
Global Proteome Changes in Liver Tissue 6 Weeks after FOLFOX Treatment of Colorectal Cancer Liver Metastases

Abstract
:1. Introduction
2. Results
2.1. Clinical Data
2.2. Proteome Description
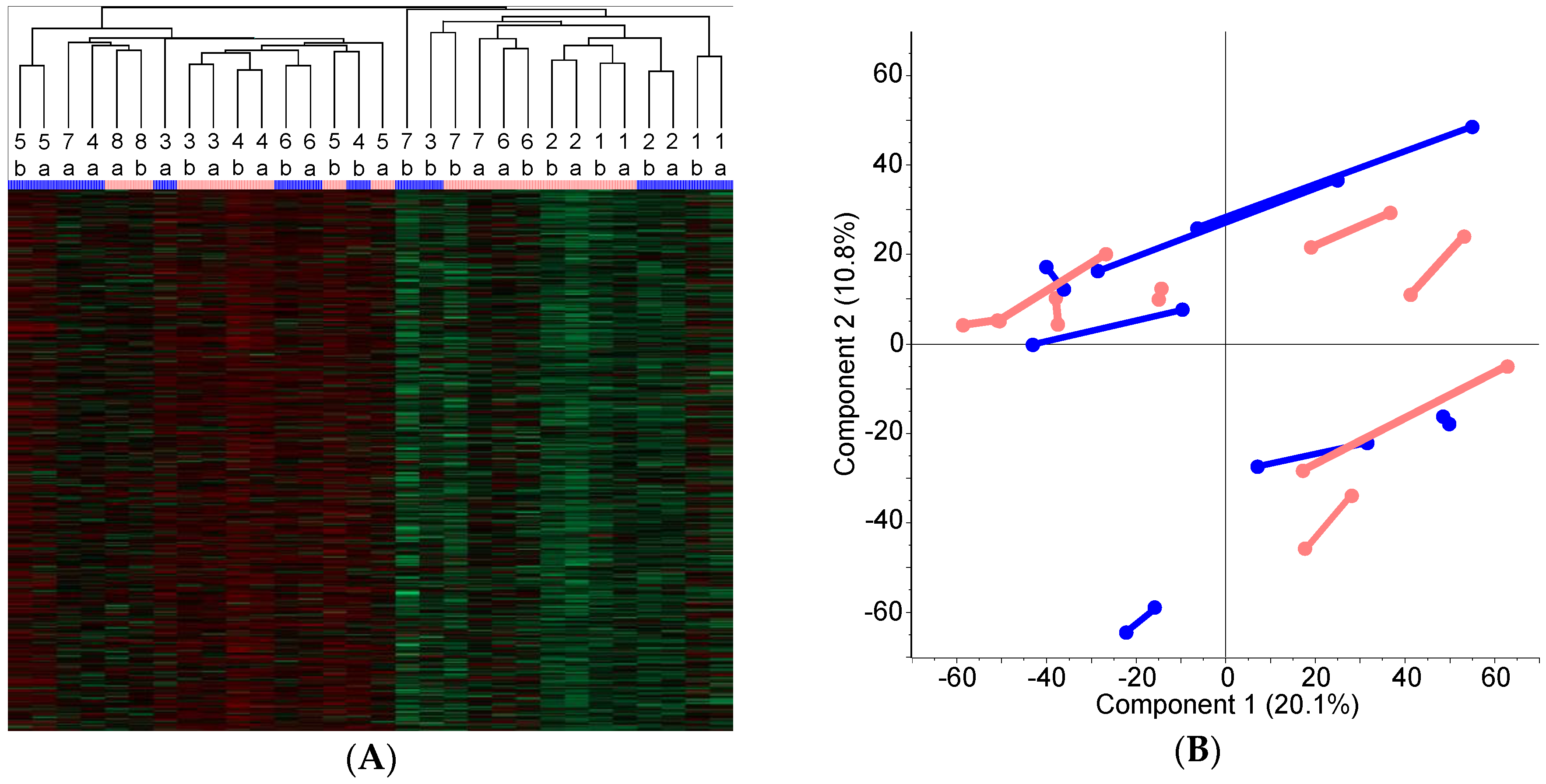
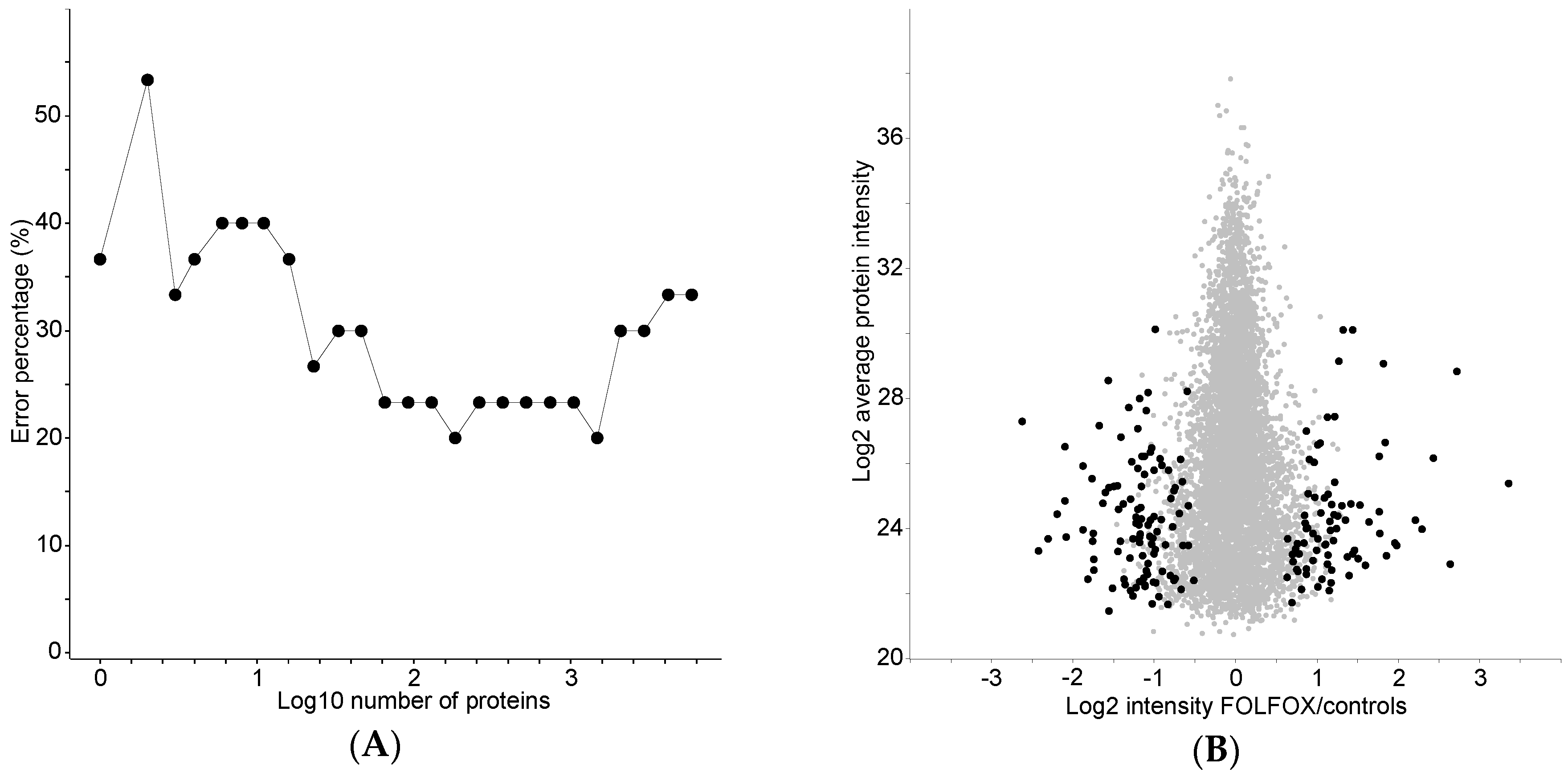
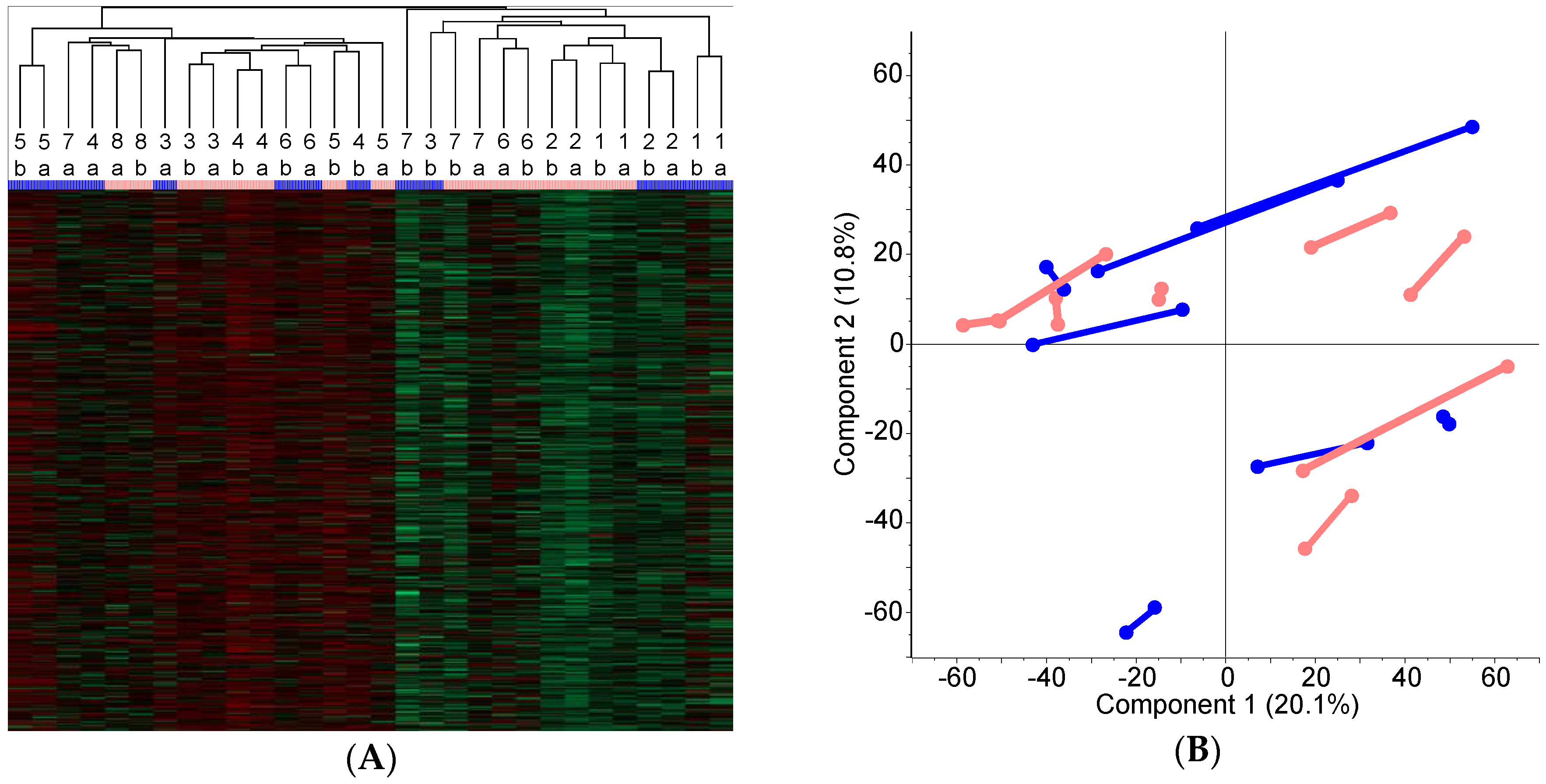
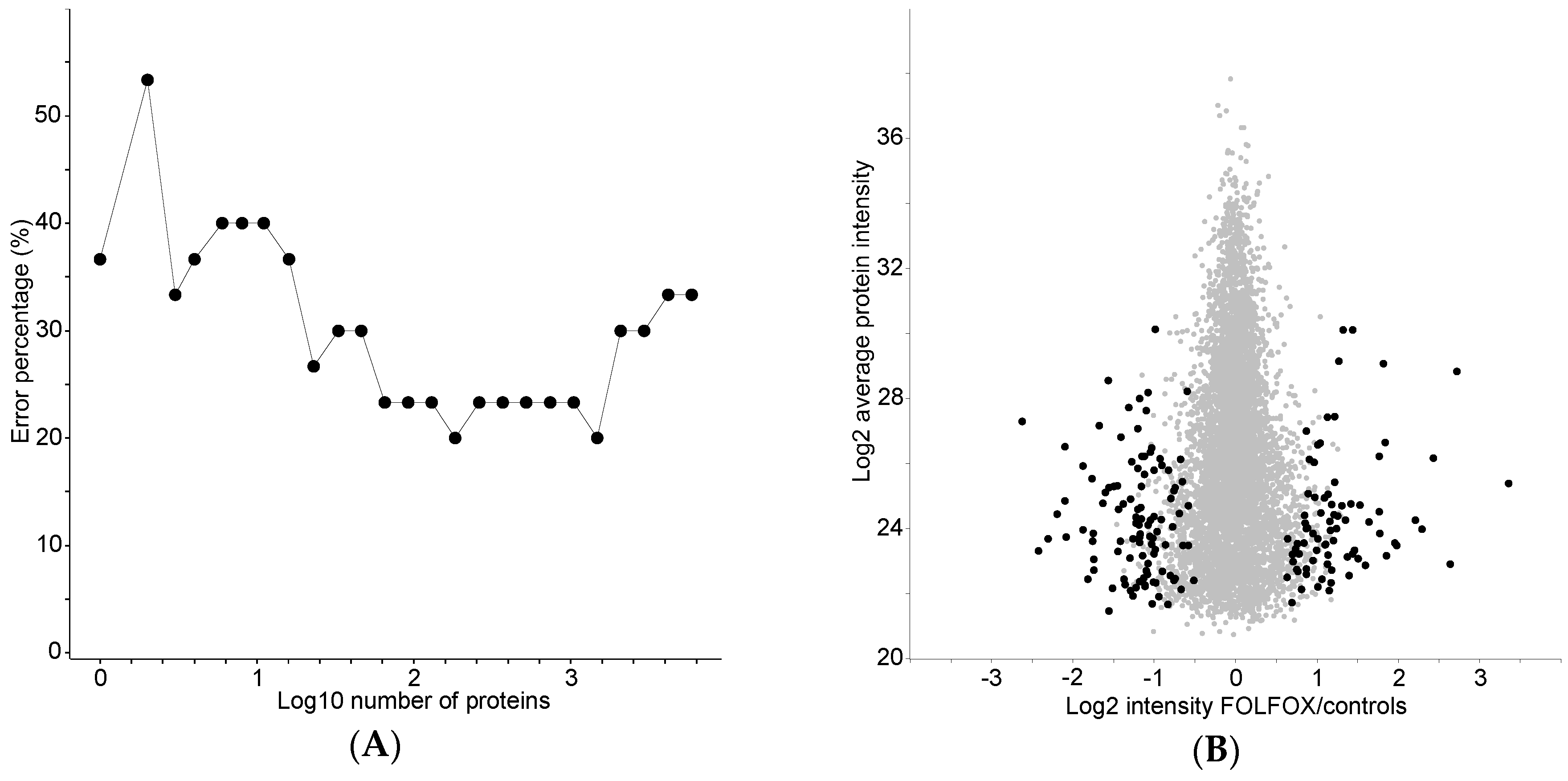
2.3. Classification of the FOLFOX-Treated and Control Group on the Basis of Protein Expression
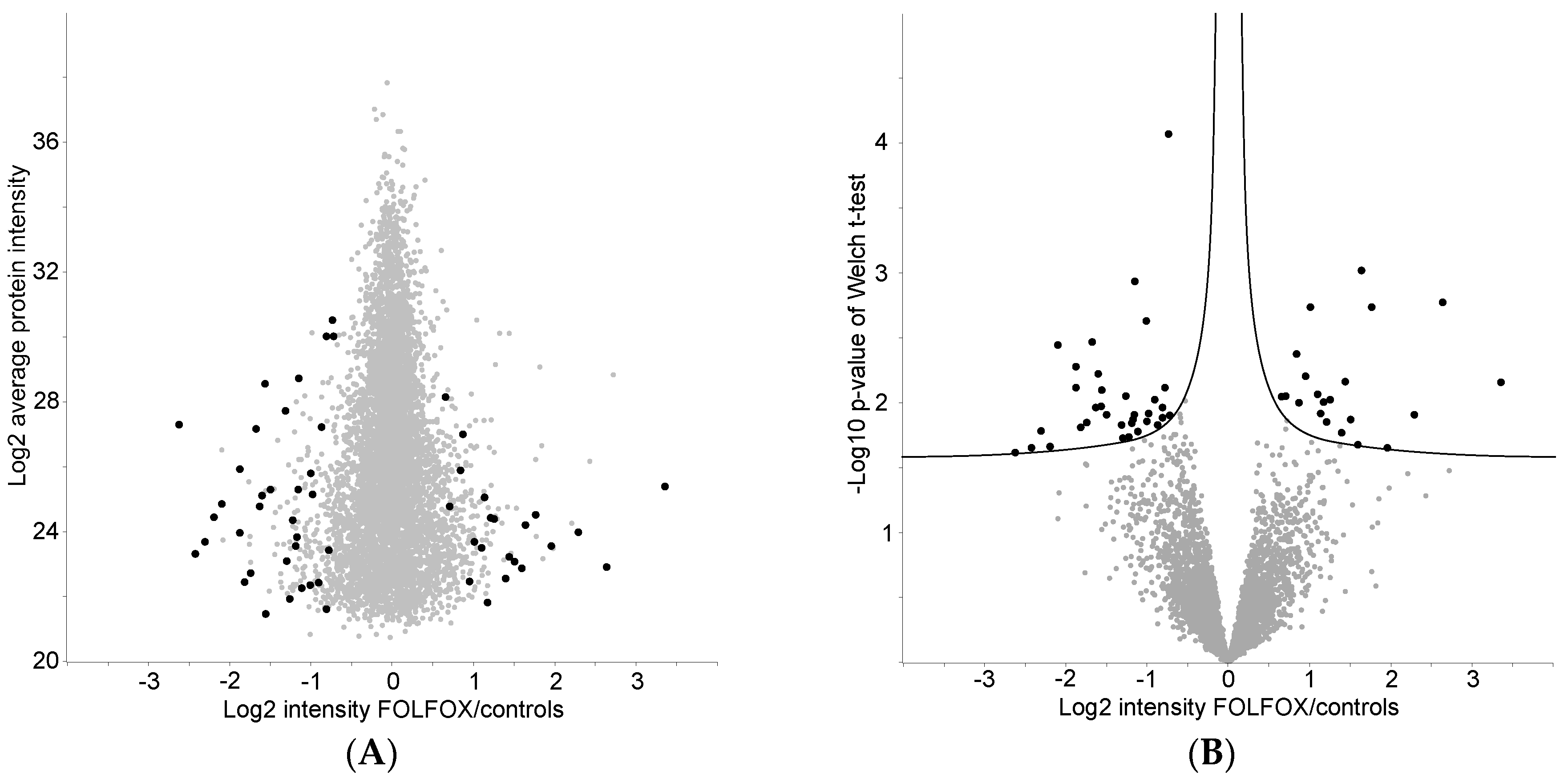
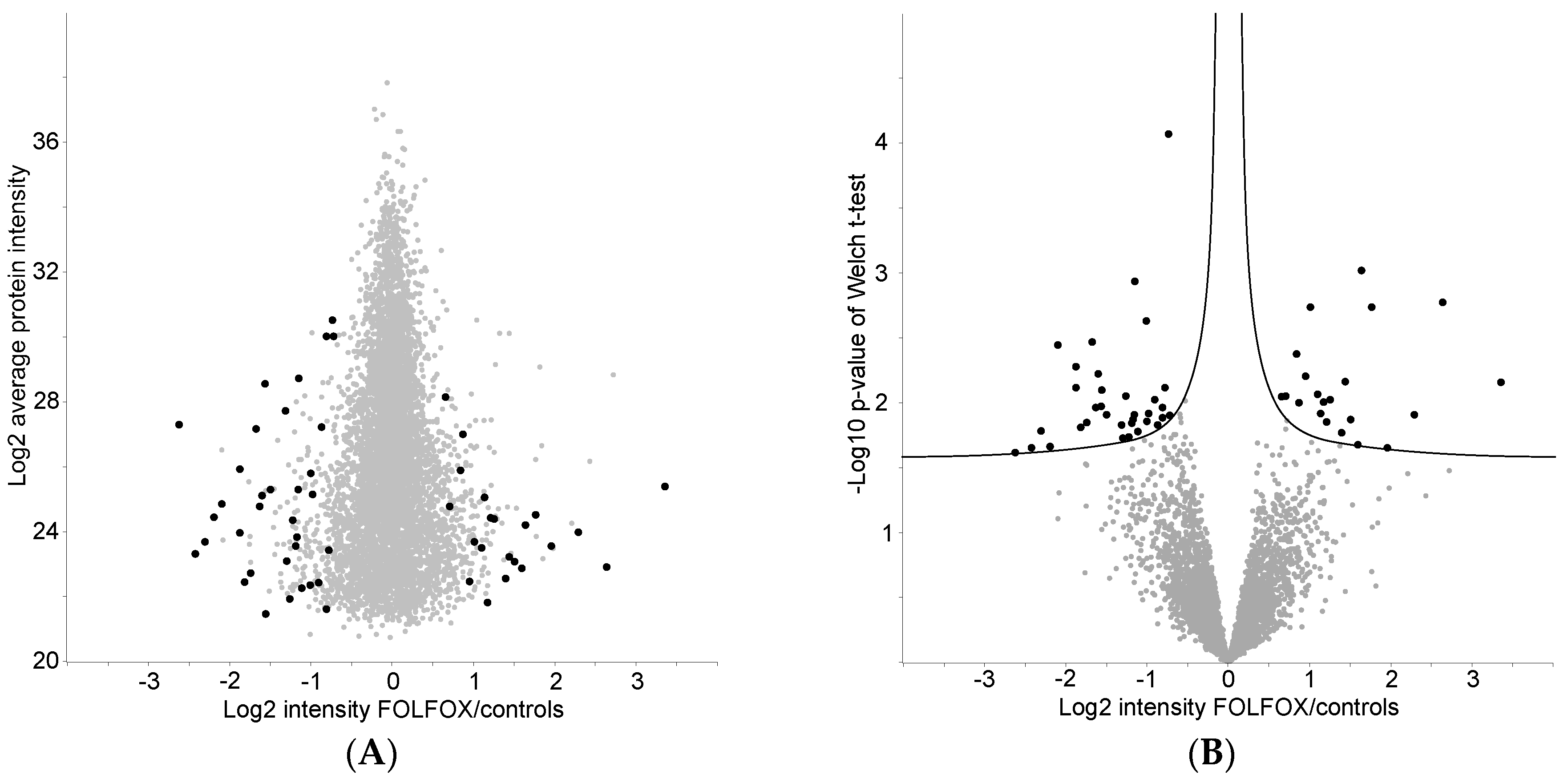
2.4. Proteome Differences between FOLFOX-Treated and Control Group
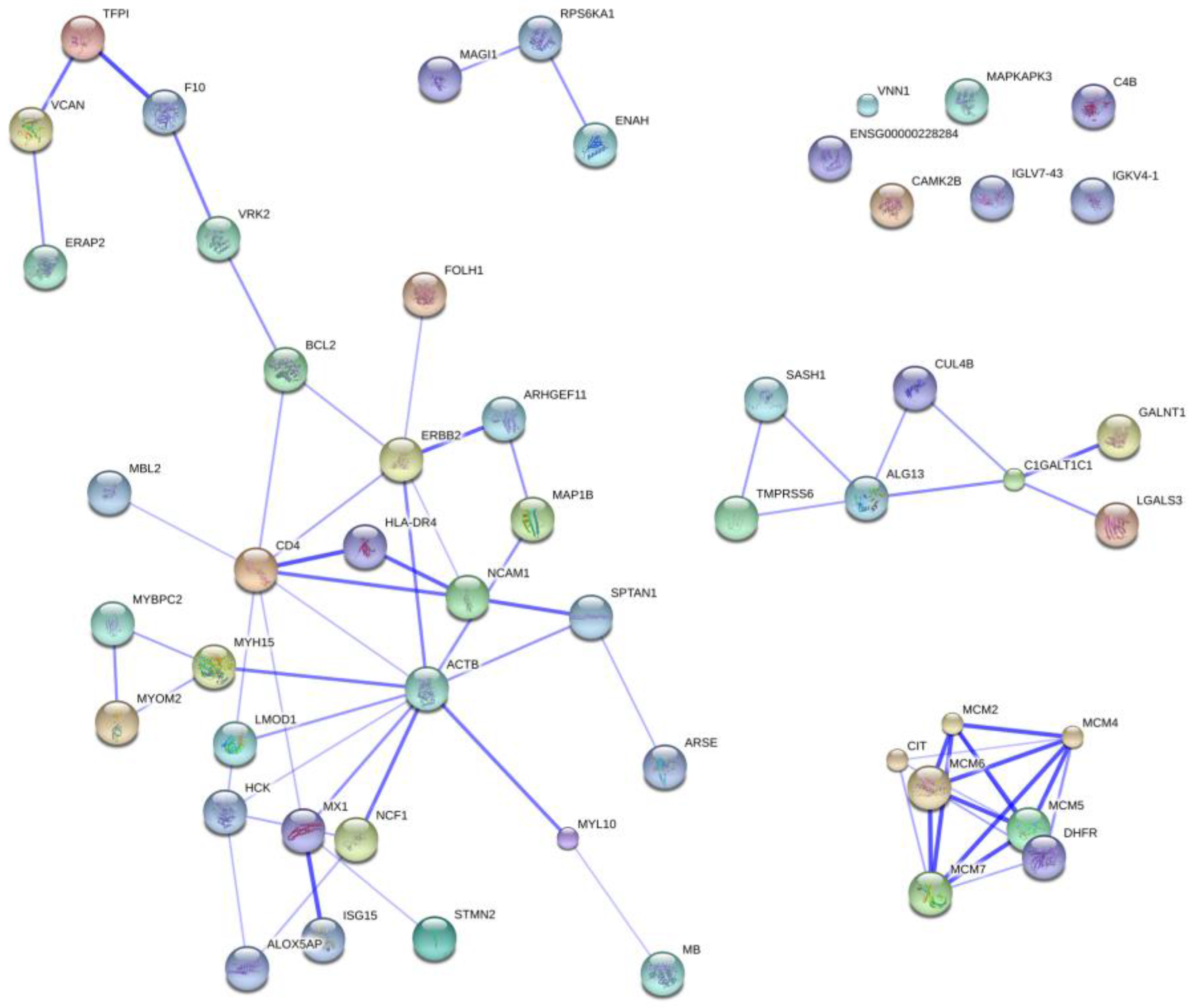
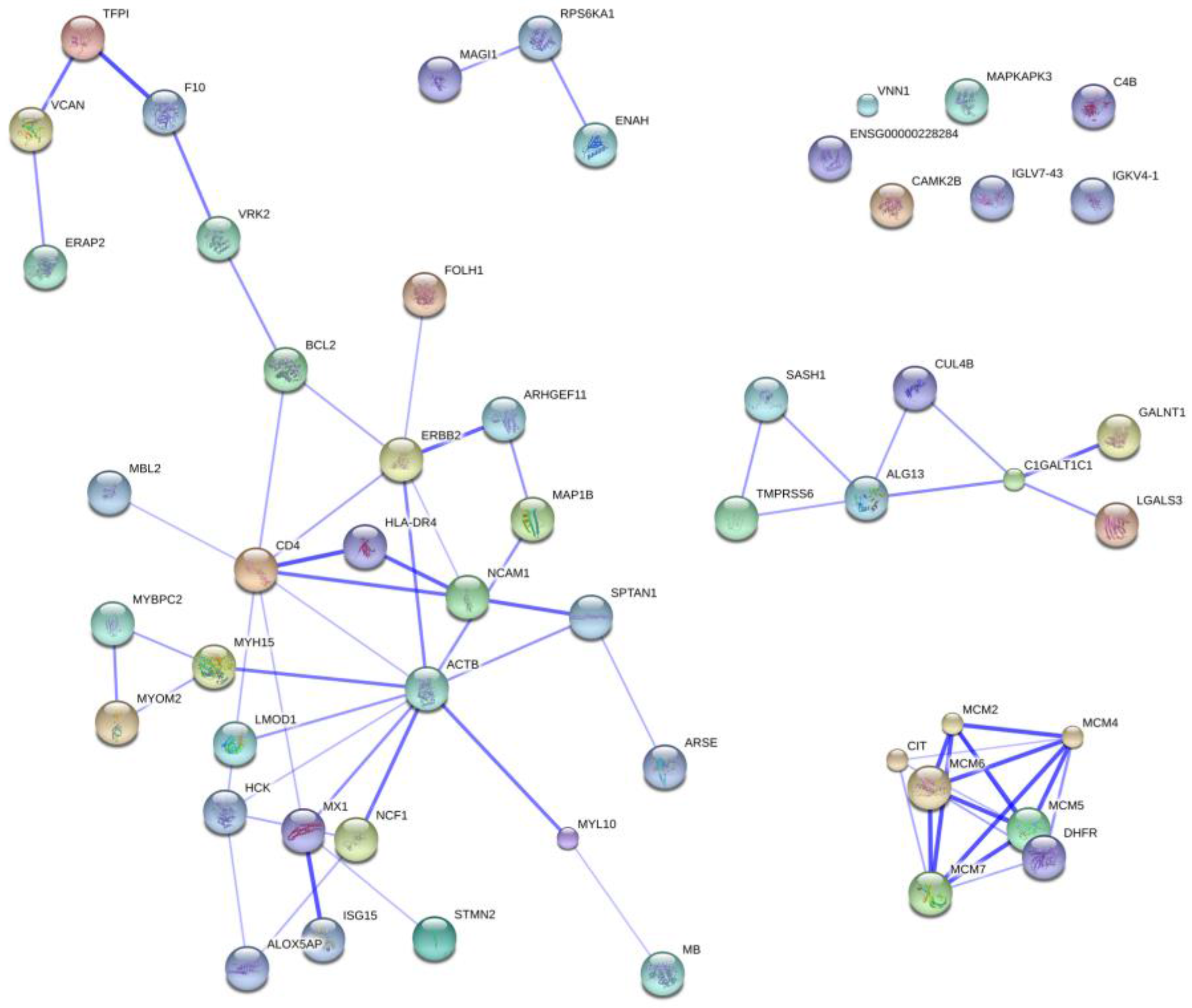
2.5. Protein Ontology and Pathway Analysis
3. Discussion
4. Materials and Methods
4.1. Patients and Liver Tissue Samples
4.2. Preparation of Tissue Lysates
4.3. Sample Processing
4.4. Removal of Substances Affecting Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS)
4.5. LC–MS/MS and Raw Data Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CRLM | Colo-Rectal cancer Liver Metastases |
| FOLFOX | Fluorouracil Leucovorin Oxaliplatin treatment |
| SI | Sinusoidal Injury |
| PCA | Principal Component Analysis |
| RFE-SVM | Recursive Feature Elimination-Support Vector Machine |
| FDR | False Discovery Ratio |
| MCM | minichromosome maintenance complex |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
References
- Adam, R.; Delvart, V.; Pascal, G.; Valeanu, A.; Castaing, D.; Azoulay, D.; Giacchetti, S.; Paule, B.; Kunstlinger, F.; Ghemard, O.; et al. Rescue surgery for unresectable colorectal liver metastases downstaged by chemotherapy: A model to predict long-term survival. Ann. Surg. 2004, 240, 644–657; discussion 657–658. [Google Scholar] [CrossRef] [PubMed]
- Nordlinger, B.; Sorbye, H.; Glimelius, B.; Poston, G.J.; Schlag, P.M.; Rougier, P.; Bechstein, W.O.; Primrose, J.N.; Walpole, E.T.; Finch-Jones, M.; et al. Perioperative chemotherapy with folfox4 and surgery versus surgery alone for resectable liver metastases from colorectal cancer (eortc intergroup trial 40983): A randomised controlled trial. Lancet 2008, 371, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Vauthey, J.N.; Pawlik, T.M.; Ribero, D.; Wu, T.T.; Zorzi, D.; Hoff, P.M.; Xiong, H.Q.; Eng, C.; Lauwers, G.Y.; Mino-Kenudson, M.; et al. Chemotherapy regimen predicts steatohepatitis and an increase in 90-day mortality after surgery for hepatic colorectal metastases. J. Clin. Oncol. 2006, 24, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Rubbia-Brandt, L.; Audard, V.; Sartoretti, P.; Roth, A.D.; Brezault, C.; Le Charpentier, M.; Dousset, B.; Morel, P.; Soubrane, O.; Chaussade, S.; et al. Severe hepatic sinusoidal obstruction associated with oxaliplatin-based chemotherapy in patients with metastatic colorectal cancer. Ann. Oncol. 2004, 15, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Rubbia-Brandt, L.; Lauwers, G.Y.; Wang, H.; Majno, P.E.; Tanabe, K.; Zhu, A.X.; Brezault, C.; Soubrane, O.; Abdalla, E.K.; Vauthey, J.N.; et al. Sinusoidal obstruction syndrome and nodular regenerative hyperplasia are frequent oxaliplatin-associated liver lesions and partially prevented by bevacizumab in patients with hepatic colorectal metastasis. Histopathology 2010, 56, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, K.; Rickenbacher, A.; Weber, A.; Pestalozzi, B.C.; Clavien, P.A. Chemotherapy before liver resection of colorectal metastases: Friend or foe? Ann. Surg. 2012, 255, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Aloia, T.; Sebagh, M.; Plasse, M.; Karam, V.; Levi, F.; Giacchetti, S.; Azoulay, D.; Bismuth, H.; Castaing, D.; Adam, R. Liver histology and surgical outcomes after preoperative chemotherapy with fluorouracil plus oxaliplatin in colorectal cancer liver metastases. J. Clin. Oncol. 2006, 24, 4983–4990. [Google Scholar] [CrossRef] [PubMed]
- Vietor, N.O.; George, B.J. Oxaliplatin-induced hepatocellular injury and ototoxicity: A review of the literature and report of unusual side effects of a commonly used chemotherapeutic agent. J. Oncol. Pharm. Pract. 2012, 18, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Maru, D.M.; Charnsangavej, C.; Loyer, E.M.; Wang, H.; Pathak, P.; Eng, C.; Hoff, P.M.; Vauthey, J.N.; Wolff, R.A.; et al. Oxaliplatin-mediated increase in spleen size as a biomarker for the development of hepatic sinusoidal injury. J. Clin. Oncol. 