
“Omics”-Informed Drug and Biomarker Discovery: Opportunities, Challenges and Future Perspectives

{kind=link}
Abstract
:1. Introduction
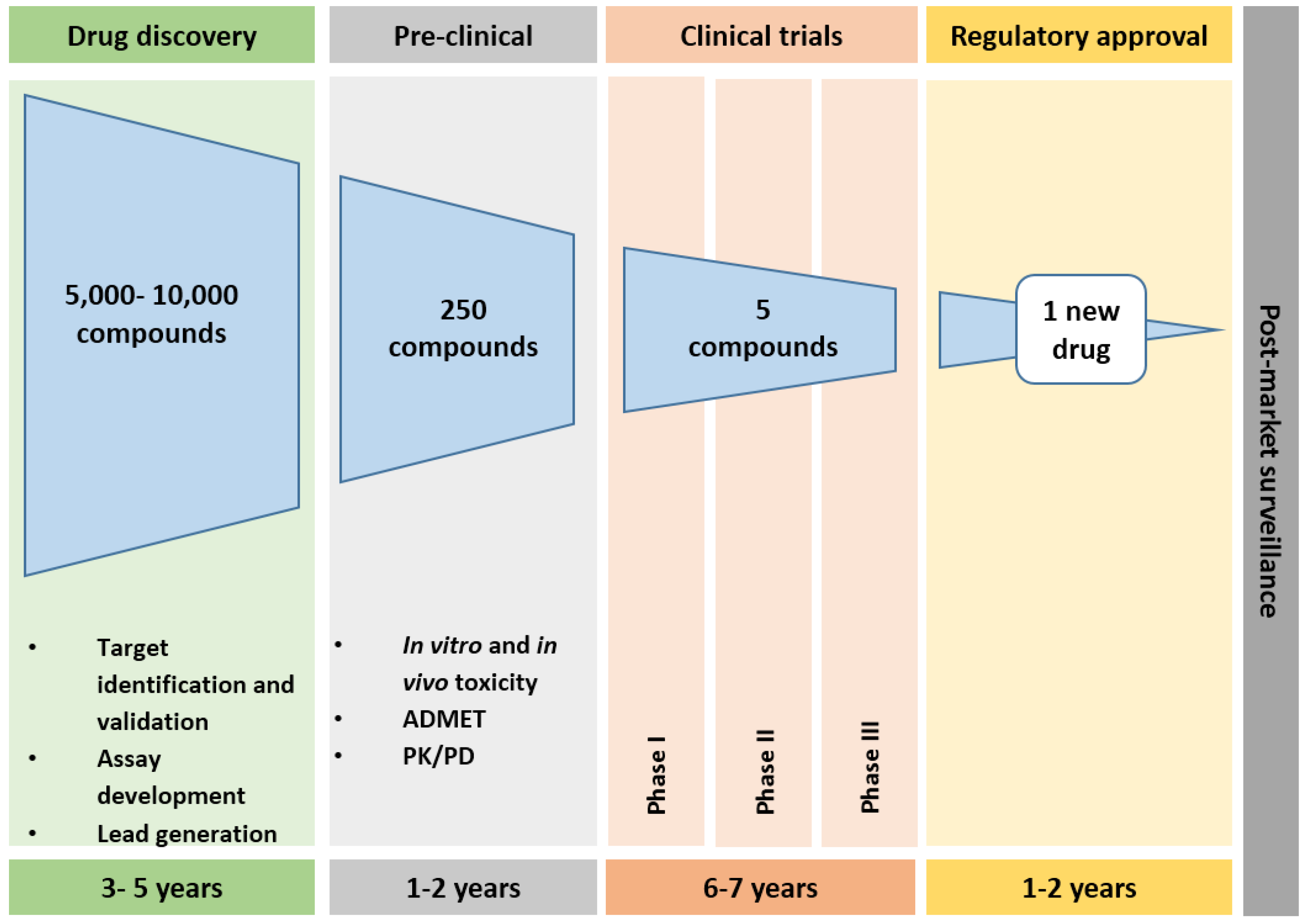
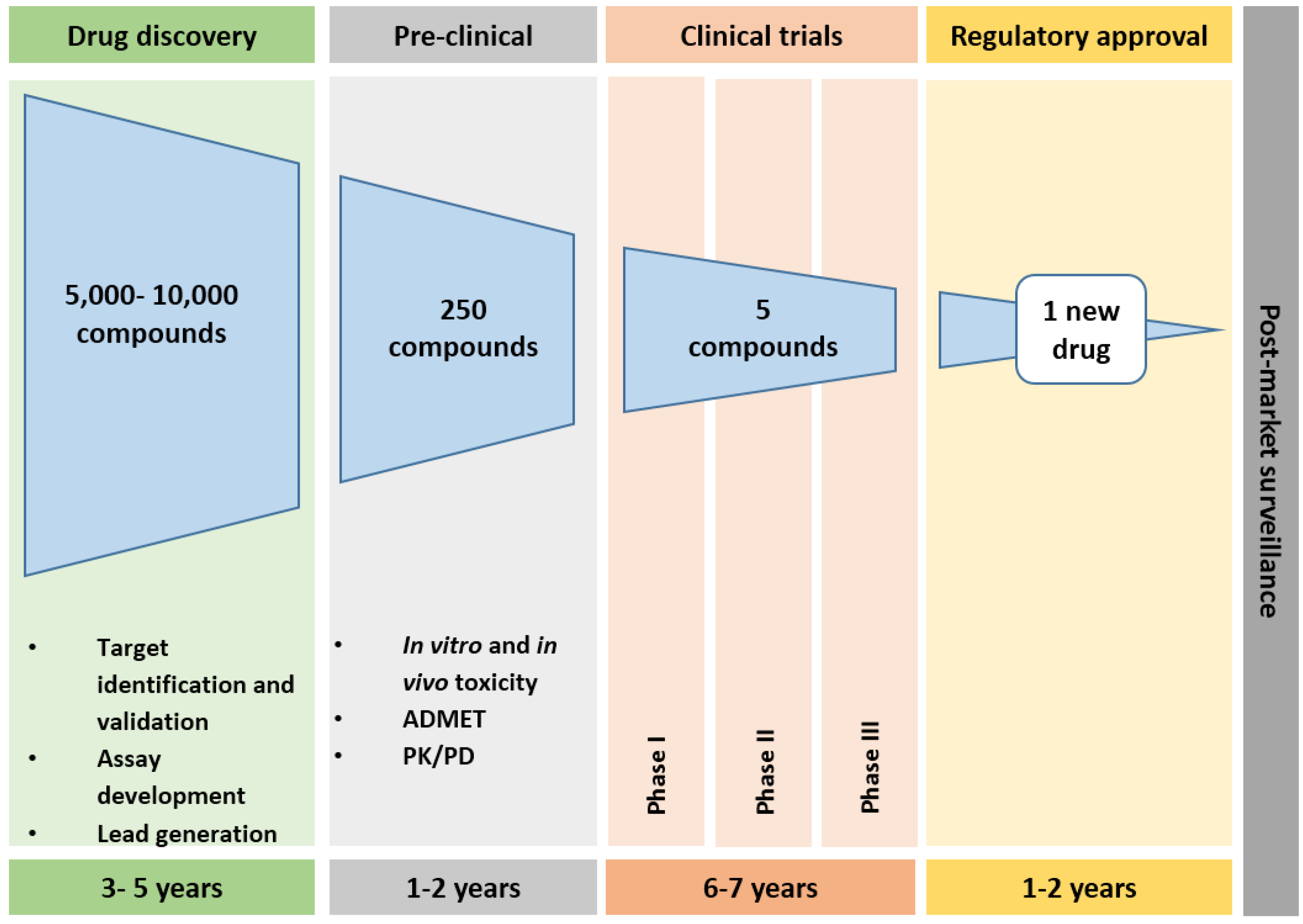
2. Drug Development Pathways
3. Challenges to Drug Discovery
4. Genomics in Drug Discovery
5. Proteomics in Drug Discovery
6. Metabolomics in Drug Discovery
7. Bioinformatics in Drug and Biomarker Discovery
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaitin, K.I.; DiMasi, J.A. Pharmaceutical innovation in the 21st century: New drug approvals in the first decade, 2000–2009. Clin. Pharmacol. Ther. 2011, 89, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Munos, B. Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discov. 2009, 8, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Breyer, M.D. Improving productivity of modern day drug discovery. Exp. Opin. Drug Discov. 2014, 9, 115–118. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thompson, S.; Craven, R.A.; Nirmalan, N.J.; Harnden, P.; Selby, S.J.; Banks, R.E. Impact of pre-analytical factors on the proteomic analysis of formalin-fixed paraffin-embedded tissue. Proteom. Clin. Appl. 2012. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Casadevall, A. Increasing disparities between resource inputs and outcomes, as measured by certain health deliverables in biomedical research. Proc. Natl. Acad. Sci. USA 2015, 112, 11335–11340. [Google Scholar]
- Ioannidis, J. Why most published research findings are false. PLoS Med. 2005, 2, 124. [Google Scholar] [CrossRef] [PubMed]
- Prinz, F.; Schlange, T.; Asadullah, K. Believe it or not: How much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 2011, 10, 328–329. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.; Usman-Idris, M.; Khan, F.; Read, M.; Nirmalan, N.J. Drug repositioning as a route to anti-malarial drug discovery: Preliminary investigation of the in vitro anti-malarial efficacy of emetine dihydrochloride hydrate. Malar. J. 2013, 12, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Publication Guidelines for the Analysis and Documentation of Peptide and Protein Identifications. Available online: http://www.mcponline.org/site/misc/ParisReport_Final.xhtml (accessed on 8 August 2016).
