilization in plants: Challenges for sustainable and productive agriculture. Ann. Bot. 2010, 105, 1141–1157. [Google Scholar] [CrossRef]
- Miller, A.J.; Fan, X.; Orsel, M.; Smith, S.J.; Wells, D.M. Nitrate transport and signalling. J. Exp. Bot. 2007, 58, 2297–2306. [Google Scholar] [CrossRef]
- Bi, Y.M.; Meyer, A.; Downs, G.S.; Shi, X.; El-Kereamy, A.; Lukens, L.; Rothstein, S.J. High throughput RNA sequencing of a hybrid maize and its parents shows different mechanisms responsive to nitrogen limitation. BMC Genom. 2014, 15, 77. [Google Scholar] [CrossRef]
- Scheible, W.R.; Morcuende, R.; Czechowski, T.; Fritz, C.; Osuna, D.; Palacios-Rojas, N.; Schindelasch, D.; Thimm, O.; Udvardi, M.K.; Stitt, M. Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of Arabidopsis in response to nitrogen. Plant Physiol. 2004, 136, 2483–2499. [Google Scholar] [CrossRef]
- Islam, N.; Li, G.; Garrett, W.M.; Lin, R.; Sriram, G.; Cooper, B.; Coleman, G.D. Proteomics of nitrogen remobilization in poplar bark. J. Proteome Res. 2015, 14, 1112–1126. [Google Scholar] [CrossRef]
- Wang, X.; Bian, Y.; Cheng, K.; Zou, H.; Sun, S.S.; He, J.X. A comprehensive differential proteomic study of nitrate deprivation in Arabidopsis reveals complex regulatory networks of plant nitrogen responses. J. Proteome Res. 2012, 11, 2301–2315. [Google Scholar] [CrossRef]
- Ma, L.; Sun, X.; Kong, X.; Galvan, J.V.; Li, X.; Yang, S.; Yang, Y.; Yang, Y.; Hu, X. Physiological, biochemical and proteomics analysis reveals the adaptation strategies of the alpine plant Potentilla saundersiana at altitude gradient of the Northwestern Tibetan Plateau. J. Proteom. 2015, 112, 63–82. [Google Scholar] [CrossRef]
- Jespersen, D.; Huang, B. Proteins associated with heat-induced leaf senescence in creeping bentgrass as affected by foliar application of nitrogen, cytokinins, and an ethylene inhibitor. Proteomics 2015, 15, 798–812. [Google Scholar] [CrossRef]
- Tyers, M.; Mann, M. From genomics to proteomics. Nature 2003, 422, 193–197. [Google Scholar] [CrossRef]
- Baginsky, S.; Gruissem, W. Arabidopsis thaliana proteomics: From proteome to genome. J. Exp. Bot. 2006, 57, 1485–1491. [Google Scholar] [CrossRef]
- Griffin, T.J.; Gygi, S.P.; Ideker, T.; Rist, B.; Eng, J.; Hood, L.; Aebersold, R. Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol. Cell. Proteom. 2002, 1, 323–333. [Google Scholar] [CrossRef]
- Tian, Q.; Stepaniants, S.B.; Mao, M.; Weng, L.; Feetham, M.C.; Doyle, M.J.; Yi, E.C.; Dai, H.; Thorsson, V.; Eng, J.; et al. Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol. Cell. Proteom. 2004, 3, 960–969. [Google Scholar] [CrossRef]
- Washburn, M.P.; Koller, A.; Oshiro, G.; Ulaszek, R.R.; Plouffe, D.; Deciu, C.; Winzeler, E.; Yates, J.R., 3rd. Protein pathway and complex clustering of correlated mRNA and protein expression analyses in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2003, 100, 3107–3112. [Google Scholar] [CrossRef]
- Hajduch, M.; Hearne, L.B.; Miernyk, J.A.; Casteel, J.E.; Joshi, T.; Agrawal, G.K.; Song, Z.; Zhou, M.; Xu, D.; Thelen, J.J. Systems analysis of seed filling in Arabidopsis: Using general linear modeling to assess concordance of transcript and protein expression. Plant Physiol. 2010, 152, 2078–2087. [Google Scholar] [CrossRef]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef]
- Byers, P.L.; Thomas, A.L. ‘Bob Gordon’ Elderberry. J. Am. Pomol. Soc. 2011, 65, 52–55. [Google Scholar]
- Byers, P.L.; Thomas, A.L.; Millican, M. ‘Wyldewood’ Elderberry. Hortscience 2010, 45, 312–313. [Google Scholar] [CrossRef]
