
The Role of Clinical Proteomics, Lipidomics, and Genomics in the Diagnosis of Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
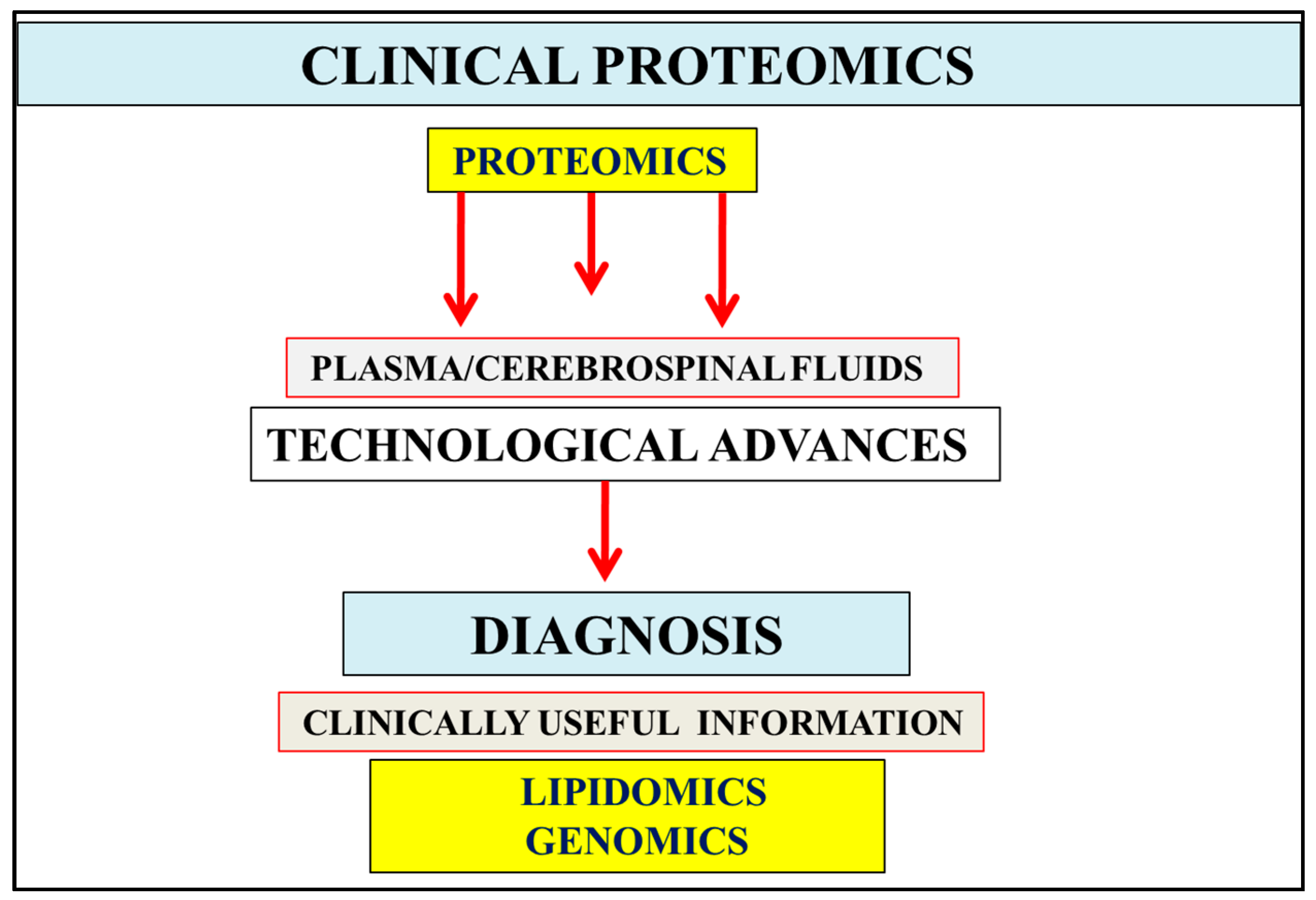
