Disseminating Metaproteomic Informatics Capabilities and Knowledge Using the Galaxy-P Framework

 ,
,  , , , , , and
, , , , , and 
Abstract
1. Introduction
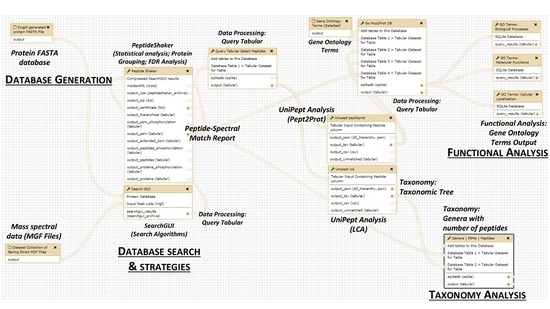
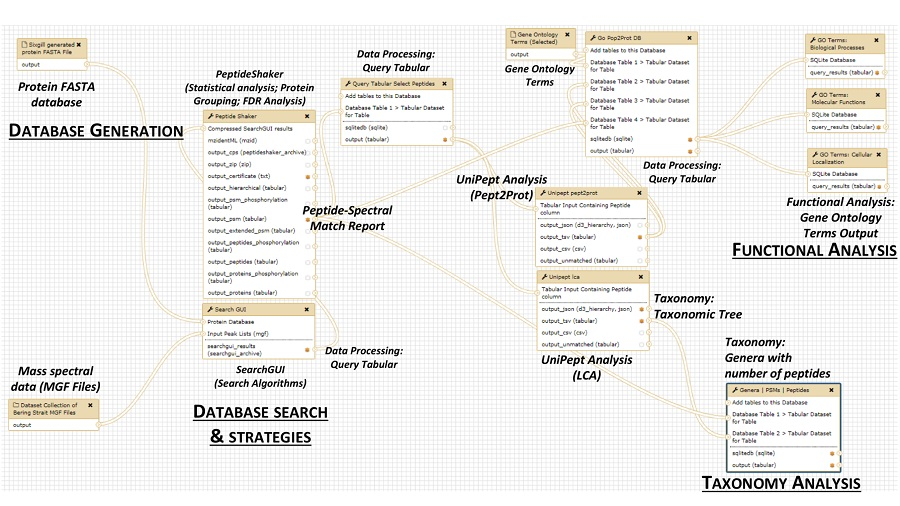
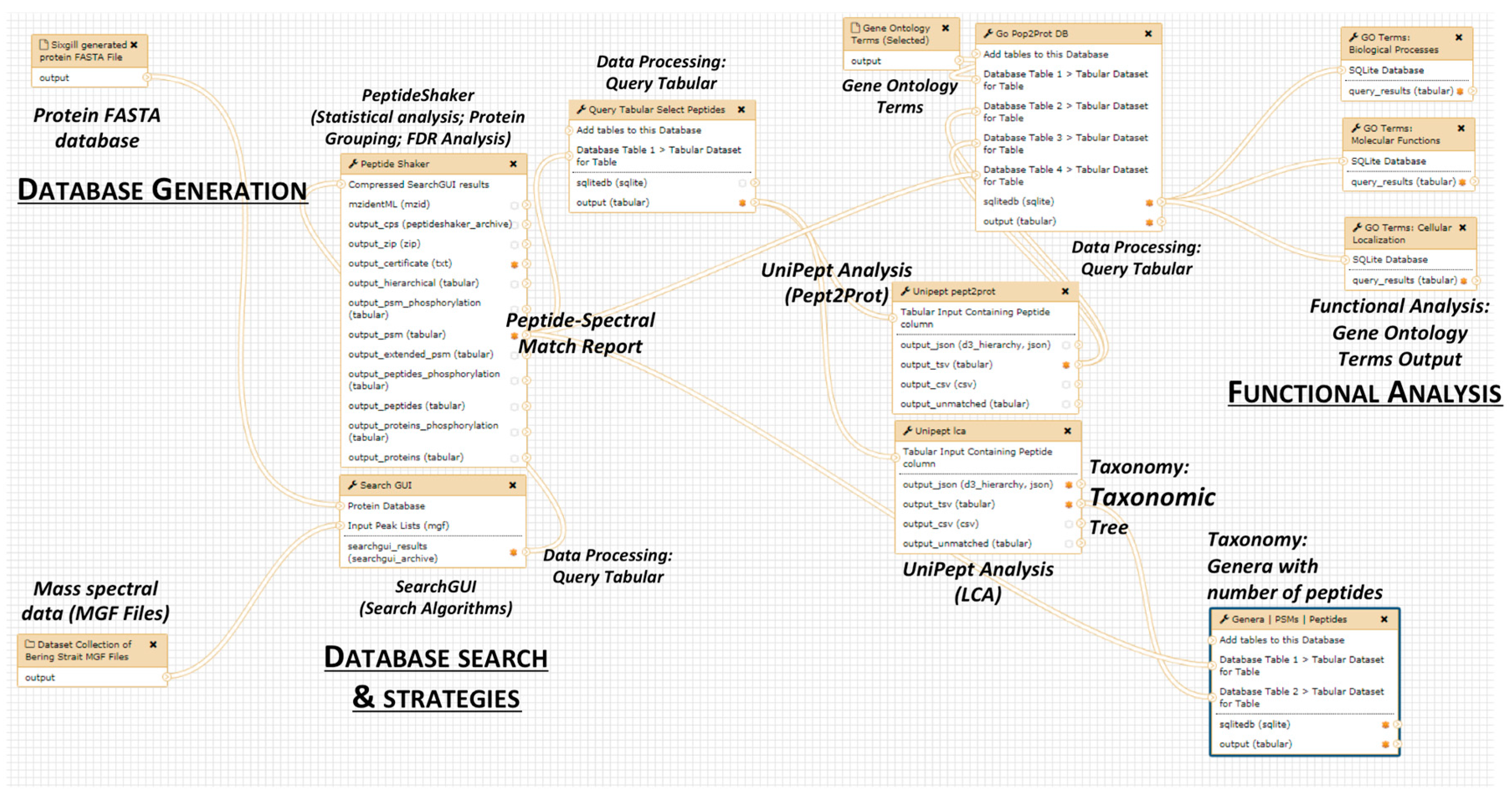
2. The Metaproteomics Gateway
2.1. Description of the Accessible Resources
2.2. The Playground: The Galaxy-P Platform
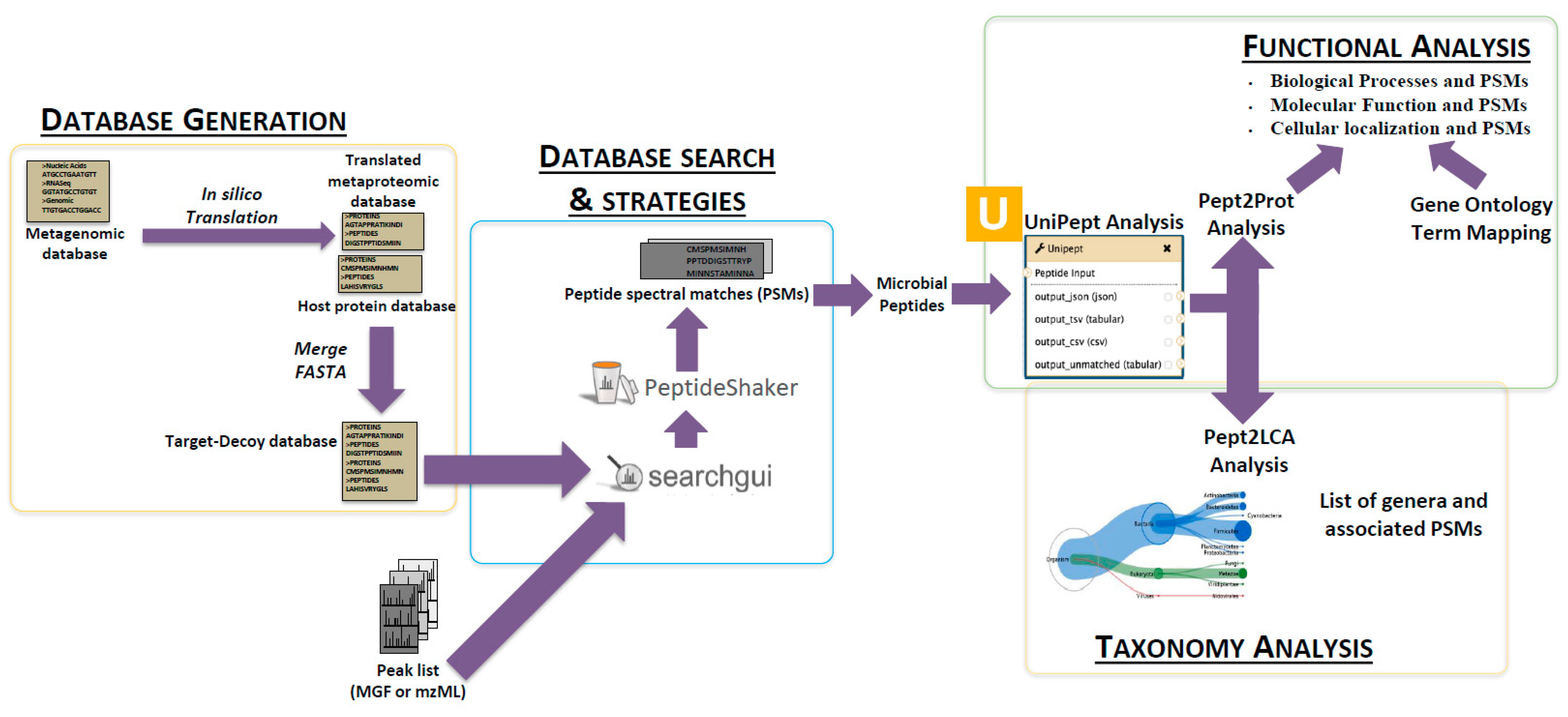
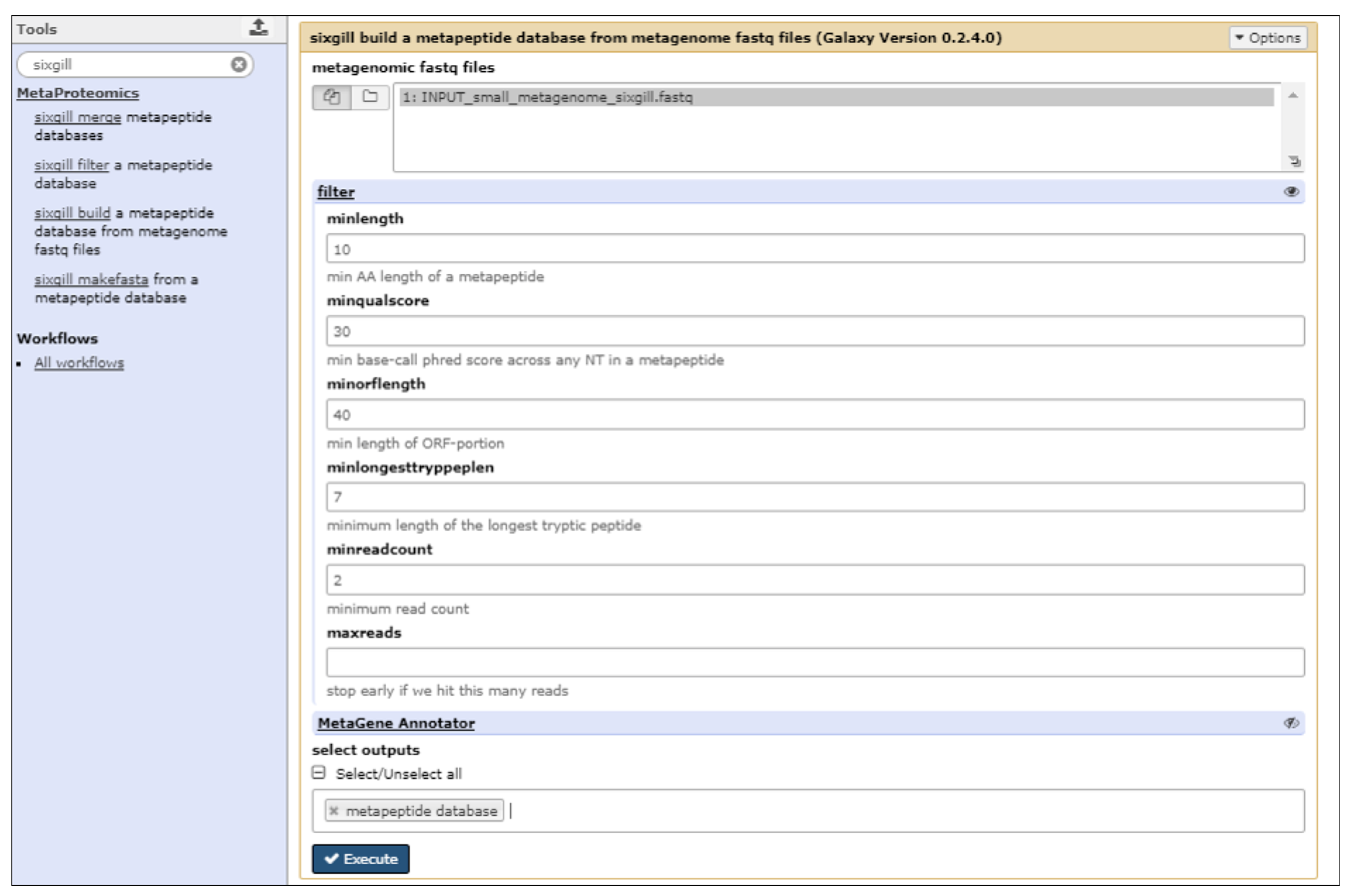
2.3. The First Step: Protein Sequence Database Generation Using a Galaxy-Based Tool
2.4. The Next Steps: Using a Galaxy Workflow
2.5. The Second Step: Spectral Matching
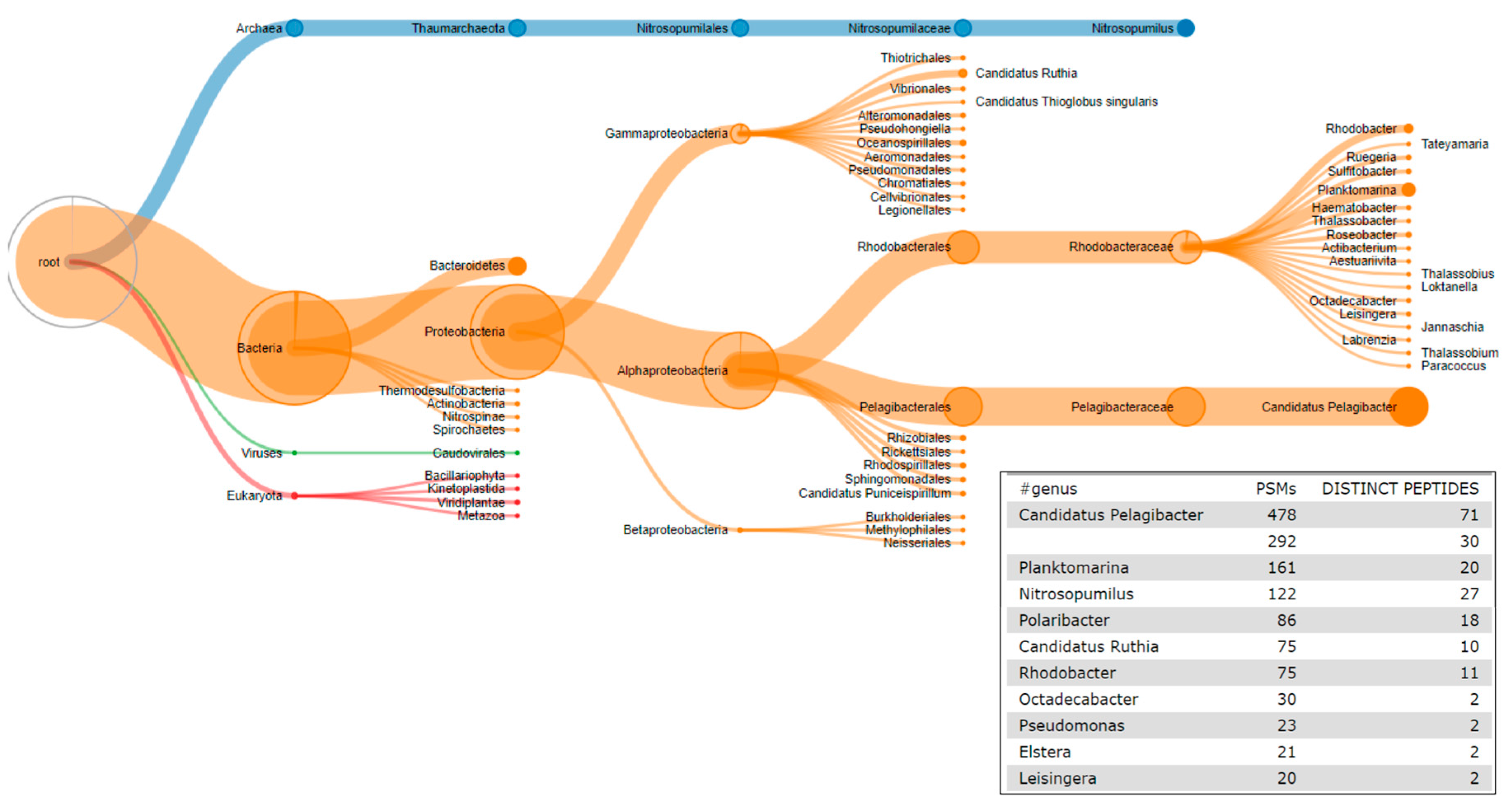
2.6. The Third Step: Taxonomic Classification
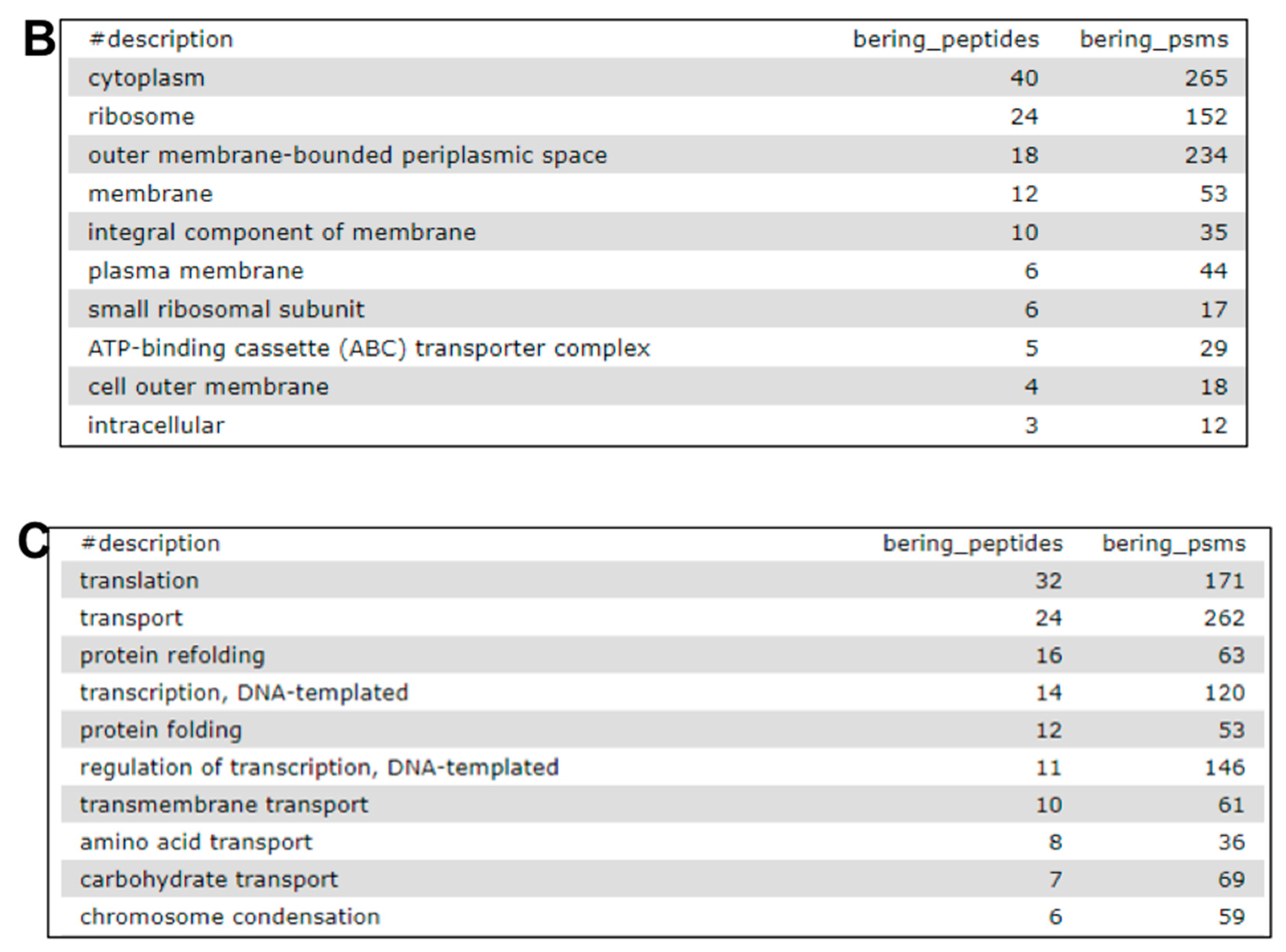
2.7. The Fourth Step: Functional Analysis
2.8. Links to Accessible Resources for Training
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Knight, R.; Callewaert, C.; Marotz, C.; Hyde, E.R.; Debelius, J.W.; McDonald, D.; Sogin, M.L. The Microbiome and Human Biology. Annu. Rev. Genom. Hum. Genet. 2017, 31, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.L.; Ling, H.; Lee, Y.S.; Chang, M.W. Microbiome engineering: Current applications and its future. Biotechnol. J. 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.W.; Roach, J.; Azcarate-Peril, M.A. Emerging Technologies for Gut Microbiome Research. Trends Microbiol. 2016, 24, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Siegwald, L.; Touzet, H.; Lemoine, Y.; Hot, D.; Audebert, C.; Caboche, S. Assessment of Common and Emerging Bioinformatics Pipelines for Targeted Metagenomics. PLoS ONE 2017, 12, e0169563. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.V.; Lucio, M.; Lee, L.H.; VerBerkmoes, N.C.; Brislawn, C.J.; Bernhardt, J.; Lamendella, R.; McDermott, J.E.; Bergeron, N.; Heinzmann, S.S.; et al. Impact of Dietary Resistant Starch on the Human Gut Microbiome, Metaproteome, and Metabolome. mBio 2017, 8, 1343–1417. [Google Scholar] [CrossRef] [PubMed]