- Loomis, R.S. On the utility of nitrogen in leaves. Proc. Natl. Acad. Sci. USA 1997, 94, 13378–13379. [Google Scholar] [CrossRef]
- Johnson, M.C.; Thomas, A.L.; Greenlief, C.M. Impact of frozen storage on the anthocyanin and polyphenol contents of American elderberry fruit juice. J. Agric. Food Chem. 2015, 63, 5653–5659. [Google Scholar] [CrossRef]
- Hurkman, W.J.; Tanaka, C.K. Solubilization of plant membrane proteins for analysis by two-dimensional gel electrophoresis. Plant Physiol. 1986, 81, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Mooney, B.P.; Miernyk, J.A.; Greenlief, C.M.; Thelen, J.J. Using quantitative proteomics of Arabidopsis roots and leaves to predict metabolic activity. Physiol. Plant. 2006, 128, 237–250. [Google Scholar] [CrossRef]
- Hajduch, M.; Ganapathy, A.; Stein, J.W.; Thelen, J.J. A systematic proteomic study of seed filling in soybean. Establishment of high-resolution two-dimensional reference maps, expression profiles, and an interactive proteome database. Plant Physiol. 2005, 137, 1397–1419. [Google Scholar] [CrossRef] [PubMed]
- Dahal, D.; Newton, K.J.; Mooney, B.P. Quantitative proteomics of Zea mays hybrids exhibiting different levels of heterosis. J. Proteome Res. 2016, 15, 2445–2454. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef]
- Caraux, G.; Pinloche, S. PermutMatrix: A graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics 2005, 21, 1280–1281. [Google Scholar] [CrossRef]
- Usadel, B.; Nagel, A.; Thimm, O.; Redestig, H.; Blaesing, O.E.; Palacios-Rojas, N.; Selbig, J.; Hannemann, J.; Piques, M.C.; Steinhauser, D.; et al. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 2005, 138, 1195–1204. [Google Scholar] [CrossRef]
- Heazlewood, J.L.; Verboom, R.E.; Tonti-Filippini, J.; Small, I.; Millar, A.H. SUBA: The Arabidopsis subcellular database. Nucleic Acids Res. 2007, 35, D213–D218. [Google Scholar] [CrossRef]
- Shang, C.; Van Damme, E.J. Comparative analysis of carbohydrate binding properties of Sambucus nigra lectins and ribosome-inactivating proteins. Glycoconj. J. 2014, 31, 345–354. [Google Scholar] [CrossRef]
- Finn, C.E.; Thomas, A.L.; Byers, P.L.; Serce, S. Evaluation of American (Sambucus canadensis) and European (S. nigra) elderberry genotypes grown in diverse environments and implications for cultivar development. Hortscience 2008, 43, 1385–1391. [Google Scholar] [CrossRef]
- Wase, N.; Black, P.N.; Stanley, B.A.; Di Russo, C.C. Integrated quantitative analysis of nitrogen stress response in Chlamydomonas reinhardtii using metabolite and protein profiling. J. Proteome Res. 2014, 13, 1373–1396. [Google Scholar] [CrossRef] [PubMed]
- Bahrman, N.; Le Gouis, J.; Negroni, L.; Amilhat, L.; Leroy, P.; Laine, A.L.; Jaminon, O. Differential protein expression assessed by two-dimensional gel electrophoresis for two wheat varieties grown at four nitrogen levels. Proteomics 2004, 4, 709–719. [Google Scholar] [CrossRef]
- Yousuf, P.Y.; Ganie, A.H.; Khan, I.; Qureshi, M.I.; Ibrahim, M.M.; Sarwat, M.; Iqbal, M.; Ahmad, A. Nitrogen-efficient and nitrogen-inefficient Indian mustard showed differential expression pattern of proteins in response to elevated CO2 and low nitrogen. Front. Plant Sci. 2016, 7, 1074. [Google Scholar] [CrossRef]
- Turkina, M.V.; Kargul, J.; Blanco-Rivero, A.; Villarejo, A.; Barber, J.; Vener, A.V. Environmentally modulated phosphoproteome of photosynthetic membranes in the green alga Chlamydomonas reinhardtii. Mol. Cell. Proteom. 2006, 5, 1412–1425. [Google Scholar] [CrossRef]
