Pilot Study on Mass Spectrometry–Based Analysis of the Proteome of CD34+CD123+ Progenitor Cells for the Identification of Potential Targets for Immunotherapy in Acute Myeloid Leukemia

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients with AML and Healthy Donors
2.2. Sample Preparation
2.3. Fluorescence Activated Cell Sorting (FACS)
2.4. Proteomic Sample Preparation and Mass Spectrometric Analyses
2.5. Evaluation of Mass Spectrometric Data and Statistics
3. Results
3.1. FACS-Sorting of CD34+CD123+ Cells
3.2. Identification of Proteins in Significantly Altered Abundance from Proteomic Data
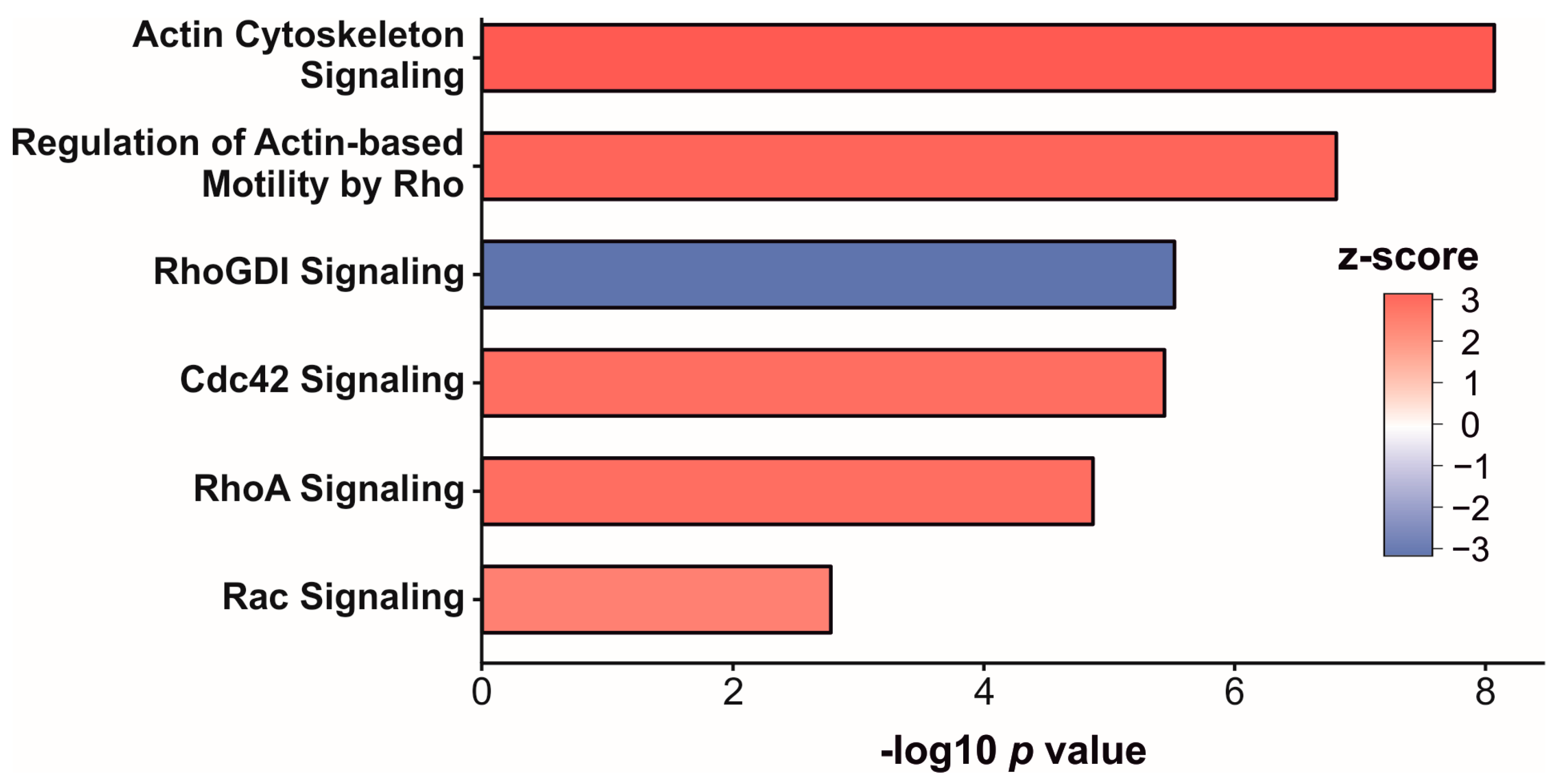
3.3. Enrichment Analysis of Significantly Altered Proteins
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ota, J.; Yamashita, Y.; Okawa, K.; Kisanuki, H.; Fujiwara, S.-I.; Ishikawa, M.; Lim Choi, Y.; Ueno, S.; Ohki, R.; Koinuma, K.; et al. Proteomic analysis of hematopoietic stem cell-like fractions in leukemic disorders. Oncogene 2003, 22, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Luczak, M.; Kaźmierczak, M.; Handschuh, L.; Lewandowski, K.; Komarnicki, M.; Figlerowicz, M. Comparative proteome analysis of acute myeloid leukemia with and without maturation. J. Proteom. 2012, 75, 5734–5748. [Google Scholar] [CrossRef] [PubMed]
- Bonardi, F.; Fusetti, F.; Deelen, P.; van Gosliga, D.; Vellenga, E.; Schuringa, J.J. A proteomics and transcriptomics approach to identify leukemic stem cell (LSC) markers. Mol. Cell. Proteom. 2013, 12, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Strassberger, V.; Gutbrodt, K.L.; Krall, N.; Roesli, C.; Takizawa, H.; Manz, M.G.; Fugmann, T.; Neri, D. A comprehensive surface proteome analysis of myeloid leukemia cell lines for therapeutic antibody development. J. Proteom. 2014, 99, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, E.; Vaudel, M.; Mjaavatten, O.; Gausdal, G.; van der Burgh, A.; Gjertsen, B.T.; Døskeland, S.O.; Bruserud, Ø.; Berven, F.S.; Selheim, F. Performance of super-SILAC based quantitative proteomics for comparison of different acute myeloid leukemia (AML) cell lines. Proteomics 2014, 14, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.; Gerrits, B.; Schmidt, A.; Bock, T.; Bausch-Fluck, D.; Aebersold, R.; Wollscheid, B. Proteomic cell surface phenotyping of differentiating acute myeloid leukemia cells. Blood 2010, 116. