A Comprehensive Guide for Performing Sample Preparation and Top-Down Protein Analysis

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Why Analyse Intact Proteoforms?
3. Defining ‘What’ Proteome Analysis Actually Is
4. Two-Dimensional Gel Electrophoresis Using Isoelectric Focusing in Immobilised pH Gradients and SDS-PAGE
5. Blue and Clear Native PAGE
6. Label-Free and DiGE-Based Relative Quantitation in PAGE
7. Affinity-Based Separations for the Top-Down Analysis of Complexes and Interactions
8. The Detection of Low Abundance Proteoforms Using SDS-PAGE and Immunoblotting
9. Top-Down Mass Spectrometry Methods for Proteoform Quantitation
10. Ion Fragmentation and Selection
11. Relative and Absolute Quantitation
12. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smith, L.M.; Kelleher, N.L. Consortium for Top Down, P. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, N.L.; Lin, H.Y.; Valaskovic, G.A.; Aaserud, D.J.; Fridriksson, E.K.; McLafferty, F.W. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J. Am. Chem. Soc. 1999, 121, 806–812. [Google Scholar] [CrossRef]
- Anderson, S. Shotgun DNA sequencing using cloned dnase i-generated fragments. Nucleic Acids Res. 1981, 9, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R., 3rd. Mass spectrometry and the age of the proteome. J. Mass Spectrom. 1998, 33, 1–19. [Google Scholar] [CrossRef]
- Kelleher, N.L. Top-down proteomics. Anal. Chem. 2004, 76, 197A–203A. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Mann, M. Proteomics to study genes and genomes. Nature 2000, 405, 837–846. [Google Scholar] [PubMed]
- Tran, J.C.; Zamdborg, L.; Ahlf, D.R.; Lee, J.E.; Catherman, A.D.; Durbin, K.R.; Tipton, J.D.; Vellaichamy, A.; Kellie, J.F.; Li, M.; et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.S.; Geiger, T.; Chatterjee, B.; Bandilla, P.; Frohlich, F.; Cox, J.; Mann, M. Deep and highly sensitive proteome coverage by lc-ms/ms without prefractionation. Mol. Cell. Proteom. 2011, 10, M110.003699. [Google Scholar] [CrossRef] [PubMed]
- Hosp, F.; Scheltema, R.A.; Eberl, H.C.; Kulak, N.A.; Keilhauer, E.C.; Mayr, K.; Mann, M. A double-barrel liquid chromatography-tandem mass spectrometry (lc-ms/ms) system to quantify 96 interactomes per day. Mol. Cell. Proteom. 2015, 14, 2030–2041. [Google Scholar] [CrossRef] [PubMed]
- Grassl, N.; Kulak, N.A.; Pichler, G.; Geyer, P.E.; Jung, J.; Schubert, S.; Sinitcyn, P.; Cox, J.; Mann, M. Ultra-deep and quantitative saliva proteome reveals dynamics of the oral microbiome. Genome Med. 2016, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Hebert, A.S.; Ulbrich, A.; Bailey, D.J.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. One-hour proteome analysis in yeast. Nat. Protoc. 2015, 10, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.F.; Paton, N.W.; Lilley, K.S.; Binz, P.A.; Julian, R.K., Jr.; Jones, A.R.; Zhu, W.; Apweiler, R.; Aebersold, R.; Deutsch, E.W.; et al. The minimum information about a proteomics experiment (miape). Nat. Biotechnol. 2007, 25, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Lluch-Senar, M.; Delgado, J.; Chen, W.H.; Llorens-Rico, V.; O'Reilly, F.J.; Wodke, J.A.; Unal, E.B.; Yus, E.; Martinez, S.; Nichols, R.J.; et al. Defining a minimal cell: Essentiality of small orfs and ncrnas in a genome-reduced bacterium. Mol. Syst. Biol. 2015, 11, 780. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Lescuyer, P. The proteomic to biology inference, a frequently overlooked concern in the interpretation of proteomic data: A plea for functional validation. Proteomics 2014, 14, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Aebersold, R. Interpretation of shotgun proteomic data: The protein inference problem. Mol. Cell. Proteom. 2005, 4, 1419–1440. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Merrill, A.E.; Coon, J.J. Proteome sequencing goes deep. Curr. Opin. Chem. Biol. 2015, 24, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Moghaddas Gholami, A.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Ezkurdia, I.; Vazquez, J.; Valencia, A.; Tress, M. Analyzing the first drafts of the human proteome. J. Proteome Res. 2014, 13, 3854–3855. [Google Scholar] [CrossRef] [PubMed]
- Durbin, K.R.; Fornelli, L.; Fellers, R.T.; Doubleday, P.F.; Narita, M.; Kelleher, N.L. Quantitation and identification of thousands of human proteoforms below 30 kda. J. Proteome Res. 2016, 15, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.P.; Partridge, M.A.; Padula, M.P.; Gauci, V.J.; Malladi, C.S.; Coorssen, J.R. Top-down proteomics: Enhancing 2d gel electrophoresis from tissue processing to high-sensitivity protein detection. Proteomics 2014, 14, 872–889. [Google Scholar] [CrossRef] [PubMed]
- Kuljanin, M.; Dieters-Castator, D.Z.; Hess, D.A.; Postovit, L.M.; Lajoie, G.A. Comparison of sample preparation techniques for large scale proteomics. Proteomics 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Issaq, H.J.; Conrads, T.P.; Janini, G.M.; Veenstra, T.D. Methods for fractionation, separation and profiling of proteins and peptides. Electrophoresis 2002, 23, 3048–3061. [Google Scholar] [CrossRef]
- Pieper, R.; Gatlin, C.L.; Makusky, A.J.; Russo, P.S.; Schatz, C.R.; Miller, S.S.; Su, Q.; McGrath, A.M.; Estock, M.A.; Parmar, P.P.; et al. The human serum proteome: Display of nearly 3700 chromatographically separated protein spots on two-dimensional electrophoresis gels and identification of 325 distinct proteins. Proteomics 2003, 3, 1345–1364. [Google Scholar] [CrossRef] [PubMed]
- Grandori, R. Origin of the conformation dependence of protein charge-state distributions in electrospray ionization mass spectrometry. J. Mass Spectrom. 2003, 38, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.G.; Senko, M.W.; Li, W.; Li, M.; Dillon, S.; Guan, S.; Logan, T.M. Protein molecular mass to 1 da by 13c, 15n double-depletion and ft-icr mass spectrometry. J. Am. Chem. Soc. 1997, 119, 433–434. [Google Scholar] [CrossRef]