2010, 28, 2549–2555. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Agostini, J.; Benoist, S.; Seman, M.; Julie, C.; Imbeaud, S.; Letourneur, F.; Cagnard, N.; Rougier, P.; Brouquet, A.; Zucman-Rossi, J.; et al. Identification of molecular pathways involved in oxaliplatin-associated sinusoidal dilatation. J. Hepatol. 2012, 56, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Pilgrim, C.H.; Brettingham-Moore, K.; Pham, A.; Murray, W.; Link, E.; Smith, M.; Usatoff, V.; Evans, P.M.; Banting, S.; Thomson, B.N.; et al. Mrna gene expression correlates with histologically diagnosed chemotherapy-induced hepatic injury. HPB 2011, 13, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.M.; Mann, J.; Manas, D.M.; Mann, D.A.; White, S.A. An experimental study to identify the potential role of pharmacogenomics in determining the occurrence of oxaliplatin-induced liver injury. HPB 2013, 15, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Vreuls, C.P.; Olde Damink, S.W.; Koek, G.H.; Winstanley, A.; Wisse, E.; Cloots, R.H.; van den Broek, M.A.; Dejong, C.H.; Bosman, F.T.; Driessen, A. Glutathione s-transferase m1-null genotype as risk factor for sos in oxaliplatin-treated patients with metastatic colorectal cancer. Br. J. Cancer 2013, 108, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Rubbia-Brandt, L.; Tauzin, S.; Brezault, C.; Delucinge-Vivier, C.; Descombes, P.; Dousset, B.; Majno, P.E.; Mentha, G.; Terris, B. Gene expression profiling provides insights into pathways of oxaliplatin-related sinusoidal obstruction syndrome in humans. Mol. Cancer Ther. 2011, 10, 687–696. [Google Scholar] [CrossRef] [PubMed]
- DeLeve, L.D.; Shulman, H.M.; McDonald, G.B. Toxic injury to hepatic sinusoids: Sinusoidal obstruction syndrome (veno-occlusive disease). Semin. Liver Dis. 2002, 22, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.; Nicco, C.; Chereau, C.; Goulvestre, C.; Alexandre, J.; Alves, A.; Levy, E.; Goldwasser, F.; Panis, Y.; Soubrane, O.; et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005, 65, 948–956. [Google Scholar] [PubMed]