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, E.; Forthun, R.B.; Berven, F.; Selheim, F.; Hernandez-Valladares, M. Global Cell Proteome Profiling, Phospho-signaling and Quantitative Proteomics for Identification of New Biomarkers in Acute Myeloid Leukemia Patients. Curr. Pharm. Biotechnol. 2016, 17, 52–70. [Google Scholar] [CrossRef] [PubMed]
- Luczak, M.; Kaźmierczak, M.; Hadschuh, L.; Lewandowski, K.; Komarnicki, M.; Figlerowicz, M. Comparative proteomics in acute myeloid leukemia. Contemp. Oncol. (Pozn. Pol.) 2012, 16, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Lee, E.M.; Ramshaw, H.S.; Busfield, S.J.; Peoppl, A.G.; Wilkinson, L.; Guthridge, M.A.; Thomas, D.; Barry, E.F.; Boyd, A.; et al. Monoclonal Antibody-Mediated Targeting of CD123, IL-3 Receptor α Chain, Eliminates Human Acute Myeloid Leukemic Stem Cells. Cell Stem Cell 2009, 5, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawali, A.; Gillis, D.; Lewis, I. Immunoprofiling of leukemic stem cells CD34+/CD38−/CD123+ delineate FLT3/ITD-positive clones. J. Hematol. Oncol. 2016, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Attar, A. Changes in the Cell Surface Markers During Normal Hematopoiesis: A Guide to Cell Isolation. Glob. J. Hematol. Blood Transfus. 2014, 1, 20–28. [Google Scholar] [CrossRef]
- Taussig, D.C.; Pearce, D.J.; Simpson, C.; Rohatiner, A.Z.; Lister, T.A.; Kelly, G.; Luongo, J.L.; Danet-Desnoyers, G.A.H.; Bonnet, D. Hematopoietic stem cells express multiple myeloid markers: Implications for the origin and targeted therapy of acute myeloid leukemia. Blood 2005, 106, 4086–4092. [Google Scholar] [CrossRef] [PubMed]
- Jouy, F.; Lohmann, N.; Wandel, E.; Ruiz-Gómez, G.; Pisabarro, M.T.; Beck-Sickinger, A.G.; Schnabelrauch, M.; Möller, S.; Simon, J.C.; Kalkhof, S.; et al. Sulfated hyaluronan attenuates inflammatory signaling pathways in macrophages involving induction of antioxidants. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J. Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawali, A.; Pinto, A.D.; Al-Zadjali, S. CD34+CD38−CD123+ Cells Are Present in Virtually All Acute Myeloid Leukaemia Blasts: A Promising Single Unique Phenotype for Minimal Residual Disease Detection. Acta Haematol. 