- Heintz-Buschart, A.; May, P.; Laczny, C.C.; Lebrun, L.A.; Bellora, C.; Krishna, A.; Wampach, L.; Schneider, J.G.; Hogan, A.; de Beaufort, C.; et al. Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat. Microbiol. 2016, 2, 16180. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, P.; Bond, P.L. Metaproteomics: Studying functional gene expression in microbial ecosystems. Trends Microbiol. 2006, 14, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Heintz-Buschart, A.; Wilmes, P. Human Gut Microbiome: Function Matters. Trends Microbiol. 2017, 17, 30251–30252. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, P.; Heintz-Buschart, A.; Bond, P.L. A decade of metaproteomics: Where we stand and what the future holds. Proteomics 2015, 15, 3409–3417. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Abbondio, M.; Palomba, A.; Fraumene, C.; Manghina, V.; Cucca, F.; Fiorillo, E.; Uzzau, S. Potential and active functions in the gut microbiota of a healthy human cohort. Microbiome 2017, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar]
- Tanca, A.; Palomba, A.; Fraumene, C.; Pagnozzi, D.; Manghina, V.; Deligios, M.; Muth, T.; Rapp, E.; Martens, L.; Addis, M.F.; et al. The impact of sequence database choice on metaproteomic results in gut microbiota studies. Microbiome 2016, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Palomba, A.; Deligios, M.; Cubeddu, T.; Fraumene, C.; Biosa, G.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. Evaluating the impact of different sequence databases on metaproteome analysis: Insights from a lab-assembled microbial mixture. PLoS ONE 2013, 8, e82981. [Google Scholar] [CrossRef] [PubMed]