- Alexandre, J.; Nicco, C.; Chereau, C.; Laurent, A.; Weill, B.; Goldwasser, F.; Batteux, F. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J. Natl. Cancer Inst. 2006, 98, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Deleve, L.D.; Wang, X.; Tsai, J.; Kanel, G.; Strasberg, S.; Tokes, Z.A. Sinusoidal obstruction syndrome (veno-occlusive disease) in the rat is prevented by matrix metalloproteinase inhibition. Gastroenterology 2003, 125, 882–890. [Google Scholar] [CrossRef]
- Mikalauskas, S.; Mikalauskiene, L.; Bruns, H.; Nickkholgh, A.; Hoffmann, K.; Longerich, T.; Strupas, K.; Buchler, M.W.; Schemmer, P. Dietary glycine protects from chemotherapy-induced hepatotoxicity. Amino. Acids 2011, 40, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.H.; Lu, J.F.; Wang, K. The effect of cisplatin and transplatin on the conformation and association of f-actin. Cell. Biol. Int. 1995, 19, 491–497. [Google Scholar] [PubMed]
- Robinson, S.M.; Mann, J.; Vasilaki, A.; Mathers, J.; Burt, A.D.; Oakley, F.; White, S.A.; Mann, D.A. Pathogenesis of folfox induced sinusoidal obstruction syndrome in a murine chemotherapy model. J. Hepatol. 2013, 59, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Mejias, M.; Garcia-Pras, E.; Mendez, R.; Garcia-Pagan, J.C.; Bosch, J. Reversal of portal hypertension and hyperdynamic splanchnic circulation by combined vascular endothelial growth factor and platelet-derived growth factor blockade in rats. Hepatology 2007, 46, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.M.; Mann, D.A.; Manas, D.M.; Oakley, F.; Mann, J.; White, S.A. The potential contribution of tumour-related factors to the development of folfox-induced sinusoidal obstruction syndrome. Br. J. Cancer 2013, 109, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Singh, T.S.; Lee, H.C.; Moon, P.G.; Lee, J.E.; Lee, M.H.; Choi, E.C.; Chen, Y.J.; Kim, S.H.; Baek, M.C. In-depth identification of pathways related to cisplatin-induced hepatotoxicity through an integrative method based on an informatics-assisted