- Drug Discovery and Development, Understanding the R&D Process. Available online: http://cmidd.northwestern.edu/files/2015/10/Drug_RD_Brochure-12e7vs6.pdf (accessed on 8 August 2016).
- Biopharmaceutical Research and Development, the Process Behind New Medicines. Available online: http://www.phrma.org/sites/default/files/pdf/rd_brochure_022307.pdf (accessed on 8 August 2016).
- Norris, S.M.P.; Pankevich, D.; Davis, M.; Altevogt, B.M. Improving and Accelerating Therapeutic Development for Neurosystem Disorders; National Academic Press: Washington, DC, USA, 2014. [Google Scholar]
- Bunnage, M.E. Getting pharmaceutical R&D back on target. Nat. Chem. Biol. 2011, 7, 335–339. [Google Scholar] [PubMed]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Young, J.E.; Goldstein, L.S. Alzheimer’s disease in a dish: Promises and challenges of human stem cell models. Hum. Mol. Genet. 2012, 21, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Orloff, J.; Douglas, F.; Pinheiro, J.; Levinson, S.; Branson, M.; Chaturvedi, P.; Ette, E.; Gallo, P.; Hirsch, G.; Mehta, C.; et al. The future of drug development: Advancing clinical trial design. Nat. Rev. Drug Discov. 2009, 8, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Elebring, T.; Gill, A.; Plowright, A.T. What is the most important approach in current drug discovery: Doing the right things or doing things right? Drug Discov. Today. 2012, 17, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Debouck, C.; Metcalf, B. The impact of genomics on drug discovery. Ann. Rev. Pharmacol. Toxicol. 2000, 40, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.G. From gene to screen with yeast. Curr. Opin. Genet. Dev. 1997, 7, 405–409. [Google Scholar] [CrossRef]
- Helali, A.M.; Iti, F.M.; Mohamed, I.N. Cathepsin K inhibitors: A novel target but promising approach in the treatment of osteoporosis. Curr. Drug Targets 2013, 14, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Land, M.; Hauser, L.; Jun, S.; Nookaew, I.; Leuze, M.R.; Ahn, T.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.H.; Neefs, J.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Wasinger, V.C.; Cordwell, S.J.; Cerpa-Poljak, A.; Yan, J.X.; Gooley, A.A.; Wilkins, M.R.; Duncan, M.W.; Harris, R.; Williams, K.L.; Humphery-Smith, I. Progress with gene-product mapping of the Mollicutes: Mycoplasma genitalium. Electrophoresis 1995, 16, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M. Proteomics data mining. Exp. Rev. Prot. 2009, 6, 599–603. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Chiu, J. Proteomics in Biomarker discovery and drug development. J. Cell. Biochem. 2003, 89, 868–886. [Google Scholar] [CrossRef] [PubMed]
- Bichsel, V.E.; Liotta, L.A.; Petricoin, E.F. Cancer proteomics: from biomarker discovery to signal pathway profiling. Cancer J. 2001, 7, 69–78. [Google Scholar] [PubMed]
- Wang, K.; Huang, C.; Nice, H. Recent advances in proteomics: Towards the human proteome. Biomed. Chromatogr. 2014, 28, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Garbis, S.; Lubec, G.; Fountoulakis, M. Limitations of current proteomics technologies. J. Chromatogr. A 2005, 1077, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Moxona, J.V.; Padulab, M.P.; Herbert, B.R.; Golledgea, J. Challenges, current status and future perspectives of proteomics in improving understanding, diagnosis and treatment of vascular disease. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Hood, L.E.; Omenn, G.S.; Moritz, R.L.; Aebersold, R.; Yamamoto, K.R.; Amos, M.; Hunter-Cevera, J.; Locascio, L. New and improved proteomics technologies for understanding complex biological systems: Addressing a grand challenge in the life sciences. Proteomics 2012, 12, 2773–2783. [Google Scholar] [CrossRef] [PubMed]