- Timmins-Schiffman, E.; May, D.H.; Mikan, M.; Riffle, M.; Frazar, C.; Harvey, H.R.; Noble, W.S.; Nunn, B.L. Critical decisions in metaproteomics: Achieving high confidence protein annotations in a sea of unknowns. ISME J. 2017, 11, 309–314. [Google Scholar] [CrossRef] [PubMed]
- May, D.H.; Timmins-Schiffman, E.; Mikan, M.P.; Harvey, H.R.; Borenstein, E.; Nunn, B.L.; Noble, W.S. An Alignment-Free “Metapeptide” Strategy for Metaproteomic Characterization of Microbiome Samples Using Shotgun Metagenomic Sequencing. J. Proteome Res. 2016, 15, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Li, S.; Ye, Y. A Graph-Centric Approach for Metagenome-Guided Peptide and Protein Identification in Metaproteomics. PLoS Comput. Biol. 2016, 12, 1005224. [Google Scholar] [CrossRef] [PubMed]
- Muth, T.; Renard, B.Y.; Martens, L. Metaproteomic data analysis at a glance: Advances in computational microbial community proteomics. Expert Rev. Proteom. 2016, 13, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Muth, T.; Kolmeder, C.A.; Salojärvi, J.; Keskitalo, S.; Varjosalo, M.; Verdam, F.J.; Rensen, S.S.; Reichl, U.; de Vos, W.M.; Rapp, E.; et al. Navigating through metaproteomics data: A logbook of database searching. Proteomics 2015, 15, 3439–3453. [Google Scholar] [CrossRef] [PubMed]
- Mesuere, B.; Debyser, G.; Aerts, M.; Devreese, B.; Vandamme, P.; Dawyndt, P. The Unipept metaproteomics analysis pipeline. Proteomics 2015, 15, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Brown, C.T.; Morowitz, M.J.; Banfield, J.F.; Hettich, R.L. Genome-resolved metaproteomic characterization of preterm infant gut microbiota development reveals species-specific metabolic shifts and variabilities during early life. Microbiome 2017, 5, 72. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, 1004957. [Google Scholar] [CrossRef] [PubMed]