- Lomeli, S.H.; Yin, S.; Ogorzalek Loo, R.R.; Loo, J.A. Increasing charge while preserving noncovalent protein complexes for esi-ms. J. Am. Soc. Mass Spectrom. 2009, 20, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Sterling, H.J.; Daly, M.P.; Feld, G.K.; Thoren, K.L.; Kintzer, A.F.; Krantz, B.A.; Williams, E.R. Effects of supercharging reagents on noncovalent complex structure in electrospray ionization from aqueous solutions. J. Am. Soc. Mass Spectrom. 2010, 21, 1762–1774. [Google Scholar] [CrossRef] [PubMed]
- Zenaidee, M.A.; Donald, W.A. Extremely supercharged proteins in mass spectrometry: Profiling the ph of electrospray generated droplets, narrowing charge state distributions, and increasing ion fragmentation. Analyst 2015, 140, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, S.P.; Cordwell, S.J.; Djordjevic, M.A.; Wilton, J.; Minion, F.C. Proteolytic processing of the mycoplasma hyopneumoniae cilium adhesin. Infect. Immun. 2004, 72, 2791–2802. [Google Scholar] [CrossRef] [PubMed]
- Tacchi, J.L.; Raymond, B.B.; Jarocki, V.M.; Berry, I.J.; Padula, M.P.; Djordjevic, S.P. Cilium adhesin p216 (mhj_0493) is a target of ectodomain shedding and aminopeptidase activity on the surface of mycoplasma hyopneumoniae. J. Proteome Res. 2014, 13, 2920–2930. [Google Scholar] [CrossRef] [PubMed]
- Raymond, B.B.; Jenkins, C.; Seymour, L.M.; Tacchi, J.L.; Widjaja, M.; Jarocki, V.M.; Deutscher, A.T.; Turnbull, L.; Whitchurch, C.B.; Padula, M.P.; et al. Proteolytic processing of the cilium adhesin mhj_0194 (p123j ) in mycoplasma hyopneumoniae generates a functionally diverse array of cleavage fragments that bind multiple host molecules. Cell Microbiol. 2015, 17, 425–444. [Google Scholar] [CrossRef] [PubMed]
- Raymond, B.B.; Tacchi, J.L.; Jarocki, V.M.; Minion, F.C.; Padula, M.P.; Djordjevic, S.P. P159 from mycoplasma hyopneumoniae binds porcine cilia and heparin and is cleaved in a manner akin to ectodomain shedding. J. Proteome Res. 2013, 12, 5891–5903. [Google Scholar] [CrossRef] [PubMed]
- Tacchi, J.L.; Raymond, B.B.; Haynes, P.A.; Berry, I.J.; Widjaja, M.; Bogema, D.R.; Woolley, L.K.; Jenkins, C.; Minion, F.C.; Padula, M.P.; et al. Post-translational processing targets functionally diverse proteins in mycoplasma hyopneumoniae. Open. Biol. 2016, 6, 150210. [Google Scholar] [CrossRef] [PubMed]
- Catherman, A.D.; Durbin, K.R.; Ahlf, D.R.; Early, B.P.; Fellers, R.T.; Tran, J.C.; Thomas, P.M.; Kelleher, N.L. Large-scale top-down proteomics of the human proteome: Membrane proteins, mitochondria, and senescence. Mol. Cell. Proteomics 2013, 12, 3465–3473. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Braun, H.P.; Schagger, H. Blue native page. Nat. Protoc. 2006, 1, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Schagger, H. Native electrophoretic techniques to identify protein-protein interactions. Proteomics 2009, 9, 5214–5223. [Google Scholar] [CrossRef] [PubMed]
- Zickermann, V.; Wumaier, Z.; Wrzesniewska, B.; Hunte, C.; Schagger, H. Native immunoblotting of blue native gels to identify conformation-specific antibodies. Proteomics 2010, 10, 159–163. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [PubMed]
- Westermeier, R. Looking at proteins from two dimensions: A review on five decades of 2d electrophoresis. Arch. Physiol. Biochem. 2014, 120, 168–172. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.Z.; Goodman, H.M.; O’Farrell, P.H. High resolution two-dimensional electrophoresis of basic as well as acidic proteins. Cell 1977, 12, 1133–1141. [Google Scholar] [CrossRef]
- Gorg, A.; Postel, W.; Westermeier, R. Ultrathin-layer isoelectric focusing in polyacrylamide gels on cellophane. Anal. Biochem. 1978, 89, 60–70. [Google Scholar] [CrossRef]
- Bjellqvist, B.; Ek, K.; Righetti, P.G.; Gianazza, E.; Gorg, A.; Westermeier, R.; Postel, W. Isoelectric focusing in immobilized ph gradients: Principle, methodology and some applications. J. Biochem. Biophys. Methods 1982, 6, 317–339. [Google Scholar] [CrossRef]
- Churchward, M.A.; Butt, R.H.; Lang, J.C.; Hsu, K.K.; Coorssen, J.R. Enhanced detergent extraction for analysis of membrane proteomes by two-dimensional gel electrophoresis. Proteome Sci. 2005, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Pfeifer, T.A.; Delaney, A.; Grigliatti, T.A.; Tetzlaff, W.G.; Coorssen, J.R. Enabling coupled quantitative genomics and proteomics analyses from rat spinal cord samples. Mol. Cell. Proteom. 2007, 6, 1574–1588. [Google Scholar] [CrossRef] [PubMed]
- Molloy, M.P.; Herbert, B.R.; Williams, K.L.; Gooley, A.A. Extraction of escherichia coli proteins with organic solvents prior to two-dimensional electrophoresis. Electrophoresis 1999, 20, 701–704. [Google Scholar] [CrossRef]
- Herbert, B.R.; Harry, J.L.; Packer, N.H.; Gooley, A.A.; Pedersen, S.K.; Williams, K.L. What place for polyacrylamide in proteomics? Trends Biotechnol. 2001, 19, S3–S9. [Google Scholar] [CrossRef]
- Herbert, B.R.; Grinyer, J.; McCarthy, J.T.; Isaacs, M.; Harry, E.J.; Nevalainen, H.; Traini, M.D.; Hunt, S.; Schulz, B.; Laver, M.; et al. Improved 2-de of microorganisms after acidic extraction. Electrophoresis 2006, 27, 1630–1640. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T. Variations on a theme: Changes to electrophoretic separations that can make a difference. J. Proteomics 2010, 73, 1562–1572. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Lelong, C. Two-dimensional gel electrophoresis in proteomics: A tutorial. J. Proteomics 2011, 74, 1829–1841. [Google Scholar] [CrossRef] [PubMed]
- Peaks amino acid mass table. Available online: http://www.bioinfor.com/wp-content/uploads/2017/01/2015-massref-web.pdf (accessed on 6 April 2017).