2017, 138, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, Z.; Yu, J.F.; Young, D.; Bashey, A.; Ho, A.D.; Law, P. Correlation between IL-3 receptor expression and growth potential of human CD34+ hematopoietic cells from different tissues. Stem Cells 1999, 17, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Blalock, W.L.; Weinstein-Oppenheimer, C.; Chang, F.; Hoyle, P.E.; Wang, X.Y.; Algate, P.A.; Franklin, R.A.; Oberhaus, S.M.; Steelman, L.S.; McCubrey, J.A. Signal transduction, cell cycle regulatory, and anti-apoptotic pathways regulated by IL-3 in hematopoietic cells: Possible sites for intervention with anti-neoplastic drugs. Leukemia 1999, 13, 1109–1166. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Frankel, A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark. Res. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Lichtenegger, F.S.; Krupka, C.; Haubner, S.; Köhnke, T.; Subklewe, M. Recent developments in immunotherapy of acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Barrett, A.J. Novel immunotherapeutic approaches for the treatment of acute leukemia (myeloid and lymphoblastic). Ther. Adv. Hematol. 2016, 7, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, K.-H.; Park, J.S.; Jung, J.W.; Kim, Y.H.; Kim, S.K.; Kim, W.-S.; Goh, H.; Kim, S.; Yoo, J.-S.; et al. Comparative analysis of cell surface proteins in chronic and acute leukemia cell lines. Biochem. Biophys. Res. Commun. 2007, 357, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Rockstroh, M.; Müller, S.A.; Jende, C.; Kerzhner, A.; Von Bergen, M.; Tomm, J.M. Cell fractionation—An important tool for compartment proteomics. Jiomics 2011, 1, 135–143. [Google Scholar] [CrossRef]
- Hernandez-Valladares, M.; Aasebø, E.; Selheim, F.; Berven, F.S.; Bruserud, Ø. Selecting Sample Preparation Workflows for Mass Spectrometry-Based Proteomic and Phosphoproteomic Analysis of Patient Samples with Acute Myeloid Leukemia. Proteomes 2016. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Berlin, C.; Kowalewski, D.J.; Schuster, H.; Mirza, N.; Walz, S.; Handel, M.; Schmid-Horch, B.; Salih, H.R.; Kanz, L.; Rammensee, H.-G.; et al. Mapping the HLA ligandome landscape of acute myeloid leukemia: a targeted approach toward peptide-based immunotherapy. Leukemia 2015, 29, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, W.; Zou, D.; Chen, G.; Wan, T.; Zhang, M.; Cao, X. Cloning and functional characterization of GMPR2, a novel human guanosine monophosphate reductase, which promotes the monocytic differentiation of HL-60 leukemia cells. J. Cancer Res. Clin. Oncol. 