Quantification of Cardiovascular Disease Biomarkers in Human Platelets by Targeted Mass Spectrometry


Abstract
:1. Introduction
1.1. Platelets Are Key Players in the Generation and Progression of Cardiovascular Diseases
1.2. Targeted Mass Spectrometry as a Clinical Tool-Kit for Future Diagnostics
2. Materials and Methods
2.1. Platelet Isolation
2.2. Sample Preparation
2.3. Synthesis of SIL Peptides
2.4. Mass Spectrometry Analysis
2.5. Data Evaluation Using Skyline and R
3. Results and Discussion
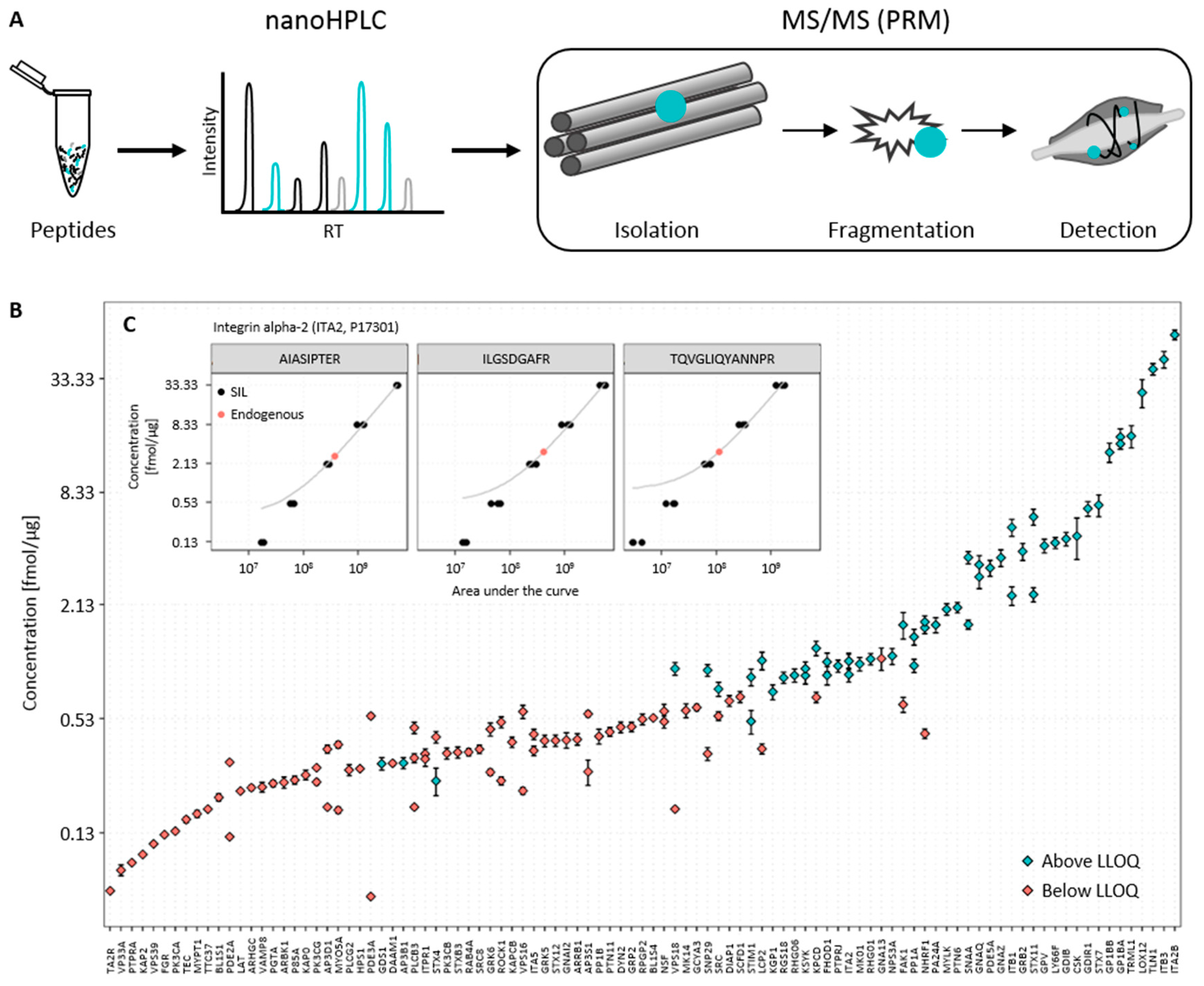