- Wu, X.; Xiong, E.; Wang, W.; Scali, M.; Cresti, M. Universal sample preparation method integrating trichloroacetic acid/acetone precipitation with phenol extraction for crop proteomic analysis. Nat. Protoc. 2014, 9, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Coorssen, J.R. Pre-extraction sample handling by automated frozen disruption significantly improves subsequent proteomic analyses. J. Proteome Res. 2006, 5, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Molloy, M.P.; Herbert, B.R.; Walsh, B.J.; Tyler, M.I.; Traini, M.; Sanchez, J.C.; Hochstrasser, D.F.; Williams, K.L.; Gooley, A.A. Extraction of membrane proteins by differential solubilization for separation using two-dimensional gel electrophoresis. Electrophoresis 1998, 19, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Blisnick, T.; Morales-Betoulle, M.E.; Vuillard, L.; Rabilloud, T.; Braun Breton, C. Non-detergent sulphobetaines enhance the recovery of membrane and/or cytoskeleton-associated proteins and active proteases from erythrocytes infected by plasmodium falciparum. Eur. J. Biochem. 1998, 252, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Chevallet, M.; Santoni, V.; Poinas, A.; Rouquie, D.; Fuchs, A.; Kieffer, S.; Rossignol, M.; Lunardi, J.; Garin, J.; Rabilloud, T. New zwitterionic detergents improve the analysis of membrane proteins by two-dimensional electrophoresis. Electrophoresis 1998, 19, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.E.; Expert-Bezancon, N.; Vuillard, L.; Rabilloud, T. Non-detergent sulphobetaines: A new class of molecules that facilitate in vitro protein renaturation. Fold Des. 1996, 1, 21–27. [Google Scholar] [CrossRef]
- Rabilloud, T.; Gianazza, E.; Catto, N.; Righetti, P.G. Amidosulfobetaines, a family of detergents with improved solubilization properties: Application for isoelectric focusing under denaturing conditions. Anal. Biochem. 1990, 185, 94–102. [Google Scholar] [CrossRef]
- Chertov, O.; Biragyn, A.; Kwak, L.W.; Simpson, J.T.; Boronina, T.; Hoang, V.M.; Prieto, D.A.; Conrads, T.P.; Veenstra, T.D.; Fisher, R.J. Organic solvent extraction of proteins and peptides from serum as an effective sample preparation for detection and identification of biomarkers by mass spectrometry. Proteomics 2004, 4, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Herbert, B.R.; Molloy, M.P.; Gooley, A.A.; Walsh, B.J.; Bryson, W.G.; Williams, K.L. Improved protein solubility in two-dimensional electrophoresis using tributyl phosphine as reducing agent. Electrophoresis 1998, 19, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.A.; Jencks, W.P. The relationship of structure to the effectiveness of denaturing agents for proteins. Biochemistry 1963, 2, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Gronow, M.; Griffiths, G. Rapid isolation and separation of the non-histone proteins of rat liver nuclei. FEBS Lett. 1971, 15, 340–344. [Google Scholar] [CrossRef]
- Rabilloud, T.; Adessi, C.; Giraudel, A.; Lunardi, J. Improvement of the solubilization of proteins in two-dimensional electrophoresis with immobilized ph gradients. Electrophoresis 1997, 18, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T. Use of thiourea to increase the solubility of membrane proteins in two-dimensional electrophoresis. Electrophoresis 1998, 19, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T. Solubilization of proteins in 2-d electrophoresis. An outline. Methods Mol. Biol. 1999, 112, 9–19. [Google Scholar] [PubMed]
- Zhou, R.; Li, J.; Hua, L.; Yang, Z.; Berne, B.J. Comment on “urea-mediated protein denaturation: A consensus view”. J. Phys. Chem. B 2011, 115, 1323–1326, discussion 1327–1328. [Google Scholar] [CrossRef] [PubMed]
- Perdew, G.H.; Schaup, H.W.; Selivonchick, D.P. The use of a zwitterionic detergent in two-dimensional gel electrophoresis of trout liver microsomes. Anal. Biochem. 1983, 135, 453–455. [Google Scholar] [CrossRef]
- Gianazza, E.; Rabilloud, T.; Quaglia, L.; Caccia, P.; Astrua-Testori, S.; Osio, L.; Grazioli, G.; Righetti, P.G. Additives for immobilized ph gradient two-dimensional separation of particulate material: Comparison between commercial and new synthetic detergents. Anal. Biochem. 1987, 165, 247–257. [Google Scholar] [CrossRef]
- Rabilloud, T.; Blisnick, T.; Heller, M.; Luche, S.; Aebersold, R.; Lunardi, J.; Braun-Breton, C. Analysis of membrane proteins by two-dimensional electrophoresis: Comparison of the proteins extracted from normal or plasmodium falciparum-infected erythrocyte ghosts. Electrophoresis 1999, 20, 3603–3610. [Google Scholar] [CrossRef]
- Luche, S.; Santoni, V.; Rabilloud, T. Evaluation of nonionic and zwitterionic detergents as membrane protein solubilizers in two-dimensional electrophoresis. Proteomics 2003, 3, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Herbert, B.; Hopwood, F.; Oxley, D.; McCarthy, J.; Laver, M.; Grinyer, J.; Goodall, A.; Williams, K.; Castagna, A.; Righetti, P.G. Beta-elimination: An unexpected artefact in proteome analysis. Proteomics 2003, 3, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Luche, S.; Diemer, H.; Tastet, C.; Chevallet, M.; Van Dorsselaer, A.; Leize-Wagner, E.; Rabilloud, T. About thiol derivatization and resolution of basic proteins in two-dimensional electrophoresis. Proteomics 2004, 4, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Sechi, S.; Chait, B.T. Modification of cysteine residues by alkylation. A tool in peptide mapping and protein identification. Anal. Chem. 1998, 70, 5150–5158. [Google Scholar] [CrossRef] [PubMed]
- Aitken, A.; Learmonth, M. Carboxymethylation of cysteine using iodoacetamide/ iodoacetic acid. In The Protein Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2002; pp. 455–456. [Google Scholar]
- Patterson, S.D.; Aebersold, R. Mass spectrometric approaches for the identification of gel-separated proteins. Electrophoresis 1995, 16, 1791–1814. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.D. Matrix-assisted laser-desorption/ionization mass spectrometric approaches for the identification of gel-separated proteins in the 5–50 pmol range. Electrophoresis 1995, 16, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.; Hopwood, F.; Oxley, D.; Laver, M.; Castagna, A.; Righetti, P.G.; Williams, K.; Herbert, B. Carbamylation of proteins in 2-d electrophoresis—Myth or reality? J. Proteome Res. 2003, 2, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Righetti, P.G.; Castagna, A.; Antonioli, P.; Boschetti, E. Prefractionation techniques in proteome analysis: The mining tools of the third millennium. Electrophoresis 2005, 26, 297–319. [Google Scholar] [CrossRef] [PubMed]