- Muth, T.; Behne, A.; Heyer, R.; Kohrs, F.; Benndorf, D.; Hoffmann, M.; Lehtevä, M.; Reichl, U.; Martens, L.; Rapp, E. The MetaProteomeAnalyzer: A powerful open-source software suite for metaproteomics data analysis and interpretation. J. Proteome Res. 2015, 14, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.D.; Johnson, J.E.; Onsongo, G.; Sadler, F.W.; Murray, K.; Wang, Y.; Shenykman, G.M.; Bandhakavi, S.; Smith, L.M.; Griffin, T.J. Flexible and accessible workflows for improved proteogenomic analysis using the Galaxy framework. J. Proteome Res. 2014, 13, 5898–5908. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.D.; Blakely, A.; Murray, K.; Stewart, S.; Kooren, J.; Johnson, J.E.; Rhodus, N.L.; Rudney, J.; Griffin, T.J. Metaproteomic analysis using the Galaxy framework. Proteomics 2015, 15, 3553–3565. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, P.; Bond, P.L. The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ. Microbiol. 2004, 6, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Klaassens, E.S.; de Vos, W.M.; Vaughan, E.E. Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Appl. Environ. Microbiol. 2007, 73, 1388–1392. [Google Scholar] [CrossRef] [PubMed]
- Rudney, J.D.; Xie, H.; Rhodus, N.L.; Ondrey, F.G.; Griffin, T.J. A metaproteomic analysis of the human salivary microbiota by three-dimensional peptide fractionation and tandem mass spectrometry. Mol. Oral Microbiol. 