3.1. Establishment of the Targeted Mass Spectrometry-Based Assay
3.2. Determination of Endogenous Concentration of Relevant Platelet Proteins
3.3. Data Analysis and Quality Control
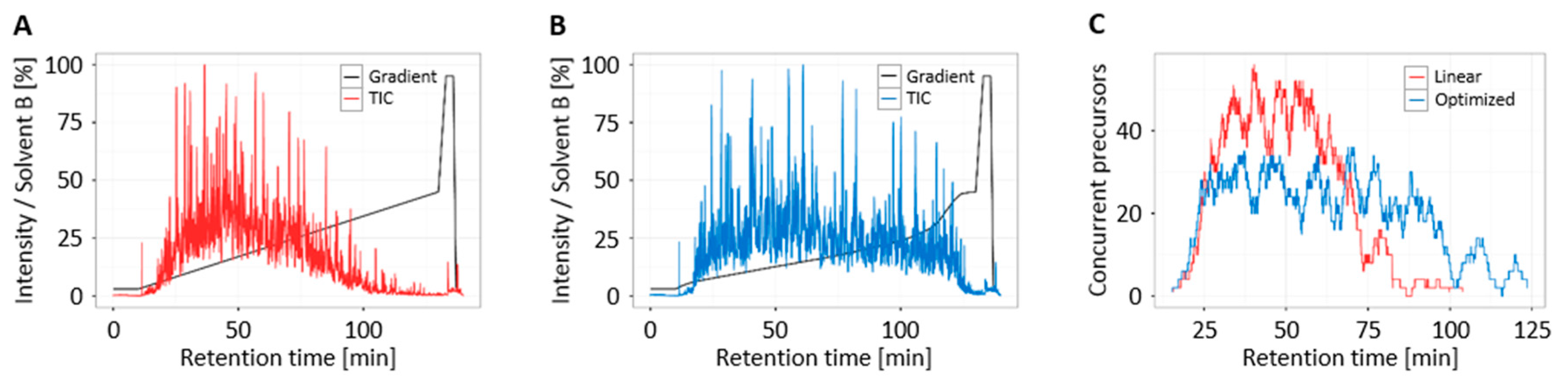
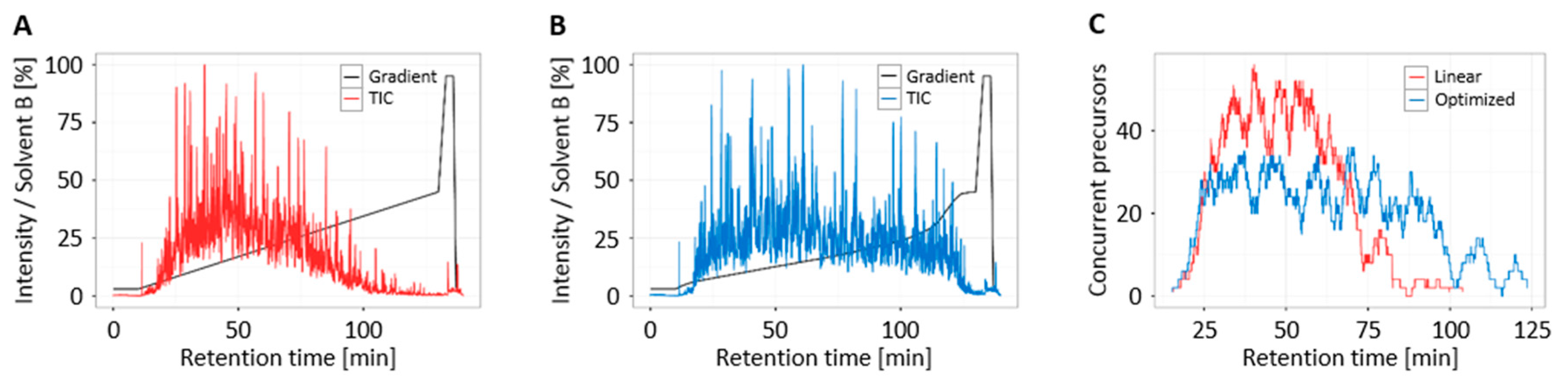
3.4. Increased Multiplexing Capability by Optimized Liquid Chromatography
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO). Cardiovascular Diseases (CVDs). Available online: http://www.who.int/mediacentre/factsheets/fs317/en/ (accessed on 1 October 2017).