2003, 129, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Gillissen, M.A.; Kedde, M.; De Jong, G.; Moiset, G.; Yasuda, E.; Levie, S.E.; Bakker, A.Q.; Claassen, Y.B.; Wagner, K.; Böhne, M.; et al. AML-specific cytotoxic antibodies in patients with durable graft-versus-leukemia responses. Blood 2018, 131, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Vey, N.; Delaunay, J.; Martinelli, G.; Fiedler, W.; Raffoux, E.; Prebet, T.; Gomez-Roca, C.; Papayannidis, C.; Kebenko, M.; Paschka, P.; et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 2016, 7, 32532–32542. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Eng, W.S.; Hu, W.; Khalaj, M.; Garrett-Bakelman, F.E.; Tavakkoli, M.; Levine, R.L.; Carroll, M.; Klimek, V.M.; Melnick, A.M.; et al. CD99 is a therapeutic target on disease stem cells in myeloid malignancies. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø. Effects of interleukin-13 on cytokine secretion by human acute myelogenous leukemia blasts. Leukemia 1996, 10, 1497–1503. [Google Scholar] [PubMed]
- Kornblau, S.M.; McCue, D.; Singh, N.; Chen, W.; Estrov, Z.; Coombes, K.R. Recurrent expression signatures of cytokines and chemokines are present and are independently prognostic in acute myelogenous leukemia and myelodysplasia. Blood 2010, 116, 4251–4261. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.D.; Lee, K.E.; Williams, D.A.; Gu, Y. Cross-talk between RhoH and Rac1 in regulation of actin cytoskeleton and chemotaxis of hematopoietic progenitor cells. Blood 2008, 111, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Jasti, A.C.; Jansen, M.; Siefring, J.E. RhoH, a hematopoietic-specific Rho GTPase, regulates proliferation, survival, migration, and engraftment of hematopoietic progenitor cells. Blood 2005, 105, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liu, S.; Wang, J.; Sun, M.Z.; Greenaway, F.T. ACTB in cancer. Clin. Chim. Acta 2013, 417, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Verbiest, T.; Bouffler, S.; L.Nutt, S.; Badie, C. PU.1 downregulation in murine radiation-induced acute myeloid leukaemia (AML): From molecular mechanism to human AML. Carcinogenesis 2014, 36, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Valladares, M.; Vaudel, M.; Selheim, F.; Berven, F.; Bruserud, Ø. Proteogenomics approaches for studying cancer biology and their potential in the identification of acute myeloid leukemia biomarkers. Expert Rev. Proteom. 