- Righetti, P.G.; Castagna, A.; Herbert, B.; Reymond, F.; Rossier, J.S. Prefractionation techniques in proteome analysis. Proteomics 2003, 3, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, R.A. The challenge of the proteome dynamic range and its implications for in-depth proteomics. Proteomics 2013, 13, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, C.; Fialka, I.; Huber, L.A. Preparative two-dimensional gel electrophoresis of membrane proteins. Electrophoresis 1997, 18, 2573–2581. [Google Scholar] [CrossRef] [PubMed]
- Padula, M.P. The Development of Proteomic Techniques to Study the Australian Paralysis Tick, Ixodes Holocyclus: The Application of Proteomic Technology to an Organism with Poor Bioinformatic Information; University of Technology Sydney: Sydney, Australia, 2009. [Google Scholar]
- D’Amici, G.M.; Timperio, A.M.; Zolla, L. Coupling of native liquid phase isoelectrofocusing and blue native polyacrylamide gel electrophoresis: A potent tool for native membrane multiprotein complex separation. J. Proteome Res. 2008, 7, 1326–1340. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Parrado, J.; Machado, A. Use of rotofor preparative isoelectrofocusing cell in protein purification procedure. Appl. Biochem. Biotechnol. 1998, 69, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Speicher, D.W. A method for global analysis of complex proteomes using sample prefractionation by solution isoelectrofocusing prior to two-dimensional electrophoresis. Anal. Biochem. 2000, 284, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Speicher, D.W. Comprehensive analysis of complex proteomes using microscale solution isoelectrofocusing prior to narrow ph range two-dimensional electrophoresis. Proteomics 2002, 2, 58–68. [Google Scholar] [CrossRef]
- Ros, A.; Faupel, M.; Mees, H.; Oostrum, J.; Ferrigno, R.; Reymond, F.; Michel, P.; Rossier, J.S.; Girault, H.H. Protein purification by off-gel electrophoresis. Proteomics 2002, 2, 151–156. [Google Scholar] [CrossRef]
- Heller, M.; Michel, P.E.; Morier, P.; Crettaz, D.; Wenz, C.; Tissot, J.D.; Reymond, F.; Rossier, J.S. Two-stage off-gel isoelectric focusing: Protein followed by peptide fractionation and application to proteome analysis of human plasma. Electrophoresis 2005, 26, 1174–1188. [Google Scholar] [CrossRef] [PubMed]
- Bogema, D.R.; Scott, N.E.; Padula, M.P.; Tacchi, J.L.; Raymond, B.B.; Jenkins, C.; Cordwell, S.J.; Minion, F.C.; Walker, M.J.; Djordjevic, S.P. Sequence ttkf downward arrow qe defines the site of proteolytic cleavage in mhp683 protein, a novel glycosaminoglycan and cilium adhesin of mycoplasma hyopneumoniae. J. Biol. Chem. 2011, 286, 41217–41229. [Google Scholar] [CrossRef] [PubMed]
- Edman, P.; Begg, G. A protein sequenator. Eur. J. Biochem. 1967, 1, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Edman, P. A method for the determination of amino acid sequence in peptides. Arch. Biochem. 1949, 22, 475. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, K.; Goethals, M.; Martens, L.; Van Damme, J.; Staes, A.; Thomas, G.R.; Vandekerckhove, J. Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted n-terminal peptides. Nat. Biotechnol. 2003, 21, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Percent semi-tryptic. Available online: http://massqc.proteomesoftware.com/help/metrics/percent_semi_tryptic (accessed on 6 April 2017).
- Kleifeld, O.; Doucet, A.; Prudova, A.; auf dem Keller, U.; Gioia, M.; Kizhakkedathu, J.N.; Overall, C.M. Identifying and quantifying proteolytic events and the natural n terminome by terminal amine isotopic labeling of substrates. Nat. Protoc. 2011, 6, 1578–1611. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T. Membrane proteins and proteomics: Love is possible, but so difficult. Electrophoresis 2009, 30 (Suppl. 1), S174–S180. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Agnoletto, C.; De Marchi, E.; Marchi, S.; Patergnani, S.; Bonora, M.; Giorgi, C.; Missiroli, S.; Poletti, F.; Rimessi, A.; et al. Protein kinases and phosphatases in the control of cell fate. Enzyme Res. 2011, 2011, 26. [Google Scholar] [CrossRef] [PubMed]
- Reisinger, V.; Eichacker, L.A. Solubilization of membrane protein complexes for blue native page. J. Proteomics 2008, 71, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Krause, F. Detection and analysis of protein-protein interactions in organellar and prokaryotic proteomes by native gel electrophoresis: (membrane) Protein complexes and supercomplexes. Electrophoresis 2006, 27, 2759–2781. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, A.; Grimm, V.; Arakaki, A.K.; Skolnick, J. Prediction of physical protein-protein interactions. Phys. Biol. 2005, 2, S1–16. [Google Scholar] [CrossRef] [PubMed]
- Le Maire, M.; Champeil, P.; Møller, J.V. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim. Biophys. Acta (BBA) Biomembr. 2000, 1508, 86–111. [Google Scholar] [CrossRef]
- Fiala, G.J.; Schamel, W.W.A.; Blumenthal, B. Blue native polyacrylamide gel electrophoresis (bn-page) for analysis of multiprotein complexes from cellular lysates. J. Vis. Exp. JoVE 2011, 2164. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wei, C.; Zhao, L.; Liu, L.; Leng, W.; Li, W.; Jin, Q. Combining blue native polyacrylamide gel electrophoresis with liquid chromatography tandem mass spectrometry as an effective strategy for analyzing potential membrane protein complexes of mycobacterium bovis bacillus calmette-guérin. BMC Genom. 2011, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.W.; Buchtmann, K.A.; Jenkins, C.; Tacchi, J.L.; Raymond, B.B.; To, J.; Roy Chowdhury, P.; Woolley, L.K.; Labbate, M.; Turnbull, L.; et al. Mhj_0125 is an m42 glutamyl aminopeptidase that moonlights as a multifunctional adhesin on the surface of mycoplasma hyopneumoniae. Open Biol. 2013, 3, 130017. [Google Scholar] [CrossRef] [PubMed]
- Dudkina, N.V.; Eubel, H.; Keegstra, W.; Boekema, E.J.; Braun, H.P. Structure of a mitochondrial supercomplex formed by respiratory-chain complexes i and iii. Proc. Natl. Acad. Sci. USA 2005, 102, 3225–3229. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, L.; Wittig, I.; Barth, H.D.; Schagger, H.; Brutschy, B.; Brandt, U. Laser-induced liquid bead ion desorption-ms of protein complexes from blue-native gels, a sensitive top-down proteomic approach. Proteomics 2010, 10, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Gault, J.; Donlan, J.A.; Liko, I.; Hopper, J.T.; Gupta, K.; Housden, N.G.; Struwe, W.B.; Marty, M.T.; Mize, T.; Bechara, C.; et al. High-resolution mass spectrometry of small molecules bound to membrane proteins. Nat. Methods 2016, 13, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Hopper, J.T.; Robinson, C.V. Mass spectrometry quantifies protein interactions—From molecular chaperones to membrane porins. Angew. Chem. Int. Ed. Engl. 2014, 53, 14002–14015. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Robinson, C.V. A comparative cross-linking strategy to probe conformational changes in protein complexes. Nat. Protoc. 2014, 9, 2224–2236. [Google Scholar] [CrossRef] [PubMed]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilising the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Unlu, M.; Morgan, M.E.; Minden, J.S. Difference gel electrophoresis: A single gel method for detecting changes in protein extracts. Electrophoresis 1997, 18, 2071–2077. [Google Scholar] [CrossRef] [PubMed]