- Patel, S.R.; Hartwig, J.H.; Italiano, J.E. The biogenesis of platelets from megakaryocyte proplatelets. J. Clin. Investig. 2005, 115, 3348–3354. [Google Scholar] [CrossRef] [PubMed]
- Gachet, C. P2 receptors, platelet function and pharmacological implications. Thromb. Haemost. 2008, 99, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Hewing, B.; Landmesser, U. LDL, HDL, VLDL, and CVD Prevention: Lessons from Genetics? Curr. Cardiol. Rep. 2015, 17, 610. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.A.E.; Singhateh, Y.; Mackay, D.; Huxley, R.R.; Woodward, M. Total cholesterol as a risk factor for coronary heart disease and stroke in women compared with men: A systematic review and meta-analysis. Atherosclerosis 2016, 248, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Adukauskienė, D.; Čiginskienė, A.; Adukauskaitė, A.; Pentiokinienė, D.; Šlapikas, R.; Čeponienė, I. Clinical relevance of high sensitivity C-reactive protein in cardiology. Medicina 2016, 52, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, B.J.; Boekholdt, S.M.; Kastelein, J.J.P. Lipid parameters for measuring risk of cardiovascular disease. Nat. Rev. Cardiol. 2011, 8, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tan, G.-J.; Han, L.-N.; Bai, Y.-Y.; He, M.; Liu, H.-B. Novel biomarkers for cardiovascular risk prediction. J. Geriatr. Cardiol. 2017, 14, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef] [PubMed]
- Beck, F.; Geiger, J.; Gambaryan, S.; Solari, F.A.; Dell’Aica, M.; Loroch, S.; Mattheij, N.J.; Mindukshev, I.; Pötz, O.; Jurk, K.; et al. Temporal quantitative phosphoproteomics of ADP stimulation reveals novel central nodes in platelet activation and inhibition. Blood 2017, 129, e1–e12. [Google Scholar] [CrossRef] [PubMed]
- Beck, F.; Geiger, J.; Gambaryan, S.; Veit, J.; Vaudel, M.; Nollau, P.; Kohlbacher, O.; Martens, L.; Walter, U.; Sickmann, A.; et al. Time-resolved characterization of cAMP/PKA-dependent signaling reveals that platelet inhibition is a concerted process involving multiple signaling pathways. Blood 2014, 123, e1–e10. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Yang, W.; Sun, S.; Pasay, J.; Faraday, N.; Zhang, H. Platelet glycoproteins associated with aspirin-treatment upon platelet activation. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Lewandrowski, U.; Moebius, J.; Walter, U.; Sickmann, A. Elucidation of N-glycosylation sites on human platelet proteins: A glycoproteomic approach. Mol. Cell. Proteom. 2006, 5, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Lewandrowski, U.; Lohrig, K.; Zahedi, R.P.; Wolters, D.; Sickmann, A. Glycosylation Site Analysis of Human Platelets by Electrostatic Repulsion Hydrophilic Interaction Chromatography. Clin. Proteom. 2008, 4, 25–36. [Google Scholar] [CrossRef]