2017, 14, 649–663. [Google Scholar] [CrossRef] [PubMed]




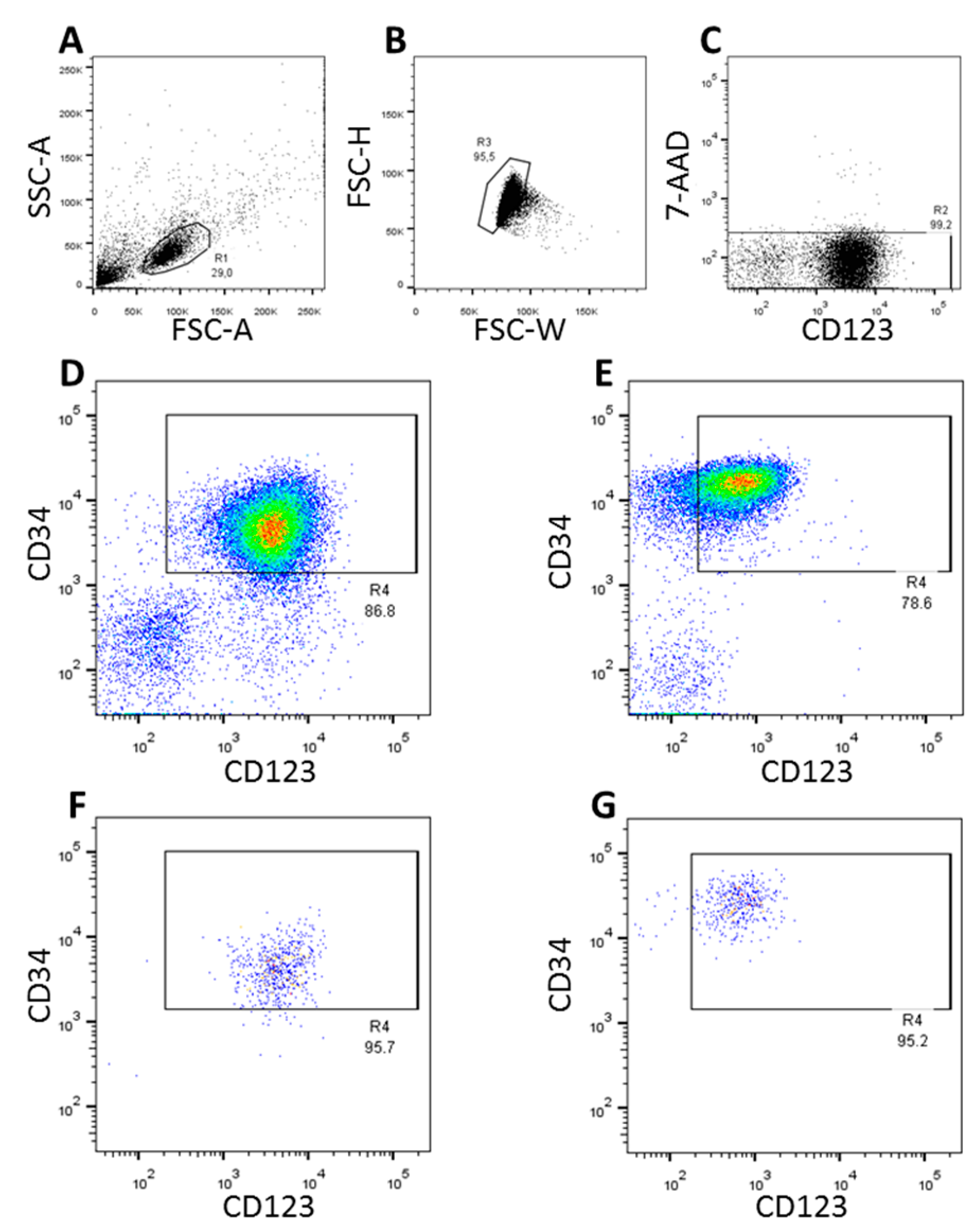{kind=link}
{kind=link}
{kind=link}
| Patient/Donor ID | Sex | Age | Disease Characteristics/Donor Information |
|---|---|---|---|
| AML_033 | f | 61 | AML FAB M4, Complex aberrant karyotype, negative for FLT3-ITD, NPM1, CEBPA |
| AML_036 | m | 66 | sAML from CMML, trisomy 8, deletion 21q, duplication 15q11/15q26, negative for FLT3-ITD, NPM1, CEBPA |
| AML_037 | f | 60 | AML FAB M5, normal karyotype, FLT3-ITD+, NPM1 mutation type D |
| AML_044 | m | 71 | AML FAB M1, normal karyotype, negative for FLT3-ITD, NPM1, CEBPA |
| AML_049 | f | 18 | AML FAB M1, normal karyotype, FLT3-ITDlow, negative for NPM1, CEBPA |
| DON_004 | m | 39 | all donors passed a medical work-up and were released for stem cell donation; the following conditions were excluded: active infections such as viral hepatitis, AIDS, syphilis, malaria; history of malignant tumors or systemic autoimmune diseases; medically significant abnormalities of the differential blood count |
| DON_005 | f | 25 | |
| DON_011 | f | 40 | |
| DON_012 | m | 44 | |
| DON_013 | m | 20 |
| Cytosolic/Membrane Fraction | Accession ID | Gene ID | Description | Number of Peptides | LOG2 [FC (AML/DON)] | p-Value |
|---|---|---|---|---|---|---|
| CF | P63261 | ACTG1 | Actin, cytoplasmic 2 | 67 | 1.08 | <0.0001 |
| CF | Q9P2T1 | GMPR2 | GMP reductase 2 | 3 | −0.90 | <0.0001 |
| CF | Q9UNH7 | SNX6 | Sorting nexin-6 | 3 | 0.77 | <0.0001 |
| CF | P11766 | ADH5 | Alcohol dehydrogenase class-3 | 2 | −0.53 | <0.0001 |
| CF | Q9H2H8 | PPIL3 | Peptidyl-prolyl cis-trans isomerase-like 3 | 3 | −0.72 | <0.0001 |
| CF | P0CG34 | TMSB15A | Thymosin beta-15A | 2 | −1.31 | <0.0001 |
| MF | P30626 | SRI | Sorcin | 4 | −1.57 | 0.0007 |
| MF | Q92734 | TFG | Protein TFG | 3 | −1.49 | 0.0019 |
| Cluster | Enrichment Score | Number of Proteins Sign. Higher Abundance | Number of Proteins Sign. Lower Abundance |
|---|---|---|---|