label-free protein quantitation and microarray gene expression approach. Mol. Cell. Proteomics 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.S.; Fong, B.M. Lc-ms/ms in the routine clinical laboratory: Has its time come? Anal. Bioanal. Chem. 2014, 406, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, E.; Fagerberg, L.; Klevebring, D.; Matic, I.; Geiger, T.; Cox, J.; Algenas, C.; Lundeberg, J.; Mann, M.; Uhlen, M. Defining the transcriptome and proteome in three functionally different human cell lines. Mol. Syst. Biol. 2010, 6, 450. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Paabo, S.; Mann, M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [Google Scholar] [CrossRef] [PubMed]
- Kmiec, Z. Cooperation of liver cells in health and disease. Adv. Anat. Embryol. Cell. Biol. 2001, 161, 1–151. [Google Scholar]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N.; Campbell, J.S. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech. Dev. 2003, 120, 117–130. [Google Scholar] [CrossRef]
- Waga, S.; Stillman, B. The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem. 1998, 67, 721–751. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Schwacha, A. The mcm complex: Unwinding the mechanism of a replicative helicase. Microbiol. Mol. Biol. Rev. 2009, 73, 652–683. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.; Hamid, S.; Morris, L.; Vowler, S.; Rushbrook, S.; Wight, D.G.; Coleman, N.; Alexander, G.J. Improved detection of hepatocyte proliferation using antibody to the pre-replication complex: An association with hepatic fibrosis and viral replication in chronic hepatitis c virus infection. J. Viral Hepat. 2003, 10, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Gaugaz, F.Z. Fast and sensitive total protein and peptide assays for proteomic analysis. Anal. Chem. 2015, 87, 4110–4116. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Mann, M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 2012, 84, 2631–2637. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Mann, M. Combination of fasp and stagetip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 2009, 8, 5674–5678. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. Proteomexchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Names | Protein Names | Welch t-Test p-Value | Fold Change | Coefficient of Variation | Peptides | Unique Peptides |