- Dowal, L.; Yang, W.; Freeman, M.R.; Steen, H.; Flaumenhaft, R. Proteomic analysis of palmitoylated platelet proteins. Blood 2011, 118, e62–e73. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.M. Palmitoylation of platelet proteins. Platelets 1994, 5, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Sim, D.S.; Dilks, J.R.; Flaumenhaft, R. Platelets possess and require an active protein palmitoylation pathway for agonist-mediated activation and in vivo thrombus formation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Wickham, H. The Split-Apply-Combine Strategy for Data Analysis. J. Stat. Softw. 2011. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Solari, F.A.; Mattheij, N.J.A.; Burkhart, J.M.; Swieringa, F.; Collins, P.W.; Cosemans, J.M.E.M.; Sickmann, A.; Heemskerk, J.W.M.; Zahedi, R.P. Combined Quantification of the Global Proteome, Phosphoproteome, and Proteolytic Cleavage to Characterize Altered Platelet Functions in the Human Scott Syndrome. Mol. Cell. Proteom. 2016, 15, 3154–3169. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.T.; Poole, A.W. Diverse functions of protein kinase C isoforms in platelet activation and thrombus formation. J. Thromb. Haemost. 2010, 8, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.S.; Lee, J.R.; Clements, J.L.; Turner, M.; Tybulewicz, V.L.; Findell, P.R.; Koretzky, G.A.; Watson, S.P. Tyrosine phosphorylation of SLP-76 is downstream of Syk following stimulation of the collagen receptor in platelets. J. Biol. Chem. 1999, 274, 5963–5971. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Polgar, J.; Reed, G.L. Protein kinase C phosphorylation of syntaxin 4 in thrombin-activated human platelets. J. Biol. Chem. 2000, 275, 25286–25291. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Münzer, P.; Gawaz, M.; Borst, O. Regulation of STIM1/Orai1-dependent Ca2+ signalling in platelets. Thromb. Haemost. 2013, 110, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Marjoram, R.J.; Voss, B.; Pan, Y.; Dickeson, S.K.; Zutter, M.M.; Hamm, H.E.; Santoro, S.A. Suboptimal activation of protease-activated receptors enhances alpha2beta1 integrin-mediated platelet adhesion to collagen. J. Biol. Chem. 2009, 284, 34640–34647. [Google Scholar] [CrossRef] [PubMed]
- Mischak, H.; Apweiler, R.; Banks, R.E.; Conaway, M.; Coon, J.; Dominiczak, A.; Ehrich, J.H.H.; Fliser, D.; Girolami, M.; Hermjakob, H.; et al. Clinical proteomics: A need to define the field and to begin to set adequate standards. Proteom. Clin. Appl. 2007, 1, 148–156. [Google Scholar] [CrossRef] [PubMed]