2010, 25, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Haange, S.B.; Oberbach, A.; Schlichting, N.; Hugenholtz, F.; Smidt, H.; von Bergen, M.; Till, H.; Seifert, J. Metaproteome analysis and molecular genetics of rat intestinal microbiota reveals section and localization resolved species distribution and enzymatic functionalities. J. Proteome Res. 2012, 11, 5406–5417. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.; McGowan, T.; Bandhakavi, S.; Tu, Z.J.; Seymour, S.; Griffin, T.J.; Rudney, J.D. Deep metaproteomic analysis of human salivary supernatant. Proteomics 2012, 12, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Bastida, F.; Hernández, T.; García, C. Metaproteomics of soils from semiarid environment: Functional and phylogenetic information obtained with different protein extraction methods. J. Proteom. 2014, 101, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhu, J.; Yin, H.; Liu, X.; An, M.; Pudlo, N.A.; Martens, E.C.; Chen, G.Y.; Lubman, D.M. Development of an Integrated Pipeline for Profiling Microbial Proteins from Mouse Fecal Samples by LC-MS/MS. J. Proteome Res. 2016, 15, 3635–3642. [Google Scholar] [CrossRef] [PubMed]
- Kohrs, F.; Wolter, S.; Benndorf, D.; Heyer, R.; Hoffmann, M.; Rapp, E.; Bremges, A.; Sczyrba, A.; Schlüter, A.; Reichl, U. Fractionation of biogas plant sludge material improves metaproteomic characterization to investigate metabolic activity of microbial communities. Proteomics 2015, 15, 3585–3589. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Okubo, T.; Kubota, K.; Kasahara, Y.; Tsurumaru, H.; Anda, M.; Ikeda, S.; Minamisawa, K. Metaproteomic identification of diazotrophic methanotrophs and their localization in root tissues of field-grown rice plants. Appl. Environ. Microbiol. 2014, 80, 5043–5052. [Google Scholar] [CrossRef] [PubMed]