|---|---|---|---|---|---|---|
| MAP1B | Microtubule-associated protein 1B;MAP1 light chain LC1 | 0.007 | 10.21 | 1.43 | 24 | 23 |
| HLA-DQA1 | Major histocompatibility complex, class II, DQ alpha 1 | 0.002 | 6.23 | 1.12 | 4 | 2 |
| C19orf52 | Uncharacterized protein C19orf52 | 0.012 | 4.90 | 1.92 | 3 | 3 |
| IGHD | Ig delta chain C region | 0.022 | 3.88 | 1.82 | 7 | 7 |
| MCM2 | DNA replication licensing factor MCM2 | 0.002 | 3.40 | 0.80 | 12 | 12 |
| MLIP | Muscular LMNA-interacting protein | 0.001 | 3.11 | 0.74 | 4 | 4 |
| STMN2 | Stathmin-2 | 0.021 | 3.02 | 0.93 | 1 | 1 |
| Q7Z7K6 | Centromere protein V | 0.013 | 2.84 | 1.37 | 8 | 3 |
| MCM4 | DNA replication licensing factor MCM4 | 0.007 | 2.71 | 0.82 | 7 | 7 |
| EMG1 | Ribosomal RNA small subunit methyltransferase NEP1 | 0.017 | 2.63 | 1.17 | 4 | 4 |
| NUDT12 | Peroxisomal NADH pyrophosphatase NUDT12 | 0.009 | 2.38 | 0.73 | 8 | 8 |
| DHFR; DHFRL1 | Dihydrofolate reductase;Dihydrofolate reductase, mitochondrial | 0.014 | 2.31 | 0.62 | 2 | 2 |
| OSBPL6 | Oxysterol-binding protein-related protein 6 | 0.010 | 2.25 | 0.84 | 3 | 3 |
| MCM7 | DNA replication licensing factor MCM7 | 0.012 | 2.20 | 0.71 | 11 | 11 |
| ANGPTL3 | Angiopoietin-related protein 3 | 0.009 | 2.14 | 0.62 | 5 | 5 |
| TMEM2 | Transmembrane protein 2 | 0.002 | 2.02 | 0.51 | 4 | 4 |
| DDX20 | Probable ATP-dependent RNA helicase DDX20 | 0.006 | 1.94 | 0.54 | 4 | 4 |
| ISG15 | Ubiquitin-like protein ISG15 | 0.010 | 1.82 | 0.67 | 5 | 5 |
| CCDC25 | Coiled-coil domain-containing protein 25 | 0.004 | 1.79 | 0.59 | 9 | 9 |
| NBEAL1 | Neurobeachin-like protein 1 | 0.009 | 1.63 | 0.52 | 10 | 10 |
| BCO2 | Beta,beta-carotene 9,10-oxygenase | 0.009 | 1.57 | 0.43 | 22 | 22 |
| HAL | Histidine ammonia-lyase | 0.013 | −1.65 | 0.46 | 25 | 25 |
| ASAH1 | Acid ceramidase;Acid ceramidase subunit alpha;Acid ceramidase subunit beta | 0.000 | −1.66 | 0.38 | 15 | 15 |