- García, Á. Platelet clinical proteomics: Facts, challenges, and future perspectives. Proteom. Clin. Appl. 2016, 10, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Kettenbach, A.N.; Rush, J.; Gerber, S.A. Absolute quantification of protein and post-translational modification abundance with stable isotope-labeled synthetic peptides. Nat. Protoc. 2011, 6, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Han, J.; Pan, J.; Liu, T.; Parker, C.E.; Borchers, C.H. Current trends in quantitative proteomics—An update. J. Mass Spectrom. 2017, 52, 319–341. [Google Scholar] [CrossRef] [PubMed]
- Percy, A.J.; Michaud, S.A.; Jardim, A.; Sinclair, N.J.; Zhang, S.; Mohammed, Y.; Palmer, A.L.; Hardie, D.B.; Yang, J.; LeBlanc, A.M.; et al. Multiplexed MRM-based assays for the quantitation of proteins in mouse plasma and heart tissue. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.M.; Bollinger, J.G.; Becker, J.O.; Wallace, J.M.; Laha, T.J.; MacCoss, M.J.; Hoofnagle, A.N. Quantification by nano liquid chromatography parallel reaction monitoring mass spectrometry of human apolipoprotein A-I, apolipoprotein B, and hemoglobin A1c in dried blood spots. Proteom. Clin. Appl. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2008, 4, 222. [Google Scholar] [CrossRef] [PubMed]
- Kuzyk, M.A.; Smith, D.; Yang, J.; Cross, T.J.; Jackson, A.M.; Hardie, D.B.; Anderson, N.L.; Borchers, C.H. Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteom. 2009, 8, 1860–1877. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Aebersold, R. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.G.; Percy, A.J.; Yang, J.; Camenzind, A.G.; Borchers, C.H. Multiplexed quantitation of endogenous proteins in dried blood spots by multiple reaction monitoring-mass spectrometry. Mol. Cell. Proteom. 2013, 12, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Gallien, S.; El-Khoury, V.; Goswami, P.; Sertamo, K.; Schlesser, M.; Berchem, G.; Domon, B. Quantification of SAA1 and SAA2 in lung cancer plasma using the isotype-specific PRM assays. Proteomics 2015, 15, 3116–3125. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, A.; Michaud, S.A.; Percy, A.J.; Hardie, D.B.; Yang, J.; Sinclair, N.J.; Proudfoot, J.I.; Pistawka, A.; Smith, D.S.; Borchers, C.H. Multiplexed MRM-Based Protein Quantitation Using Two Different Stable Isotope-Labeled Peptide Isotopologues for Calibration. J. Proteome Res. 2017, 16, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Delatour, T.; Mottier, P.; Gremaud, E. Limits of suspicion, recognition and confirmation as concepts that account for the confirmation transitions at the detection limit for quantification by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2007, 1169, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Mermelekas, G.; Vlahou, A.; Zoidakis, J. SRM/MRM targeted proteomics as a tool for biomarker validation and absolute quantification in human urine. Expert Rev. Mol. Diagn. 2015, 15, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; MacLean, B.; Held, J.M.; Sahu, A.K.; Rardin, M.J.; Sorensen, D.J.; Peters, T.; Wolfe, A.J.; Hunter, C.L.; MacCoss, M.J.; et al. Multiplexed, Scheduled, High-Resolution Parallel Reaction Monitoring on a Full Scan QqTOF Instrument with Integrated Data-Dependent and Targeted Mass Spectrometric Workflows. Anal. Chem. 2015, 87, 10222–10229. [Google Scholar] [CrossRef] [PubMed]