- Marouga, R.; David, S.; Hawkins, E. The development of the dige system: 2d fluorescence difference gel analysis technology. Anal. Bioanal. Chem. 2005, 382, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Keeping, A.J.; Collins, R.A. Data variance and statistical significance in 2d-gel electrophoresis and dige experiments: Comparison of the effects of normalization methods. J. Proteome Res. 2011, 10, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Hirohashi, S. Application of highly sensitive fluorescent dyes (cydye dige fluor saturation dyes) to laser microdissection and two-dimensional difference gel electrophoresis (2d-dige) for cancer proteomics. Nat. Protoc. 2006, 1, 2940–2956. [Google Scholar] [CrossRef] [PubMed]
- Sriharshan, A.; Boldt, K.; Sarioglu, H.; Barjaktarovic, Z.; Azimzadeh, O.; Hieber, L.; Zitzelsberger, H.; Ueffing, M.; Atkinson, M.J.; Tapio, S. Proteomic analysis by silac and 2d-dige reveals radiation-induced endothelial response: Four key pathways. J. Proteom. 2012, 75, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Phizicky, E.M.; Fields, S. Protein-protein interactions: Methods for detection and analysis. Microbiol. Rev. 1995, 59, 94–123. [Google Scholar] [PubMed]
- De Gunzburg, J.; Riehl, R.; Weinberg, R.A. Identification of a protein associated with p21ras by chemical crosslinking. Proc. Natl. Acad. Sci. USA 1989, 86, 4007–4011. [Google Scholar] [CrossRef] [PubMed]
- Fields, S.; Song, O. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.G.; Alberts, B.M. F-actin affinity chromatography: Technique for isolating previously unidentified actin-binding proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 4808–4812. [Google Scholar] [CrossRef] [PubMed]
- Widjaja, M.; Berry, I.; Pont, E.; Padula, M.; Djordjevic, S. P40 and p90 from mpn142 are targets of multiple processing events on the surface of mycoplasma pneumoniae. Proteomes 2015, 3, 512–537. [Google Scholar] [CrossRef] [PubMed]
- Schiapparelli, L.M.; McClatchy, D.B.; Liu, H.H.; Sharma, P.; Yates, J.R., 3rd; Cline, H.T. Direct detection of biotinylated proteins by mass spectrometry. J. Proteome Res. 2014, 13, 3966–3978. [Google Scholar] [CrossRef] [PubMed]
- Scheurer, S.B.; Roesli, C.; Neri, D.; Elia, G. A comparison of different biotinylation reagents, tryptic digestion procedures, and mass spectrometric techniques for 2-d peptide mapping of membrane proteins. Proteomics 2005, 5, 3035–3039. [Google Scholar] [CrossRef] [PubMed]
- Elia, G. Cell surface protein biotinylation for sds-page analysis. Methods Mol. Biol. 2012, 869, 361–372. [Google Scholar] [PubMed]
- Elia, G. Biotinylation reagents for the study of cell surface proteins. Proteomics 2008, 8, 4012–4024. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, P.; Rose, C.S.; Eakin, R.E.; Snell, E.E.; Williams, R.J. Egg-white injury as the result of nonabsorption or inactivation of biotin. Science 1941, 93, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, A.; Xu, Z.; Li, R.; Chen, H.; Jin, M. Large-scale identification of bacteria-host crosstalk by affinity chromatography: Capturing the interactions of streptococcus suis proteins with host cells. J. Proteome Res. 2011, 10, 5163–5174. [Google Scholar] [CrossRef] [PubMed]
- Yalow, R.S.; Berson, S.A. Immunoassay of endogenous plasma insulin in man. J. Clin. Investig. 1960, 39, 1157–1175. [Google Scholar] [CrossRef] [PubMed]
- Liedberg, B.; Nylander, C.; Lundstrom, I. Biosensing with surface plasmon resonance—How it all started. Biosens. Bioelectron. 1995, 10, i–ix. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; Wienken, C.J.; Braun, D.; Baaske, P.; Duhr, S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 2011, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, S.A.; Horton, M.A. A nonradioactive biochemical characterization of membrane proteins using enhanced chemiluminescence. Anal. Biochem. 1992, 206, 267–272. [Google Scholar] [CrossRef]
- Ornberg, R.L.; Harper, T.F.; Liu, H. Western blot analysis with quantum dot fluorescence technology: A sensitive and quantitative method for multiplexed proteomics. Nat. Meth. 2005, 2, 79–81. [Google Scholar] [CrossRef]
- Burnett, T.A.; Dinkla, K.; Rohde, M.; Chhatwal, G.S.; Uphoff, C.; Srivastava, M.; Cordwell, S.J.; Geary, S.; Liao, X.; Minion, F.C.; et al. P159 is a proteolytically processed, surface adhesin of mycoplasma hyopneumoniae: Defined domains of p159 bind heparin and promote adherence to eukaryote cells. Mol. Microbiol. 2006, 60, 669–686. [Google Scholar] [CrossRef] [PubMed]
- Bogema, D.R.; Deutscher, A.T.; Woolley, L.K.; Seymour, L.M.; Raymond, B.B.; Tacchi, J.L.; Padula, M.P.; Dixon, N.E.; Minion, F.C.; Jenkins, C.; et al. Characterization of cleavage events in the multifunctional cilium adhesin mhp684 (p146) reveals a mechanism by which mycoplasma hyopneumoniae regulates surface topography. MBio 2012, 3, e00282-11. [Google Scholar] [CrossRef] [PubMed]
- Deutscher, A.T.; Jenkins, C.; Minion, F.C.; Seymour, L.M.; Padula, M.P.; Dixon, N.E.; Walker, M.J.; Djordjevic, S.P. Repeat regions r1 and r2 in the p97 paralogue mhp271 of mycoplasma hyopneumoniae bind heparin, fibronectin and porcine cilia. Mol. Microbiol. 2010, 78, 444–458. [Google Scholar] [CrossRef] [PubMed]
- Deutscher, A.T.; Tacchi, J.L.; Minion, F.C.; Padula, M.P.; Crossett, B.; Bogema, D.R.; Jenkins, C.; Kuit, T.A.; Walker, M.J.; Djordjevic, S.P. Mycoplasma hyopneumoniae surface proteins mhp385 and mhp384 bind host cilia and glycosaminoglycans and are endoproteolytically processed by proteases that recognize different cleavage motifs. J. Proteome Res. 2012, 11, 1924–1936. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.M.; Deutscher, A.T.; Jenkins, C.; Kuit, T.A.; Falconer, L.; Minion, F.C.; Crossett, B.; Padula, M.; Dixon, N.E.; Djordjevic, S.P.; et al. A processed multidomain mycoplasma hyopneumoniae adhesin binds fibronectin, plasminogen, and swine respiratory cilia. J. Biol. Chem. 2010, 285, 33971–33978. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.M.; Falconer, L.; Deutscher, A.T.; Minion, F.C.; Padula, M.P.; Dixon, N.E.; Djordjevic, S.P.; Walker, M.J. Mhp107 is a member of the multifunctional adhesin family of mycoplasma hyopneumoniae. J. Biol. Chem. 2011, 286, 10097–10104. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.M.; Jenkins, C.; Deutscher, A.T.; Raymond, B.B.; Padula, M.P.; Tacchi, J.L.; Bogema, D.R.; Eamens, G.J.; Woolley, L.K.; Dixon, N.E.; et al. Mhp182 (p102) binds fibronectin and contributes to the recruitment of plasmin(ogen) to the mycoplasma hyopneumoniae cell surface. Cell. Microbiol. 2012, 14, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Peng, Y.; Valeja, S.G.; Xiu, L.; Alpert, A.J.; Ge, Y. Online hydrophobic interaction chromatography-mass spectrometry for top-down proteomics. Anal. Chem. 2016, 88, 1885–1891. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.C.; Doucette, A.A. Multiplexed size separation of intact proteins in solution phase for mass spectrometry. Anal. Chem. 2009, 81, 6201–6209. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Compton, P.D.; Tran, J.C.; Kelleher, N.L. Online matrix removal platform for coupling gel-based separations to whole protein electrospray ionization mass spectrometry. J. Proteome Res. 2015, 14, 2199–2206. [Google Scholar] [CrossRef] [PubMed]