- Colatriano, D.; Ramachandran, A.; Yergeau, E.; Maranger, R.; Gélinas, Y.; Walsh, D.A. Metaproteomics of aquatic microbial communities in a deep and stratified estuary. Proteomics 2015, 15, 3566–3579. [Google Scholar] [CrossRef] [PubMed]
- Young, J.C.; Pan, C.; Adams, R.M.; Brooks, B.; Banfield, J.F.; Morowitz, M.J.; Hettich, R.L. Metaproteomics reveals functional shifts in microbial and human proteins during a preterm infant gut colonization case. Proteomics 2015, 15, 3463–3473. [Google Scholar] [CrossRef] [PubMed]
- Mattarozzi, M.; Manfredi, M.; Montanini, B.; Gosetti, F.; Sanangelantoni, A.M.; Marengo, E.; Careri, M.; Visioli, G. A metaproteomic approach dissecting major bacterial functions in the rhizosphere of plants living in serpentine soil. Anal. Bioanal. Chem. 2017, 409, 2327–2339. [Google Scholar] [CrossRef] [PubMed]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [PubMed]
- Haider, B.; Ahn, T.H.; Bushnell, B.; Chai, J.; Copeland, A.; Pan, C. Omega: An overlap-graph de novo assembler for metagenomics. Bioinformatics 2014, 30, 2717–2722. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Stupp, G.S.; Park, S.K.; Ducom, J.C.; Yates, J.R., 3rd; Su, A.I.; Wolan, D.W. A comprehensive and scalable database search system for metaproteomics. BMC Genom. 2016, 17, 642. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, Z.; Yao, Q.; Mueller, R.S.; Eng, J.K.; Tabb, D.L.; Hervey, W.J., 4th; Pan, C. Sipros Ensemble Improves Database Searching and Filtering for Complex Metaproteomics. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- Rooijers, K.; Kolmeder, C.; Juste, C.; Doré, J.; de Been, M.; Boeren, S.; Galan, P.; Beauvallet, C.; de Vos, W.M.; Schaap, P.J. An iterative workflow for mining the human intestinal metaproteome. BMC Genom. 2011, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Kertesz-Farkas, A.; Keich, U.; Noble, W.S. Tandem Mass Spectrum Identification via Cascaded Search. J. Proteome Res. 2015, 14, 3027–3038. [Google Scholar] [CrossRef] [PubMed]