- Burgess, M.W.; Keshishian, H.; Mani, D.R.; Gillette, M.A.; Carr, S.A. Simplified and Efficient Quantification of Low Abundance Proteins at Very High Multiplex by Targeted Mass Spectrometry. Mol. Cell. Proteom. 2014, 13, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Moruz, L.; Pichler, P.; Stranzl, T.; Mechtler, K.; Käll, L. Optimized Nonlinear Gradients for Reversed-Phase Liquid Chromatography in Shotgun Proteomics. Anal. Chem. 2013, 85, 7777–7785. [Google Scholar] [CrossRef] [PubMed]
- Moruz, L.; Käll, L. GradientOptimizer: An open-source graphical environment for calculating optimized gradients in reversed-phase liquid chromatography. Proteomics 2014, 14, 1464–1466. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef] [PubMed]
- Wegler, C.; Gaugaz, F.Z.; Andersson, T.B.; Wiśniewski, J.R.; Busch, D.; Gröer, C.; Oswald, S.; Norén, A.; Weiss, F.; Hammer, H.S.; et al. Variability in Mass Spectrometry-based Quantification of Clinically Relevant Drug Transporters and Drug Metabolizing Enzymes. Mol. Pharmacol. 2017, 14, 3142–3151. [Google Scholar] [CrossRef] [PubMed]
- Newman, H.; Maritz, J. Basic overview of method validation in the clinical virology laboratory. Rev. Med. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]

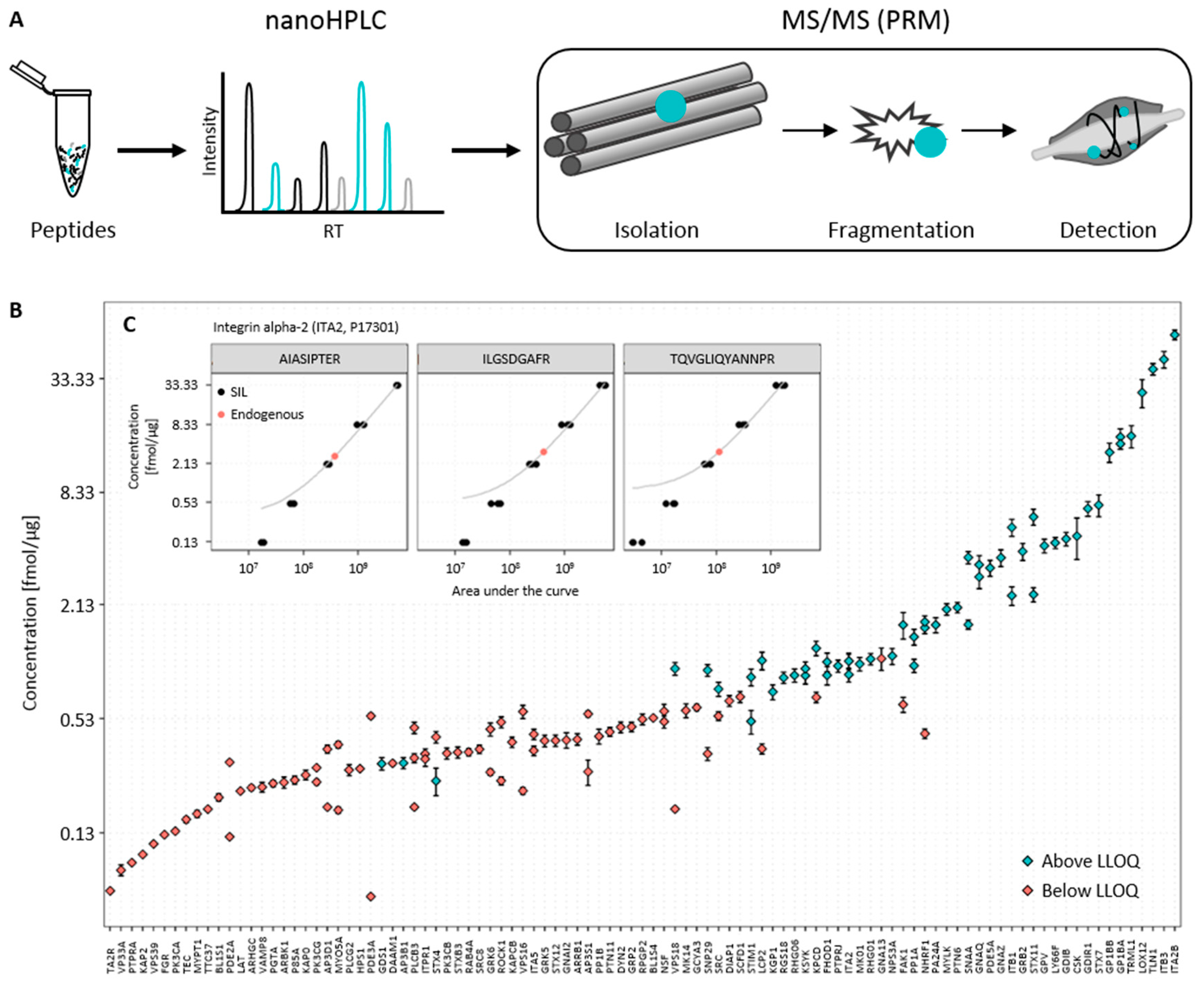

{kind=link}
{kind=link}
{kind=link}
| Protein Accession 1 | Name 2 | Short Name 3 | Peptide Sequence 4 | Concentration (fmol/μg) 5 | Standard Deviation 6 | CV (%) 7 | LLOQ (fmol/μg) 8 |
|---|---|---|---|---|---|---|---|
| Platelet activation and signaling | |||||||
| P41240 | Tyrosine-protein kinase CSK | CSK | HSNLVQLLGVIVEEK | 14.7 | 3.6 | 24.7 | 2.1 |
| Q05397 | Focal adhesion kinase 1 | FAK1 | TLLATVDETIPLLPASTHR | 5.0 | 0.8 | 16.0 | 2.1 |
| Q9Y613 | FH1/FH2 domain-containing protein 1 | FHOD1 | GDGEPVSVVTVR | 2.7 | 0.3 | 11.1 | 0.5 |
| Q9Y613 | FH1/FH2 domain-containing protein 1 | FHOD1 | HLGTAGTDVDLR | 3.2 | 0.4 | 11.3 | 2.1 |
| P50395 | Rab GDP dissociation inhibitor beta | GDIB | DLGTESQIFISR | 14.2 | 1.2 | 8.2 | 2.1 |
| P52565 | Rho GDP-dissociation inhibitor 1 | GDIR1 | AEEYEFLTPVEEAPK | 20.5 | 1.8 | 8.9 | 2.1 |
| P52306 | Rap1 GTPase-GDP dissociation stimulator 1 | GDS1 | SVAQQASLTEQR | 0.9 | 0.1 | 7.6 | 0.5 |
| P50148 | Guanine nucleotide-binding protein G(q) subunit alpha | GNAQ | VSAFENPYVDAIK | 8.9 | 1.2 | 13.3 | 2.1 |