- Juergens, M.T.; Deshpande, R.R.; Lucker, B.F.; Park, J.J.; Wang, H.; Gargouri, M.; Holguin, F.O.; Disbrow, B.; Schaub, T.; Skepper, J.N.; et al. The regulation of photosynthetic structure and function during nitrogen deprivation in Chlamydomonas reinhardtii. Plant Physiol. 2015, 167, 558–573. [Google Scholar] [CrossRef]
- Wei, S.; Wang, X.; Zhang, J.; Liu, P.; Zhao, B.; Li, G.; Dong, S. The role of nitrogen in leaf senescence of summer maize and analysis of underlying mechanisms using comparative proteomics. Plant Sci. 2015, 233, 72–81. [Google Scholar] [CrossRef]
- Cheng, L.; Fuchigami, L.H. Rubisco activation state decreases with increasing nitrogen content in apple leaves. J. Exp. Bot. 2000, 51, 1687–1694. [Google Scholar] [CrossRef]
- Chen, J.W.; Yang, Z.Q.; Zhou, P.; Hai, M.R.; Tang, T.X.; Liang, Y.L.; An, T.X. Biomass accumulation and partitioning, photosynthesis, and photosynthetic induction in field-grown maize (Zea mays L.) under low- and high-nitrogen conditions. Acta Physiol. Plant. 2013, 35, 95–105. [Google Scholar] [CrossRef]
- Ding, L.; Gao, L.; Liu, W.; Wang, M.; Gu, M.; Ren, B.; Xu, G.; Shen, Q.; Guo, S. Aquaporin plays an important role in mediating chloroplastic CO2 concentration under high-N supply in rice (Oryza sativa) plants. Physiol. Plant. 2016, 156, 215–226. [Google Scholar] [CrossRef]
- Yagi, N.; Takeda, S.; Matsumoto, N.; Okada, K. VAJ/GFA1/CLO is involved in the directional control of floral organ growth. Plant Cell Physiol. 2009, 50, 515–527. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef]
- Ursin, V.M.; Irvine, J.M.; Hiatt, W.R.; Shewmaker, C.K. Developmental analysis of elongation factor-1 alpha expression in transgenic tobacco. Plant Cell 1991, 3, 583–591. [Google Scholar] [CrossRef]
- Horiguchi, G.; Molla-Morales, A.; Perez-Perez, J.M.; Kojima, K.; Robles, P.; Ponce, M.R.; Micol, J.L.; Tsukaya, H. Differential contributions of ribosomal protein genes to Arabidopsis thaliana leaf development. Plant J. 2011, 65, 724–736. [Google Scholar] [CrossRef]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef]
- Niforou, K.; Cheimonidou, C.; Trougakos, I.P. Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biol. 2014, 2, 323–332. [Google Scholar] [CrossRef]
- Kurepa, J.; Toh, E.A.; Smalle, J.A. 26S proteasome regulatory particle mutants have increased oxidative stress tolerance. Plant J. 2008, 53, 102–114. [Google Scholar] [CrossRef]
- Pais, S.M.; Tellez-Inon, M.T.; Capiati, D.A. Serine/threonine protein phosphatases type 2A and their roles in stress signaling. Plant Signal. Behav. 2009, 4, 1013–1015. [Google Scholar] [CrossRef]
- Pratelli, R.; Pilot, G. Regulation of amino acid metabolic enzymes and transporters in plants. J. Exp. Bot. 2014, 65, 5535–5556. [Google Scholar] [CrossRef]
- Tyagi, R.; Lee, Y.T.; Guddat, L.W.; Duggleby, R.G. Probing the mechanism of the bifunctional enzyme ketol-acid reductoisomerase by site-directed mutagenesis of the active site. FEBS J. 2005, 272, 593–602. [Google Scholar] [CrossRef]
- Miesak, B.H.; Coruzzi, G.M. Molecular and physiological analysis of Arabidopsis mutants defective in cytosolic or chloroplastic aspartate aminotransferase. Plant Physiol. 2002, 129, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Peng, Y.; Ma, W.; Liu, R.; Li, C.; Li, X. Proteomic analysis revealed nitrogen-mediated metabolic, developmental, and hormonal regulation of maize (Zea mays L.) ear growth. J. Exp. Bot. 2012, 63, 5275–5288. [Google Scholar] [CrossRef]