- Poste, G. Biospecimens, biomarkers, and burgeoning data: The imperative for more rigorous research standards. Trends Mol. Med. 2012, 18, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.L.; Yang, S.N.; Taldone, T.; Chang, B.; Gerecitano, J.; Elenitoba-Johnson, K.; Shaknovich, R.; Tam, W.; Leonard, J.P.; Chiosis, G.; et al. Pharmacoproteomics identifies combinatorial therapy targets for diffuse large B cell lymphoma. J. Clin. Investig. 2015, 125, 4559–4571. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.J.; Jain, R.; Paik, Y.; Downey, R.; Ptolemy, A.S.; Ozdemir, V.; Srivastava1, S. Personalized Medicine in the Age of Pharmacoproteomics: A close up on India and need for social science engagement for responsible innovation in post-proteomic biology. Curr. Pharmacogenom. Pers. Med. 2011, 9, 67–75. [Google Scholar]
- Rabilloud, T.; Lelong, C. Two-dimensional gel electrophoresis in proteomics: A tutorial. J. Proteom. 2011, 74, 1829–1841. [Google Scholar] [CrossRef] [PubMed]
- Bunai, K.; Yamane, K. Effectiveness and limitation of two-dimensional gel electrophoresis in bacterial membrane protein proteomics and perspectives. J. Chromatogr. B 2005, 815, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Lilley, K.; Beynon, R.J.; Eyers, C.E.; Hubbard, S.J. Focus on Quantitative Proteomics. Proteomics 2015, 15, 3101–3103. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nirmalan, N.; Hughes, C.; Peng, J.; Mckenna, T.; Langridge, J.; Cairns, D.; Harnden, P.; Selby, P.; Banks, R. Initial development and validation of a novel extraction method for quantitative mining of the formalin-fixed paraffin embedded tissue proteome for biomarker investigations. J. Proteom. Res. 2011, 4, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Trinh, H.V.; Grossmann, J.; Gehrig, P.; Roschitzki, B.; Schlapbach, R.; Greber, U.F.; Hemmi, S. iTRAQ-based and label-free proteomics approaches for studies of human adenovirus infections. Int. J. Proteom. 2013, 2013, 581862. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct. Genom. 2001, 2, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Fillet, M.; Frédérich, M. The emergence of metabolomics as a key discipline in the drug discovery process. Drug Discov. Today Technol. 2015, 13, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Reily, M.; Tymiak, A. Metobolomics in the pharmaceutical industry. Drug Discov. Today Technol. 2015, 13, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Cuperlovic-Culf, M.; Culf, A.S.; Morin, P.; Touaibi, M. Application of Metabolomics in Drug Discovery, Development and Theranostics. Curr. Metabolomics 2013, 1, 41–57. [Google Scholar]
- Monteiro, M.S.; Carvalho, M.; Bastos, M.L.; Guedes de Pinho, P. Metabolomics analysis for biomarker discovery: Advances and challenges. Curr. Med. Chem. 2013, 20, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.; Daneshian, M.; Kamp, H.; Kamp, H.; Bois, F.Y.; Clench, M.R.; Coen, M.; Donley, B.; Fischer, S.M.; Ekman, D.R.; et al. Metabolomics in toxicology and preclinical research. ALTEX 2013, 30, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Desiere, D.; Deutsch, E.W.; King, N.L.; Nesvizhskii, A.; Mallick, P.; Eng, J.; Chen, S.; Eddes, J.; Loevenich, S.L.; Aebersold, R. The PeptideAtlas project. Nucleic Acid Res. 2005, 34 (Suppl. 1), D655–D658. [Google Scholar] [CrossRef] [PubMed]