| P50148 | Guanine nucleotide-binding protein G(q) subunit alpha | GNAQ | YYLNDLDR | 10.4 | 1.2 | 11.7 | 2.1 |
| P19086 | Guanine nucleotide-binding protein G(z) subunit alpha | GNAZ | IAAADYIPTVEDILR | 11.3 | 1.1 | 10.2 | 2.1 |
| P07359 | Platelet glycoprotein Ib alpha chain | GP1BA | GQDLLSTVSIR | 49.1 | 5.2 | 10.6 | 2.1 |
| P07359 | Platelet glycoprotein Ib alpha chain | GP1BA | LTSLPLGALR | 45.2 | 2.9 | 6.4 | 2.1 |
| P13224 | Platelet glycoprotein Ib beta chain | GP1BB | LSLTDPLVAER | 40.7 | 4.7 | 11.7 | 2.1 |
| P40197 | Platelet glycoprotein V | GPV | ITHLPGALLDK | 13.1 | 1.1 | 8.4 | 2.1 |
| P62993 | Growth factor receptor-bound protein 2 | GRB2 | FNSLNELVDYHR | 12.2 | 1.3 | 10.5 | 2.1 |
| P05556 | Integrin beta-1 | ITB1 | GEVFNELVGK | 7.1 | 0.8 | 11.4 | 2.1 |
| P05556 | Integrin beta-1 | ITB1 | IGFGSFVEK | 16.3 | 1.7 | 10.3 | 2.1 |
| P05106 | Integrin beta-3 | ITB3 | HVLTLTDQVTR | 126.3 | 12.7 | 10.1 | 2.1 |
| Q13976 | cGMP-dependent protein kinase 1 | KGP1 | DLKPENLILDHR | 2.2 | 0.2 | 9.0 | 2.1 |
| Q05655 | Protein kinase C delta type | KPCD | DYSNFDQEFLNEK | 3.7 | 0.3 | 8.8 | 2.1 |
| P43405 | Tyrosine-protein kinase SYK | KSYK | KPFNRPQGVQPK | 2.7 | 0.3 | 9.7 | 2.1 |
| P43405 | Tyrosine-protein kinase SYK | KSYK | NVLLVTQHYAK | 2.9 | 0.2 | 8.3 | 2.1 |
| Q13094 | Lymphocyte cytosolic protein 2 | LCP2 | IQKPPLPPTTER | 3.2 | 0.3 | 10.7 | 2.1 |
| P18054 | Arachidonate 12-lipoxygenase, 12S-type | LOX12 | GEEEEFDHDVAEDLGLLQFVR | 84.4 | 14.3 | 16.9 | 8.3 |
| Q5SQ64 | Lymphocyte antigen 6 complex locus protein G6f | LY66F | VYDVLVLK | 13.5 | 0.9 | 6.7 | 2.1 |
| P28482 | Mitogen-activated protein kinase 1 | MK01 | GQVFDVGPR | 3.1 | 0.3 | 8.3 | 0.5 |
| Q15746 | Myosin light chain kinase, smooth muscle | MYLK | VSDFYDIEER | 6.0 | 0.4 | 7.0 | 2.1 |
| O14745 | Na(+)/H(+) exchange regulatory cofactor NHE-RF1 | NHRF1 | LLVVDPETDEQLQK | 4.8 | 0.3 | 7.0 | 2.1 |
| O14745 | Na(+)/H(+) exchange regulatory cofactor NHE-RF1 | NHRF1 | SVDPDSPAEASGLR | 5.2 | 0.4 | 7.8 | 2.1 |
| Q9UFN0 | Protein NipSnap homolog 3A | NPS3A | LVGVFHTEYGALNR | 3.4 | 0.3 | 9.7 | 2.1 |
| P47712 | Cytosolic phospholipase A2 | PA24A | NVSHNPLLLLTPQK | 5.0 | 0.5 | 9.3 | 2.1 |
| O76074 | cGMP-specific 3′,5′-cyclic phosphodiesterase | PDE5A | GIVGHVAALGEPLNIK | 10.0 | 1.0 | 9.7 | 2.1 |
| P62136 | Serine/threonine-protein phosphatase PP1-alpha catalytic subunit | PP1A | LNLDSIIGR | 4.3 | 0.4 | 9.6 | 2.1 |
| P62136 | Serine/threonine-protein phosphatase PP1-alpha catalytic subunit | PP1A | NVQLTENEIR | 3.0 | 0.2 | 7.9 | 2.1 |
| P29350 | Tyrosine-protein phosphatase non-receptor type 6 | PTN6 | IQNSGDFYDLYGGEK | 6.2 | 0.4 | 6.4 | 2.1 |
| Q12913 | Receptor-type tyrosine-protein phosphatase eta | PTPRJ | VITEPIPVSDLR | 3.0 | 0.2 | 7.4 | 0.5 |
| Q07960 | Rho GTPase-activating protein 1 | RHG01 | NPEQEPIPIVLR | 3.3 | 0.2 | 6.9 | 2.1 |