- Seebauer, J.R.; Moose, S.P.; Fabbri, B.J.; Crossland, L.D.; Below, F.E. Amino acid metabolism in maize earshoots. Implications for assimilate preconditioning and nitrogen signaling. Plant Physiol. 2004, 136, 4326–4334. [Google Scholar] [CrossRef]
- Canas, R.A.; Amiour, N.; Quillere, I.; Hirel, B. An integrated statistical analysis of the genetic variability of nitrogen metabolism in the ear of three maize inbred lines (Zea mays L.). J. Exp. Bot. 2011, 62, 2309–2318. [Google Scholar] [CrossRef] [PubMed]
- Kocsy, G.; Tari, I.; Vankova, R.; Zechmann, B.; Gulyas, Z.; Poor, P.; Galiba, G. Redox control of plant growth and development. Plant Sci. 2013, 211, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Noctor, G. Ascorbate and glutathione: The heart of the redox hub. Plant Physiol. 2011, 155, 2–18. [Google Scholar] [CrossRef]
- Begara-Morales, J.C.; Sanchez-Calvo, B.; Chaki, M.; Mata-Perez, C.; Valderrama, R.; Padilla, M.N.; Lopez-Jaramillo, J.; Luque, F.; Corpas, F.J.; Barroso, J.B. Differential molecular response of monodehydroascorbate reductase and glutathione reductase by nitration and S-nitrosylation. J. Exp. Bot. 2015, 66, 5983–5996. [Google Scholar] [CrossRef]
- Wheeler, G.L.; Jones, M.A.; Smirnoff, N. The biosynthetic pathway of vitamin C in higher plants. Nature 1998, 393, 365–369. [Google Scholar] [CrossRef]
- Rhee, S.G.; Kang, S.W.; Jeong, W.; Chang, T.S.; Yang, K.S.; Woo, H.A. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr. Opin. Cell Biol. 2005, 17, 183–189. [Google Scholar] [CrossRef]
- Yang, T.; Poovaiah, B.W. Hydrogen peroxide homeostasis: Activation of plant catalase by calcium/calmodulin. Proc. Natl. Acad. Sci. USA 2002, 99, 4097–4102. [Google Scholar] [CrossRef]
- Smith, L.M.; Kelleher, N.L.; Linial, M.; Goodlett, D.; Langridge-Smith, P.; Goo, Y.A.; Safford, G.; Bonilla, L.; Kruppa, G.; Zubarev, R.; et al. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in top-down proteomics and the analysis of proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef]
- Dahal, D.; Mooney, B.P.; Newton, K.J. Specific changes in total and mitochondrial proteomes are associated with higher levels of heterosis in maize hybrids. Plant J. 2012, 72, 70–83. [Google Scholar] [CrossRef]
- Prinsi, B.; Espen, L. Mineral nitrogen sources differently affect root glutamine synthetase isoforms and amino acid balance among organs in maize. BMC Plant Biol. 2015, 15, 96. [Google Scholar] [CrossRef]
- Wang, X.; Wei, Y.; Shi, L.; Ma, X.; Theg, S.M. New isoforms and assembly of glutamine synthetase in the leaf of wheat (Triticum aestivum L.). J. Exp. Bot. 2015, 66, 6827–6834. [Google Scholar] [CrossRef]
- Colignon, B.; Raes, M.; Dieu, M.; Delaive, E.; Mauro, S. Evaluation of three-dimensional gel electrophoresis to improve quantitative profiling of complex proteomes. Proteomics 2013, 13, 2077–2082. [Google Scholar] [CrossRef]
- Buchanan, B.B.; Balmer, Y. Redox regulation: A broadening horizon. Annu. Rev. Plant Biol. 2005, 56, 187–220. [Google Scholar] [CrossRef]
- Bykova, N.V.; Rampitsch, C. Modulating protein function through reversible oxidation: Redox-mediated processes in plants revealed through proteomics. Proteomics 2013, 13, 579–596. [Google Scholar] [CrossRef]
- Racchi, M.L. Antioxidant defenses in plants with attention to prunus and citrus spp. Antioxidants 2013, 2, 340–369. [Google Scholar] [CrossRef]
- Thornalley, P.J. Pharmacology of methylglyoxal: Formation, modification of proteins and nucleic acids, and enzymatic detoxification—A role in pathogenesis and antiproliferative chemotherapy. Gen. Pharmacol. 1996, 27, 565–573. [Google Scholar] [CrossRef]