- Berglund, L.; Björling, E.; Oksvold, P.; Fagerberg, L.; Asplund, A.; Szigyarto, C.A.; Persson, A.; Ottosson, J.; Wernérus, H.; Nilsson, P.; et al. Genecentric human protein atlas for expression profiles based on antibodies. Mol. Cell. Proteom. 2008, 7, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, J.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, P.; Michel, P.; Zahn-Zabal, M.; Cusin, I.; Duek, P.D.; Evalet, O.; Gateau, A.; Gleizes, A.; Pereira, M.; Teixeira, D.; et al. The neXtProt knowledge base on human proteins: Current status. Nucl. Acids Res. 2015, 43, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Khanna, I. Drug discovery in pharmaceutical industry: Productivity challenges and trends. Drug Discov. Today 2012, 17, 1088–1102. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, J. Trial watch: Phase II failures. Nat. Rev. Drug. Discov. 2011, 10, 328–329. [Google Scholar] [CrossRef] [PubMed]
- Graul, A.I.; Cruces, E.; Stringer, M. The year’s new drugs & biologics. Drugs Today (Barc.) 2015, 51, 37–87. [Google Scholar] [PubMed]
- Shim, J.S.; Liu, J.O. Recent Advances in Drug Repositioning for the Discovery of New Anticancer Drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Corbett, A.; Pickett, J.; Burns, A.; Corcoran, J.; Dunnett, S.B.; Edison, P.; Hagan, J.J.; Holmes, C.C.; Jones, E.; Katona, C.; et al. Drug repositioning for Alzheimer’s disease. Nat. Rev. Drug Discov. 2012, 11, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Lucumi, E.; Darling, C.; Jo, H.; Napper, A.D.; Chandramohandas, R.; Fisher, N.; Shone, A.E.; Jing, H.; Ward, S.A.; Biagini, G.A.; et al. Discovery of potent small molecule inhibitors of multi drug-resistant Plasmodium falciparum using a novel miniaturised high throughput luciferase-based assay. Antimicrob. Agents Chemother. 2010, 54, 3579–3604. [Google Scholar] [CrossRef] [PubMed]
- Temesi, G.; Bolgár, B.; Arany, A.; Szalai, C.; Antal, P.; Mátyus, P. Early repositioning through compound set enrichment analysis: A knowledge-recycling strategy. Future Med. Chem. 2014, 6, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Silber, B.M. Omics in drug discovery: From small molecule leads to clinical candidates. In The Omics Applications in Neuroscience, 2nd ed.; Oxford University Press: Oxford, UK, 2014; Chapter 16. [Google Scholar]
- Silber, B.M. Driving drug Discovery: The fundamental role of academic labs. Drug Discov. 2010, 2, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M. Market watch: Pharma industry performance metrics: 2007–2012E. Nat. Rev. Drug Discov. 2009, 7, 795. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Cuatrecasas, P. Drug discovery in jeopardy. J. Clin. Investig. 2006, 116, 2837–2842. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Dugar, S. Outsourcing drug discovery to India and China: From surviving to thriving. Drug Discov. Today 2012, 17, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Garnier, J. Rebuilding the R&D engine in big pharma. Harv. Bus. Rev. 2008, 86, 68–76. [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matthews, H.; Hanison, J.; Nirmalan, N. “Omics”-Informed Drug and Biomarker Discovery: Opportunities, Challenges and Future Perspectives. Proteomes 2016, 4, 28. https://doi.org/10.3390/proteomes4030028
Matthews H, Hanison J, Nirmalan N. “Omics”-Informed Drug and Biomarker Discovery: Opportunities, Challenges and Future Perspectives. Proteomes. 2016; 4(3):28. https://doi.org/10.3390/proteomes4030028
Chicago/Turabian StyleMatthews, Holly, James Hanison, and Niroshini Nirmalan. 2016. "“Omics”-Informed Drug and Biomarker Discovery: Opportunities, Challenges and Future Perspectives" Proteomes 4, no. 3: 28. https://doi.org/10.3390/proteomes4030028
APA StyleMatthews, H., Hanison, J., & Nirmalan, N. (2016). “Omics”-Informed Drug and Biomarker Discovery: Opportunities, Challenges and Future Perspectives. Proteomes, 4(3), 28. https://doi.org/10.3390/proteomes4030028