- Rudney, J.D.; Jagtap, P.D.; Reilly, C.S.; Chen, R.; Markowski, T.W.; Higgins, L.; Johnson, J.E.; Griffin, T.J. Protein relative abundance patterns associated with sucrose-induced dysbiosis are conserved across taxonomically diverse oral microcosm biofilm models of dental caries. Microbiome 2015, 3, 69. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.; Goslinga, J.; Kooren, J.A.; McGowan, T.; Wroblewski, M.S.; Seymour, S.L.; Griffin, T.J. A two-step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies. Proteomics 2013, 13, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Vaudel, M.; Barsnes, H.; Berven, F.S.; Sickmann, A.; Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics 2011, 11, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Weber, N. Microbial community analysis using MEGAN. Methods Enzymol. 2013, 531, 465–485. [Google Scholar] [PubMed]
- Mesuere, B.; Van der Jeugt, F.; Willems, T.; Naessens, T.; Devreese, B.; Martens, L.; Dawyndt, P. High-throughput metaproteomics data analysis with Unipept: A tutorial. J. Proteom. 2017, 17, 30189–30196. [Google Scholar] [CrossRef] [PubMed]
- Mesuere, B.; Willems, T.; Van der Jeugt, F.; Devreese, B.; Vandamme, P.; Dawyndt, P. Unipept web services for metaproteomics analysis. Bioinformatics 2016, 32, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology: Enhancements for 2011. Nucleic Acids Res. 2012, 40, 559–564. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evolut. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A web-based tool for Gene Ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metaproteomics Gateway | z.umn.edu/metaproteomicsgateway |