- Vellaichamy, A.; Tran, J.C.; Catherman, A.D.; Lee, J.E.; Kellie, J.F.; Sweet, S.M.; Zamdborg, L.; Thomas, P.M.; Ahlf, D.R.; Durbin, K.R.; et al. Size-sorting combined with improved nanocapillary liquid chromatography-mass spectrometry for identification of intact proteins up to 80 kda. Anal. Chem. 2010, 82, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Jorgenson, J.W.; Lukacs, K.D. Capillary zone electrophoresis. Science 1983, 222, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Champion, M.M.; Sun, L.; Champion, P.A.; Wojcik, R.; Dovichi, N.J. Capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry as an alternative proteomics platform to ultraperformance liquid chromatography-electrospray ionization-tandem mass spectrometry for samples of intermediate complexity. Anal. Chem. 2012, 84, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, C.A.; Zhang, H.; Yeung, K.K. Recent applications of capillary electrophoresis-mass spectrometry (ce-ms): Ce performing functions beyond separation. Anal. Chim. Acta 2008, 627, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Monton, M.R.; Terabe, S. Field-enhanced sample injection for high-sensitivity analysis of peptides and proteins in capillary electrophoresis-mass spectrometry. J. Chromatogr. A 2004, 1032, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Armenta, J.M.; Gu, B.; Thulin, C.D.; Lee, M.L. Coupled affinity-hydrophobic monolithic column for on-line removal of immunoglobulin g, preconcentration of low abundance proteins and separation by capillary zone electrophoresis. J. Chromatogr. A 2007, 1148, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, R.; Li, Y.; Maccoss, M.J.; Dovichi, N.J. Capillary electrophoresis with orbitrap-velos mass spectrometry detection. Talanta 2012, 88, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Aslanian, A.; Bern, M.; Lavallee-Adam, M.; Yates, J.R., 3rd. Sheathless capillary electrophoresis-tandem mass spectrometry for top-down characterization of pyrococcus furiosus proteins on a proteome scale. Anal. Chem. 2014, 86, 11006–11012. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Knierman, M.D.; Zhu, G.; Dovichi, N.J. Fast top-down intact protein characterization with capillary zone electrophoresis-electrospray ionization tandem mass spectrometry. Anal. Chem. 2013, 85, 5989–5995. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, R.; Zhu, G.; Zhang, Z.; Yan, X.; Zhao, Y.; Sun, L.; Champion, M.M.; Dovichi, N.J. Capillary zone electrophoresis as a tool for bottom-up protein analysis. Bioanalysis 2016, 8, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Moini, M. Simplifying ce-ms operation. 2. Interfacing low-flow separation techniques to mass spectrometry using a porous tip. Anal. Chem. 2007, 79, 4241–4246. [Google Scholar] [CrossRef] [PubMed]
- Nemes, P.; Rubakhin, S.S.; Aerts, J.T.; Sweedler, J.V. Qualitative and quantitative metabolomic investigation of single neurons by capillary electrophoresis electrospray ionization mass spectrometry. Nat. Protoc. 2013, 8, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Compton, P.D.; Tran, J.C.; Ntai, I.; Kelleher, N.L. Optimizing capillary electrophoresis for top-down proteomics of 30–80 kda proteins. Proteomics 2014, 14, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, L.; Zhu, G.; Dovichi, N.J. Coupling capillary zone electrophoresis to a q exactive hf mass spectrometer for top-down proteomics: 580 proteoform identifications from yeast. J. Proteome Res. 2016, 15, 3679–3685. [Google Scholar] [CrossRef] [PubMed]
- Parks, B.A.; Jiang, L.; Thomas, P.M.; Wenger, C.D.; Roth, M.J.; Boyne, M.T., 2nd; Burke, P.V.; Kwast, K.E.; Kelleher, N.L. Top-down proteomics on a chromatographic time scale using linear ion trap fourier transform hybrid mass spectrometers. Anal. Chem. 2007, 79, 7984–7991. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Aslanian, A.; Fonslow, B.; Graczyk, B.; Davis, T.N.; Yates, J.R., 3rd. In-line separation by capillary electrophoresis prior to analysis by top-down mass spectrometry enables sensitive characterization of protein complexes. J. Proteome Res. 2014, 13, 6078–6086. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, N.L.; Senko, M.W.; Siegel, M.M.; McLafferty, F.W. Unit resolution mass spectra of 112 kda molecules with 3 da accuracy. J. Am. Soc. Mass Spectrom. 1997, 8, 380–383. [Google Scholar] [CrossRef]
- Loo, J.A.; Quinn, J.P.; Ryu, S.I.; Henry, K.D.; Senko, M.W.; McLafferty, F.W. High-resolution tandem mass spectrometry of large biomolecules. Proc. Natl. Acad. Sci. USA 1992, 89, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Henry, K.D.; McLafferty, F.W. Electrospray ionization with fourier-transform mass spectrometry. Charge state assignment from resolved isotopic peaks. Org. Mass Spectrom. 1990, 25, 490–492. [Google Scholar] [CrossRef]
- Gordon, E.F.; Mansoori, B.A.; Carroll, C.F.; Muddiman, D.C. Hydropathic influences on the quantification of equine heart cytochromec using relative ion abundance measurements by electrospray ionization fourier transform ion cyclotron resonance mass spectrometry. J. Mass Spectrom. 1999, 34, 1055–1062. [Google Scholar] [CrossRef]
- Zubarev, R.A.; Makarov, A. Orbitrap mass spectrometry. Anal. Chem. 2013, 85, 5288–5296. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Noll, R.J.; Li, H.; Makarov, A.; Hardman, M.; Graham Cooks, R. The orbitrap: A new mass spectrometer. J. Mass Spectrom. 2005, 40, 430–443. [Google Scholar] [CrossRef] [PubMed]
- McAlister, G.C.; Berggren, W.T.; Griep-Raming, J.; Horning, S.; Makarov, A.; Phanstiel, D.; Stafford, G.; Swaney, D.L.; Syka, J.E.; Zabrouskov, V.; et al. A proteomics grade electron transfer dissociation-enabled hybrid linear ion trap-orbitrap mass spectrometer. J. Proteome Res. 2008, 7, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.C.; DeHart, C.J.; Kaiser, N.K.; Fellers, R.T.; Smith, D.F.; Greer, J.B.; LeDuc, R.D.; Blakney, G.T.; Thomas, P.M.; Kelleher, N.L.; et al. Identification and characterization of human proteoforms by top-down lc-21 tesla ft-icr mass spectrometry. J. Proteome Res. 2017, 16, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Data Analysis Software Page. Available online: http://www.topdownproteomics.org/resources/software (accessed on 6 April 2017).