| O43182 | Rho GTPase-activating protein 6 | RHG06 | SVPIQSLSELER | 2.7 | 0.2 | 7.9 | 2.1 |
| P12931 | Proto-oncogene tyrosine-protein kinase Src | SRC | GPSAAFAPAAAEPK | 2.3 | 0.2 | 8.5 | 2.1 |
| Q9Y490 | Talin-1 | TLN1 | ALDGAFTEENR | 112.2 | 7.9 | 7.0 | 2.1 |
| Q86YW5 | Trem-like transcript 1 protein | TRML1 | VSLNILPPEEEEETHK | 49.7 | 6.7 | 13.5 | 2.1 |
| Platelet secretion | |||||||
| O00203 | AP-3 complex subunit beta-1 | AP3B1 | VVNVANVGAVPSGQDNIHR | 0.9 | 0.1 | 6.6 | 0.5 |
| P54920 | Alpha-soluble NSF attachment protein | SNAA | NSQSFFSGLFGGSSK | 11.3 | 0.8 | 7.4 | 2.1 |
| P54920 | Alpha-soluble NSF attachment protein | SNAA | YEELFPAFSDSR | 5.0 | 0.3 | 5.7 | 2.1 |
| O95721 | Synaptosomal-associated protein 29 | SNP29 | SVFGGLVNYFK | 2.9 | 0.2 | 7.2 | 2.1 |
| Q13586 | Stromal interaction molecule 1 | STIM1 | SHSPSSPDPDTPSPVGDSR | 1.5 | 0.2 | 14.4 | 0.1 |
| Q13586 | Stromal interaction molecule 1 | STIM1 | YAEEELEQVR | 2.6 | 0.3 | 10.1 | 2.1 |
| Q12846 | Syntaxin-4 | STX4 | VALVVHPGTAR | 0.7 | 0.1 | 16.6 | 0.5 |
| O15400 | Syntaxin-7 | STX7 | LVAEFTTSLTNFQK | 21.4 | 2.8 | 13.3 | 2.1 |
| O75558 | Syntaxin-11 | STX11 | AQYNALTLTFQR | 7.2 | 0.6 | 8.7 | 2.1 |
| O75558 | Syntaxin-11 | STX11 | LAELLDLSK | 18.6 | 1.8 | 9.8 | 2.1 |
| Q9P253 | Vacuolar protein sorting-associated protein 18 homolog | VPS18 | IEDVLPFFPDFVTIDHFK | 2.9 | 0.2 | 7.8 | 2.1 |
| Platelet aggregation | |||||||
| P17301 | Integrin alpha-2 | ITA2 | AIASIPTER | 2.7 | 0.2 | 8.6 | 2.1 |
| P17301 | Integrin alpha-2 | ITA2 | ILGSDGAFR | 3.2 | 0.3 | 9.8 | 2.1 |
| P17301 | Integrin alpha-2 | ITA2 | TQVGLIQYANNPR | 3.2 | 0.3 | 8.6 | 2.1 |
| P08514 | Integrin alpha-IIb | ITA2B | IVLLDVPVR | 170.6 | 10.2 | 6.0 | 2.1 |
| Q9NS28 | Regulator of G-protein signaling 18 | RGS18 | DGLEAFTR | 2.6 | 0.2 | 6.9 | 2.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malchow, S.; Loosse, C.; Sickmann, A.; Lorenz, C. Quantification of Cardiovascular Disease Biomarkers in Human Platelets by Targeted Mass Spectrometry. Proteomes 2017, 5, 31. https://doi.org/10.3390/proteomes5040031
Malchow S, Loosse C, Sickmann A, Lorenz C. Quantification of Cardiovascular Disease Biomarkers in Human Platelets by Targeted Mass Spectrometry. Proteomes. 2017; 5(4):31. https://doi.org/10.3390/proteomes5040031
Chicago/Turabian StyleMalchow, Sebastian, Christina Loosse, Albert Sickmann, and Christin Lorenz. 2017. "Quantification of Cardiovascular Disease Biomarkers in Human Platelets by Targeted Mass Spectrometry" Proteomes 5, no. 4: 31. https://doi.org/10.3390/proteomes5040031
APA StyleMalchow, S., Loosse, C., Sickmann, A., & Lorenz, C. (2017). Quantification of Cardiovascular Disease Biomarkers in Human Platelets by Targeted Mass Spectrometry. Proteomes, 5(4), 31. https://doi.org/10.3390/proteomes5040031