| Galaxy Training Network | http://galaxyproject.github.io/training-material/topics/proteomics/tutorials/metaproteomics/tutorial.html |
| Documentation | Supplement S1 |
| Introductory video | z.umn.edu/mpvideo2018 |
| Galaxy toolshed | https://toolshed.g2.bx.psu.edu/ |
| GitHub | https://github.com/galaxyproteomics |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blank, C.; Easterly, C.; Gruening, B.; Johnson, J.; Kolmeder, C.A.; Kumar, P.; May, D.; Mehta, S.; Mesuere, B.; Brown, Z.; et al. Disseminating Metaproteomic Informatics Capabilities and Knowledge Using the Galaxy-P Framework. Proteomes 2018, 6, 7. https://doi.org/10.3390/proteomes6010007
Blank C, Easterly C, Gruening B, Johnson J, Kolmeder CA, Kumar P, May D, Mehta S, Mesuere B, Brown Z, et al. Disseminating Metaproteomic Informatics Capabilities and Knowledge Using the Galaxy-P Framework. Proteomes. 2018; 6(1):7. https://doi.org/10.3390/proteomes6010007
Chicago/Turabian StyleBlank, Clemens, Caleb Easterly, Bjoern Gruening, James Johnson, Carolin A. Kolmeder, Praveen Kumar, Damon May, Subina Mehta, Bart Mesuere, Zachary Brown, and et al. 2018. "Disseminating Metaproteomic Informatics Capabilities and Knowledge Using the Galaxy-P Framework" Proteomes 6, no. 1: 7. https://doi.org/10.3390/proteomes6010007
APA StyleBlank, C., Easterly, C., Gruening, B., Johnson, J., Kolmeder, C. A., Kumar, P., May, D., Mehta, S., Mesuere, B., Brown, Z., Elias, J. E., Hervey, W. J., McGowan, T., Muth, T., Nunn, B. L., Rudney, J., Tanca, A., Griffin, T. J., & Jagtap, P. D. (2018). Disseminating Metaproteomic Informatics Capabilities and Knowledge Using the Galaxy-P Framework. Proteomes, 6(1), 7. https://doi.org/10.3390/proteomes6010007