- Campbell, J.L.; Le Blanc, J.C. Targeted ion parking for the quantitation of biotherapeutic proteins: Concepts and preliminary data. J. Am. Soc. Mass Spectrom. 2010, 21, 2011–2022. [Google Scholar] [CrossRef] [PubMed]
- Muddiman, D.C.; Cheng, X.; Udseth, H.R.; Smith, R.D. Charge-state reduction with improved signal intensity of oligonucleotides in electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 1996, 7, 697–706. [Google Scholar] [CrossRef]
- Reid, G.E.; Shang, H.; Hogan, J.M.; Lee, G.U.; McLuckey, S.A. Gas-phase concentration, purification, and identification of whole proteins from complex mixtures. J. Am. Chem. Soc. 2002, 124, 7353–7362. [Google Scholar] [CrossRef] [PubMed]
- McLuckey, S.A.; Reid, G.E.; Wells, J.M. Ion parking during ion/ion reactions in electrodynamic ion traps. Anal. Chem. 2002, 74, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.M.; Souda, P.; Bassilian, S.; Ujwal, R.; Zhang, J.; Abramson, J.; Ping, P.; Durazo, A.; Bowie, J.U.; Hasan, S.S.; et al. Post-translational modifications of integral membrane proteins resolved by top-down fourier transform mass spectrometry with collisionally activated dissociation. Mol. Cell. Proteom. 2010, 9, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.M.; Reid, G.E. Evaluation of gas-phase rearrangement and competing fragmentation reactions on protein phosphorylation site assignment using collision induced dissociation-ms/ms and ms3. Anal. Chem. 2008, 80, 9735–9747. [Google Scholar] [CrossRef] [PubMed]
- Ahlf, D.R.; Compton, P.D.; Tran, J.C.; Early, B.P.; Thomas, P.M.; Kelleher, N.L. Evaluation of the compact high-field orbitrap for top-down proteomics of human cells. J. Proteome Res. 2012, 11, 4308–4314. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.D.; Paša-Tolić, L.; Lipton, M.S.; Jensen, P.K.; Anderson, G.A.; Shen, Y.; Conrads, T.P.; Udseth, H.R.; Harkewicz, R.; Belov, M.E.; et al. Rapid quantitative measurements of proteomes by fourier transform ion cyclotron resonance mass spectrometry. Electrophoresis 2001, 22, 1652–1668. [Google Scholar] [CrossRef]
- Pesavento, J.J.; Mizzen, C.A.; Kelleher, N.L. Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: Human histone h4. Anal. Chem. 2006, 78, 4271–4280. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A.; Mollah, S.; Ueberheide, B.M.; Busby, S.A.; Muratore, T.L.; Shabanowitz, J.; Hunt, D.F. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007, 2, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Lin, S.; Garcia, B.A.; Zhao, Y. Quantitative proteomic analysis of histone modifications. Chem. Rev. 2015, 115, 2376–2418. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.L.; Callahan, J.H.; Monday, S.R.; Feng, P.C.; Musser, S.M. Relative quantitation of intact proteins of bacterial cell extracts using coextracted proteins as internal standards. Anal. Chem. 2004, 76, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Ntai, I.; Toby, T.K.; LeDuc, R.D.; Kelleher, N.L. A method for label-free, differential top-down proteomics. Methods Mol. Biol. 2016, 1410, 121–133. [Google Scholar] [PubMed]
- Unwin, R.D. Quantification of proteins by itraq. Methods Mol. Biol. 2010, 658, 205–215. [Google Scholar] [PubMed]
- Unwin, R.D.; Griffiths, J.R.; Whetton, A.D. Simultaneous analysis of relative protein expression levels across multiple samples using itraq isobaric tags with 2d nano lc-ms/ms. Nat. Protoc. 2010, 5, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by ms/ms. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Huang, S.Y.; Chow, N.H.; Chen, S.H. Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 2003, 75, 6843–6852. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.W.; Tholey, A. Tandem mass tag protein labeling for top-down identification and quantification. Anal. Chem. 2012, 84, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, J.M.; Vaudel, M.; Zahedi, R.P.; Martens, L.; Sickmann, A. Itraq protein quantification: A quality-controlled workflow. Proteomics 2011, 11, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Cologna, S.M.; Crutchfield, C.A.; Searle, B.C.; Blank, P.S.; Toth, C.L.; Ely, A.M.; Picache, J.A.; Backlund, P.S.; Wassif, C.A.; Porter, F.D.; et al. An efficient approach to evaluate reporter ion behavior from maldi-ms/ms data for quantification studies using isobaric tags. J. Proteome Res. 2015, 14, 4169–4178. [Google Scholar] [CrossRef] [PubMed]
- Gouw, J.W.; Krijgsveld, J.; Heck, A.J. Quantitative proteomics by metabolic labeling of model organisms. Mol. Cell. Proteom. 2010, 9, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Mann, M. A practical recipe for stable isotope labeling by amino acids in cell culture (silac). Nat. Protoc. 2006, 1, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, silac, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Waanders, L.F.; Hanke, S.; Mann, M. Top-down quantitation and characterization of silac-labeled proteins. J. Am. Soc. Mass Spectrom. 2007, 18, 2058–2064. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.S.; Hawkridge, A.M.; Georgianna, D.R.; Payne, G.A.; Muddiman, D.C. Top-down identification and quantification of stable isotope labeled proteins from aspergillus flavus using online nano-flow reversed-phase liquid chromatography coupled to a ltq-fticr mass spectrometer. Anal. Chem. 2008, 80, 4994–5001. [Google Scholar] [CrossRef] [PubMed]