| CYP2S1 | Cytochrome P450 2S1 | 0.008 | −1.72 | 0.45 | 2 | 2 |
| GPX1 | Glutathione peroxidase 1 | 0.011 | −1.76 | 0.55 | 13 | 13 |
| CLEC16A | Protein CLEC16A | 0.013 | −1.76 | 0.60 | 1 | 1 |
| ACTR1B | Beta-centractin | 0.015 | −1.83 | 0.51 | 9 | 4 |
| CHMP1A | Charged multivesicular body protein 1a | 0.009 | −1.87 | 0.56 | 1 | 1 |
| CTBS | Di-N-acetylchitobiase | 0.012 | −1.98 | 0.71 | 5 | 5 |
| FOLH1; FOLH1B | Glutamate carboxypeptidase 2;Putative N-acetylated-alpha-linked acidic dipeptidase | 0.014 | −2.01 | 0.74 | 8 | 8 |
| KHNYN | Protein KHNYN | 0.002 | −2.02 | 0.59 | 2 | 2 |
| FRG1 | Protein FRG1 | 0.017 | −2.17 | 0.92 | 3 | 3 |
| SRP72 | Signal recognition particle 72 kDa protein | 0.001 | −2.22 | 0.68 | 19 | 19 |
| PLSCR3 | Phospholipid scramblase 3 | 0.012 | −2.23 | 0.56 | 2 | 2 |
| ERF | ETS domain-containing transcription factor ERF | 0.013 | −2.25 | 0.66 | 3 | 3 |
| FNBP1 | Formin-binding protein 1 | 0.014 | −2.28 | 0.75 | 5 | 5 |
| SEPP1 | Selenoprotein P | 0.018 | −2.34 | 0.69 | 2 | 2 |
| RHPN2 | Rhophilin-2 | 0.009 | −2.40 | 0.89 | 4 | 4 |
| ELOVL1 | Elongation of very long chain fatty acids protein 1 | 0.019 | −2.46 | 1.61 | 2 | 2 |
| MRPS7 | 28S ribosomal protein S7, mitochondrial | 0.015 | −2.48 | 0.76 | 9 | 9 |
| RIN1 | Ras and Rab interactor 1 | 0.012 | −2.82 | 0.55 | 2 | 2 |
| ITIH5 | Inter-alpha-trypsin inhibitor heavy chain H5 | 0.008 | −2.95 | 1.23 | 3 | 3 |
| CAMK2G; CAMK2A; CAMK2B | Calcium/calmodulin-dependent protein kinase type II subunit gamma;Calcium/calmodulin-dependent protein kinase type II subunit alpha | 0.011 | −2.96 | 0.88 | 8 | 2 |
| NT5DC2 | 5-nucleotidase domain-containing protein 2 | 0.006 | −3.04 | 0.92 | 4 | 4 |
| FAN1 | Fanconi-associated nuclease 1 | 0.011 | −3.09 | 1.02 | 3 | 3 |
| OXNAD1 | Oxidoreductase NAD-binding domain-containing protein 1 | 0.003 | −3.20 | 1.04 | 5 | 5 |
| ALOX5AP | Arachidonate 5-lipoxygenase-activating protein | 0.014 | −3.34 | 1.25 | 2 | 2 |
| MACF1 | Microtubule-actin cross-linking factor 1, isoforms 1/2/3/5 | 0.016 | −3.52 | 1.57 | 83 | 0 |
| CPA3 | Mast cell carboxypeptidase A | 0.008 | −3.67 | 1.72 | 4 | 4 |