| cellular response to endogenous stimulus (GO:0071495) | 3.1 | 25 | 35 |
| cellular response to metal ion (GO:0071248) | 2.7 | 9 | 22 |
| isoprenoid metabolic process (GO:0006720) | 2.6 | 2 | 14 |
| tissue homeostasis (GO:0001894) | 2.6 | 6 | 7 |
| negative regulation of cytokine production (GO:0001818) | 2.5 | 6 | 15 |
| organic hydroxy compound metabolic process (GO:1901615) | 2.3 | 3 | 20 |
| transmembrane receptor protein tyrosine kinase signaling pathway (GO:0007169) | 2.3 | 20 | 16 |
| actin filament polymerization (GO:0030041) | 2.2 | 25 | 16 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, J.R.; Rücker-Braun, E.; Heidrich, K.; Von Bonin, M.; Stölzel, F.; Thiede, C.; Middeke, J.M.; Ehninger, G.; Bornhäuser, M.; Schetelig, J.; et al. Pilot Study on Mass Spectrometry–Based Analysis of the Proteome of CD34+CD123+ Progenitor Cells for the Identification of Potential Targets for Immunotherapy in Acute Myeloid Leukemia. Proteomes 2018, 6, 11. https://doi.org/10.3390/proteomes6010011
Schmidt JR, Rücker-Braun E, Heidrich K, Von Bonin M, Stölzel F, Thiede C, Middeke JM, Ehninger G, Bornhäuser M, Schetelig J, et al. Pilot Study on Mass Spectrometry–Based Analysis of the Proteome of CD34+CD123+ Progenitor Cells for the Identification of Potential Targets for Immunotherapy in Acute Myeloid Leukemia. Proteomes. 2018; 6(1):11. https://doi.org/10.3390/proteomes6010011
Chicago/Turabian StyleSchmidt, Johannes R., Elke Rücker-Braun, Katharina Heidrich, Malte Von Bonin, Friedrich Stölzel, Christian Thiede, Jan M. Middeke, Gerhard Ehninger, Martin Bornhäuser, Johannes Schetelig, and et al. 2018. "Pilot Study on Mass Spectrometry–Based Analysis of the Proteome of CD34+CD123+ Progenitor Cells for the Identification of Potential Targets for Immunotherapy in Acute Myeloid Leukemia" Proteomes 6, no. 1: 11. https://doi.org/10.3390/proteomes6010011
APA StyleSchmidt, J. R., Rücker-Braun, E., Heidrich, K., Von Bonin, M., Stölzel, F., Thiede, C., Middeke, J. M., Ehninger, G., Bornhäuser, M., Schetelig, J., Schubert, K., Von Bergen, M., & Heidenreich, F. (2018). Pilot Study on Mass Spectrometry–Based Analysis of the Proteome of CD34+CD123+ Progenitor Cells for the Identification of Potential Targets for Immunotherapy in Acute Myeloid Leukemia. Proteomes, 6(1), 11. https://doi.org/10.3390/proteomes6010011