- Picard, G.; Lebert, D.; Louwagie, M.; Adrait, A.; Huillet, C.; Vandenesch, F.; Bruley, C.; Garin, J.; Jaquinod, M.; Brun, V. Psaq standards for accurate ms-based quantification of proteins: From the concept to biomedical applications. J. Mass Spectrom. 2012, 47, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Brun, V.; Dupuis, A.; Adrait, A.; Marcellin, M.; Thomas, D.; Court, M.; Vandenesch, F.; Garin, J. Isotope-labeled protein standards: Toward absolute quantitative proteomics. Mol. Cell. Proteom. 2007, 6, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, A.; Hennekinne, J.A.; Garin, J.; Brun, V. Protein standard absolute quantification (psaq) for improved investigation of staphylococcal food poisoning outbreaks. Proteomics 2008, 8, 4633–4636. [Google Scholar] [CrossRef] [PubMed]
- Kippen, A.D.; Cerini, F.; Vadas, L.; Stocklin, R.; Vu, L.; Offord, R.E.; Rose, K. Development of an isotope dilution assay for precise determination of insulin, c-peptide, and proinsulin levels in non-diabetic and type ii diabetic individuals with comparison to immunoassay. J. Biol. Chem. 1997, 272, 12513–12522. [Google Scholar] [CrossRef] [PubMed]
- Stocklin, R.; Vu, L.; Vadas, L.; Cerini, F.; Kippen, A.D.; Offord, R.E.; Rose, K. A stable isotope dilution assay for the in vivo determination of insulin levels in humans by mass spectrometry. Diabetes 1997, 46, 44–50. [Google Scholar] [CrossRef] [PubMed]
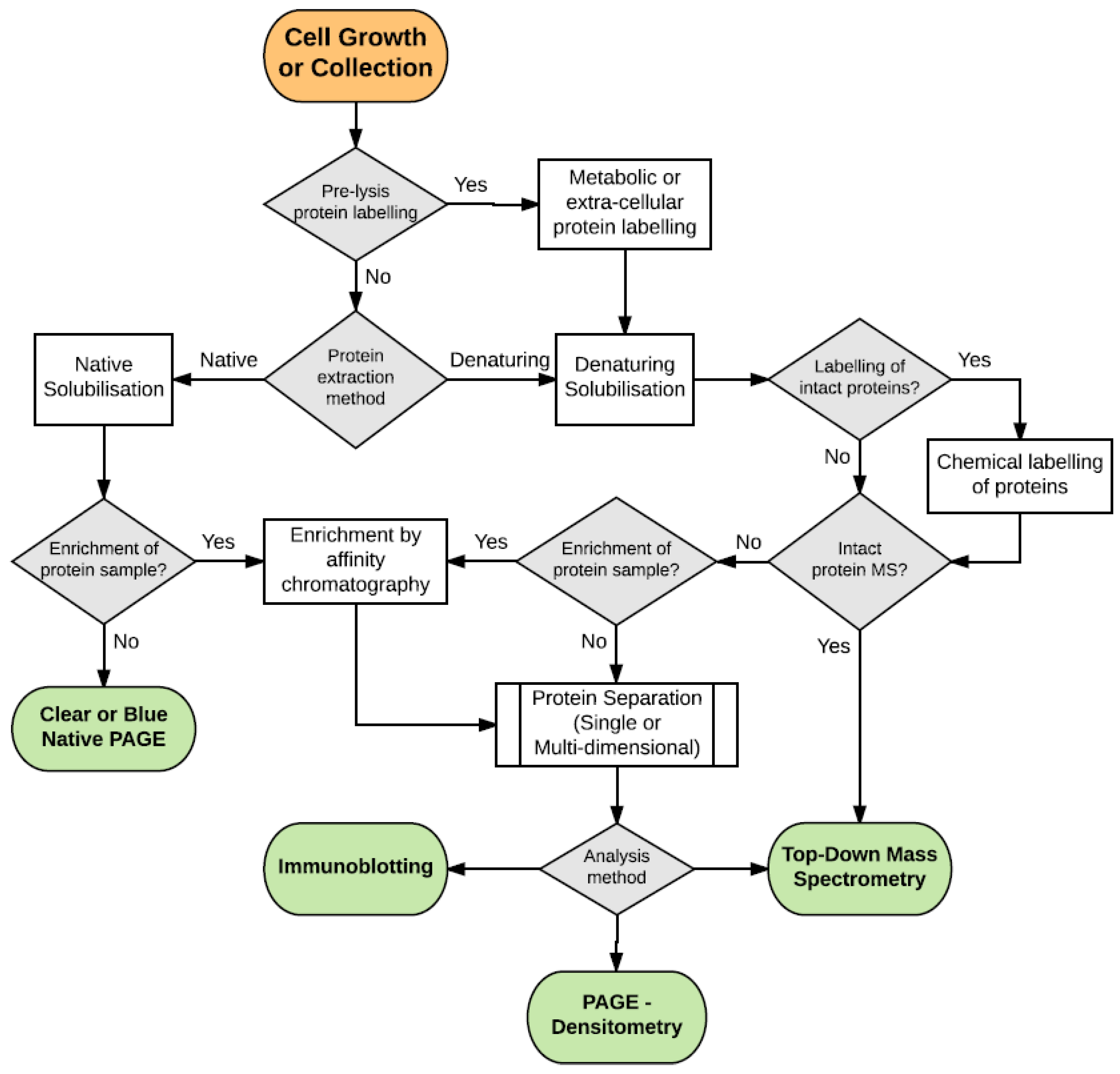

{kind=link}
| Technique | Advantages | Disadvantages | Reference |
|---|---|---|---|
| Shotgun LC/MS/MS | High proteome coverage. | Proteoform information disconnected from measured peptides unless particular peptide detected | [8,10,16,17,18] |
| 1D-PAGE/Shotgun LC/MS/MS | Proteoform size preserved allowing proteolytic cleavage of parent protein to be inferred. High proteome coverage. SDS can be used to solubilise proteins. | Proteoform information disconnected from measured peptides unless particular peptide detected. | [32,33,34] |
| 2D-PAGE | High resolution separation of intact proteoforms. Parallel processing of multiple samples. Robust quantitation of proteoforms | High amount of sample required compared to shotgun LC/MS/MS. Perception of high technical difficulty. Low proteome coverage compared to LC/MS/MS | [21] |
| GelFREE LC/MS/MS | High accuracy measurement of proteoform mass that can infer nature of PTMs or proteolytic cleavage. | Low proteome coverage compared to LC/MS/MS. Cannot parallel process multiple samples. Enormous amount of MS acquisition time required for one sample resulting in low throughput. | [20,35] |
| Native PAGE | Maintains biological context of protein-protein interaction | Sample handling needs to be controlled for temperature, pH and physical movement. Transient interactions can be easily lost if these parameters are not maintained. | [36,37,38] |
| Ligand blotting | Supportive orthogonal method to confirm interactions between 2 or more molecules | Titration of ligand to binding partner requires optimisation as in antibody western blot systems | [31] |
| Bait-prey affinity isolation | Allows for a robust labelled capture-based technique for interacting proteins | Precipitation of proteins in sample preparation can preclude them from the method. False-positive interactions can occur with binding sites of proteins folding changes in altered buffering conditions. | [31,34] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padula, M.P.; Berry, I.J.; O′Rourke, M.B.; Raymond, B.B.A.; Santos, J.; Djordjevic, S.P. A Comprehensive Guide for Performing Sample Preparation and Top-Down Protein Analysis. Proteomes 2017, 5, 11. https://doi.org/10.3390/proteomes5020011
Padula MP, Berry IJ, O′Rourke MB, Raymond BBA, Santos J, Djordjevic SP. A Comprehensive Guide for Performing Sample Preparation and Top-Down Protein Analysis. Proteomes. 2017; 5(2):11. https://doi.org/10.3390/proteomes5020011
Chicago/Turabian StylePadula, Matthew P., Iain J. Berry, Matthew B. O′Rourke, Benjamin B.A. Raymond, Jerran Santos, and Steven P. Djordjevic. 2017. "A Comprehensive Guide for Performing Sample Preparation and Top-Down Protein Analysis" Proteomes 5, no. 2: 11. https://doi.org/10.3390/proteomes5020011
APA StylePadula, M. P., Berry, I. J., O′Rourke, M. B., Raymond, B. B. A., Santos, J., & Djordjevic, S. P. (2017). A Comprehensive Guide for Performing Sample Preparation and Top-Down Protein Analysis. Proteomes, 5(2), 11. https://doi.org/10.3390/proteomes5020011