| KRT80 | Keratin, type II cytoskeletal 80 | 0.005 | −3.67 | 0.64 | 5 | 4 |
| MYBPC2 | Myosin-binding protein C, fast-type | 0.004 | −4.29 | 0.96 | 4 | 3 |
| ERAP2 | Endoplasmic reticulum aminopeptidase 2 | 0.022 | −4.59 | 0.99 | 20 | 20 |
| YOD1 | Ubiquitin thioesterase OTU1 | 0.017 | −4.95 | 1.56 | 3 | 3 |
| TLCD1 | TLC domain-containing protein 1 | 0.022 | −5.37 | 2.22 | 2 | 2 |
| UBE3B | Ubiquitin-protein ligase E3B | 0.024 | −6.17 | 0.96 | 7 | 7 |
| Category | Term | Welch t-Test Significant Proteins (n = 55) | Classifying Model Selected Proteins (n = 184) | ||||
|---|---|---|---|---|---|---|---|
| p-Value | p-Value FDR | Intersection Genes | p-Value | p-Value FDR | Intersection Genes | ||
| Biological Process | DNA unwinding involved in replication | <0.001 | 0.013 | MCM2; MCM4; MCM7 | <0.001 | 0.007 | MCM2; MCM4; MCM6; MCM7 |
| innate immune response | <0.001 | 0.029 | BCL2; C4B; CAMK2B; CD4; ENSG00000228284; HCK; HLA-DR4; IGKV4-1; IGLV7-43; ISG15; LGALS3; MAPKAPK3; MBL2; MX1; NCAM1; RPS6KA1; VNN1 | ||||
| Cellular Component | MCM complex | <0.001 | <0.001 | MCM4; MCM5; MCM6; MCM7 | |||
| Pathway | DNA replication | <0.001 | 0.013 | MCM2; MCM4; MCM7 | <0.001 | 0.021 | MCM2; MCM4; MCM5; MCM6; MCM7 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urdzik, J.; Vildhede, A.; Wiśniewski, J.R.; Duraj, F.; Haglund, U.; Artursson, P.; Norén, A. Global Proteome Changes in Liver Tissue 6 Weeks after FOLFOX Treatment of Colorectal Cancer Liver Metastases. Proteomes 2016, 4, 30. https://doi.org/10.3390/proteomes4040030
Urdzik J, Vildhede A, Wiśniewski JR, Duraj F, Haglund U, Artursson P, Norén A. Global Proteome Changes in Liver Tissue 6 Weeks after FOLFOX Treatment of Colorectal Cancer Liver Metastases. Proteomes. 2016; 4(4):30. https://doi.org/10.3390/proteomes4040030
Chicago/Turabian StyleUrdzik, Jozef, Anna Vildhede, Jacek R. Wiśniewski, Frans Duraj, Ulf Haglund, Per Artursson, and Agneta Norén. 2016. "Global Proteome Changes in Liver Tissue 6 Weeks after FOLFOX Treatment of Colorectal Cancer Liver Metastases" Proteomes 4, no. 4: 30. https://doi.org/10.3390/proteomes4040030