- Krapp, A. Plant nitrogen assimilation and its regulation: A complex puzzle with missing pieces. Curr. Opin. Plant Biol. 2015, 25, 115–122. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N Concentration Comparison | Initial Detected Protein Spots b | Differentially Abundant Protein Spots c | High Class Spots d | Effect e | MS/MS Analyzed Spots f | |||
|---|---|---|---|---|---|---|---|---|
| G | N | G and N | G × N | |||||
| 0 vs 56 | 306 | 86 | 61 | 30 | 4 | 11 | 16 | 28 |
| 0 vs 112 | 295 | 69 | 53 | 21 | 2 | 11 | 19 | 37 |
| 0 vs 169 | 281 | 68 | 51 | 19 | 4 | 8 | 20 | 36 |
| Total | 882 | 223 | 165 | 70 | 10 | 30 | 55 | 101 |
| Protein Name | Spot Number | Functional Class |
|---|---|---|
| RubisCO activase α form precursor (RCA) | 20 and 35 | Photosynthesis (34.8%) |
| Glyceraldehyde-3-phosphate dehydrogenase B (GAPB) | 26, 28 and 75 | |
| Phosphoglycerate kinase (PGK) | 27, 30 and 31 | |
| Oxygen-evolving enhancer protein 1 (OEE1) | 41, 44, 57 and 58 | |
| Chlorophyll a–b binding protein of LHCII type 1-like (LHCII type1) | 46 | |
| Chloroplast pigment-binding protein CP26 (CP26) | 48 and 52 | |
| Ferredoxin-NADP reductase, leaf isozyme (LFNR) | 64, 69, 70, 91 and 92 | |
| Glyceraldehyde-3-phosphate dehydrogenase A (GAPA) | 65, 66, 73, 76 and 101 | |
| Chaperonin 60 subunit β 1 (CPN 60) | 10 | Protein Metabolism (17.4%), |
| Elongation factor-GTP-binding domain-containing protein (EF) | 32 | |
| Photosystem II stability/assembly factor HCF136 (HCF136) | 37 and 40 | |
| 20 kDa Chaperonin family protein (CPN21) | 49, 56, 61 and 90 | |
| Catalase (CAT) | 18 and 19 | Redox Homeostasis (13.0%) |
| Monodehydroascorbate reductase family protein (MDAR) | 29 | |
| Ascorbate peroxidase (APX) | 51, 54, 55 and 59 | |
| Phosphoglucomutase (PGM) | 6, 7, 8 and 9 | Glycolysis (8.7%) |
| Glyceraldehyde-3-phosphate dehydrogenase C subunit1 (GAPC1) | 74, 77, 78, 81 and 89 | |
| Transketolase family protein (TK) | 2, 3 and 4 | OPP pathway (4.3%) |
| Glutamine synthetase cytosolic isozyme 1-1-like (GS) | 33 and 34 | N-metabolism (4.3%) |
| Putative lactoylglutathione lyase (LGL) | 36 | Biodegradation of xenobiotics (4.3%) |
| 4-Nitrophenylphosphatase (4-NPPase) | 43 | Minor CHO metabolism (4.3%) |
| Guanine nucleotide-binding protein subunit β-like protein (GNBP) | 72 | Development (4.3%) |
| Monomeric lectin SNAlm precursor (SNAlm) | 42 | Not assigned (4.3%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, B.; Thomas, A.L.; Greenlief, C.M. Comparative Proteomic Analysis Unveils Critical Pathways Underlying the Role of Nitrogen Fertilizer Treatment in American Elderberry. Proteomes 2019, 7, 10. https://doi.org/10.3390/proteomes7010010
Yang B, Thomas AL, Greenlief CM. Comparative Proteomic Analysis Unveils Critical Pathways Underlying the Role of Nitrogen Fertilizer Treatment in American Elderberry. Proteomes. 2019; 7(1):10. https://doi.org/10.3390/proteomes7010010
Chicago/Turabian StyleYang, Bo, Andrew L. Thomas, and C. Michael Greenlief. 2019. "Comparative Proteomic Analysis Unveils Critical Pathways Underlying the Role of Nitrogen Fertilizer Treatment in American Elderberry" Proteomes 7, no. 1: 10. https://doi.org/10.3390/proteomes7010010
APA StyleYang, B., Thomas, A. L., & Greenlief, C. M. (2019). Comparative Proteomic Analysis Unveils Critical Pathways Underlying the Role of Nitrogen Fertilizer Treatment in American Elderberry. Proteomes, 7(1), 10. https://doi.org/10.3390/proteomes7010010