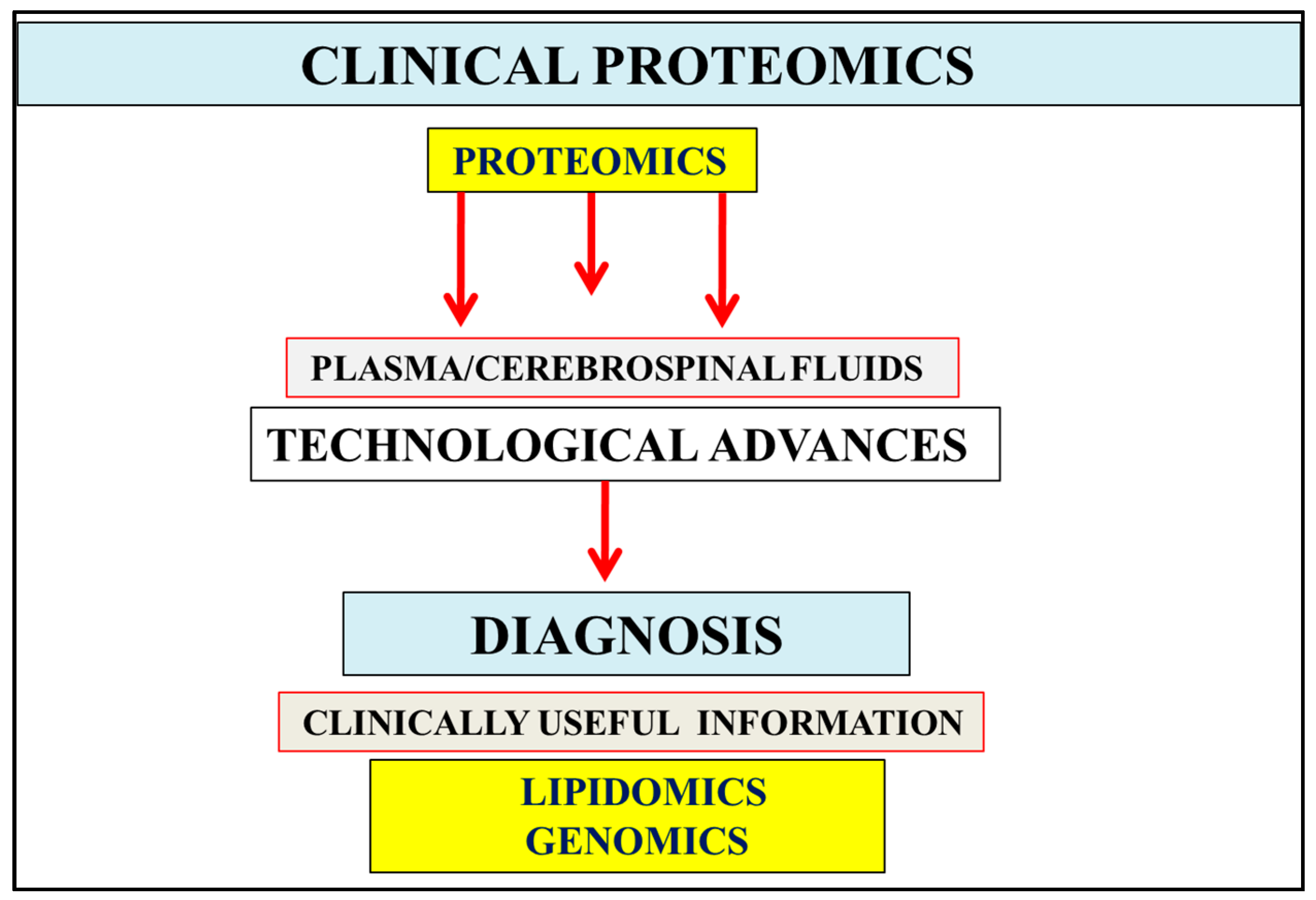
:1. Introduction
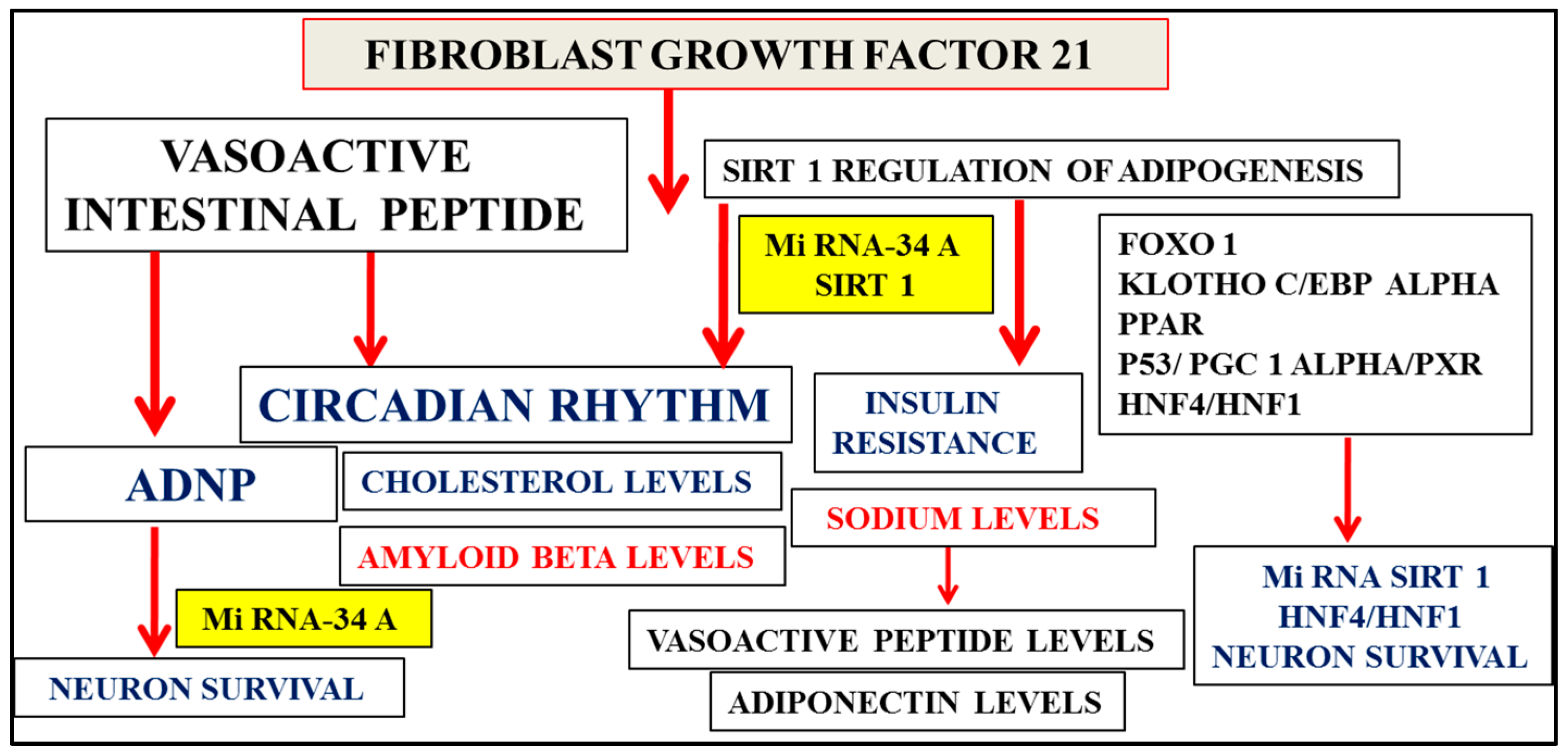
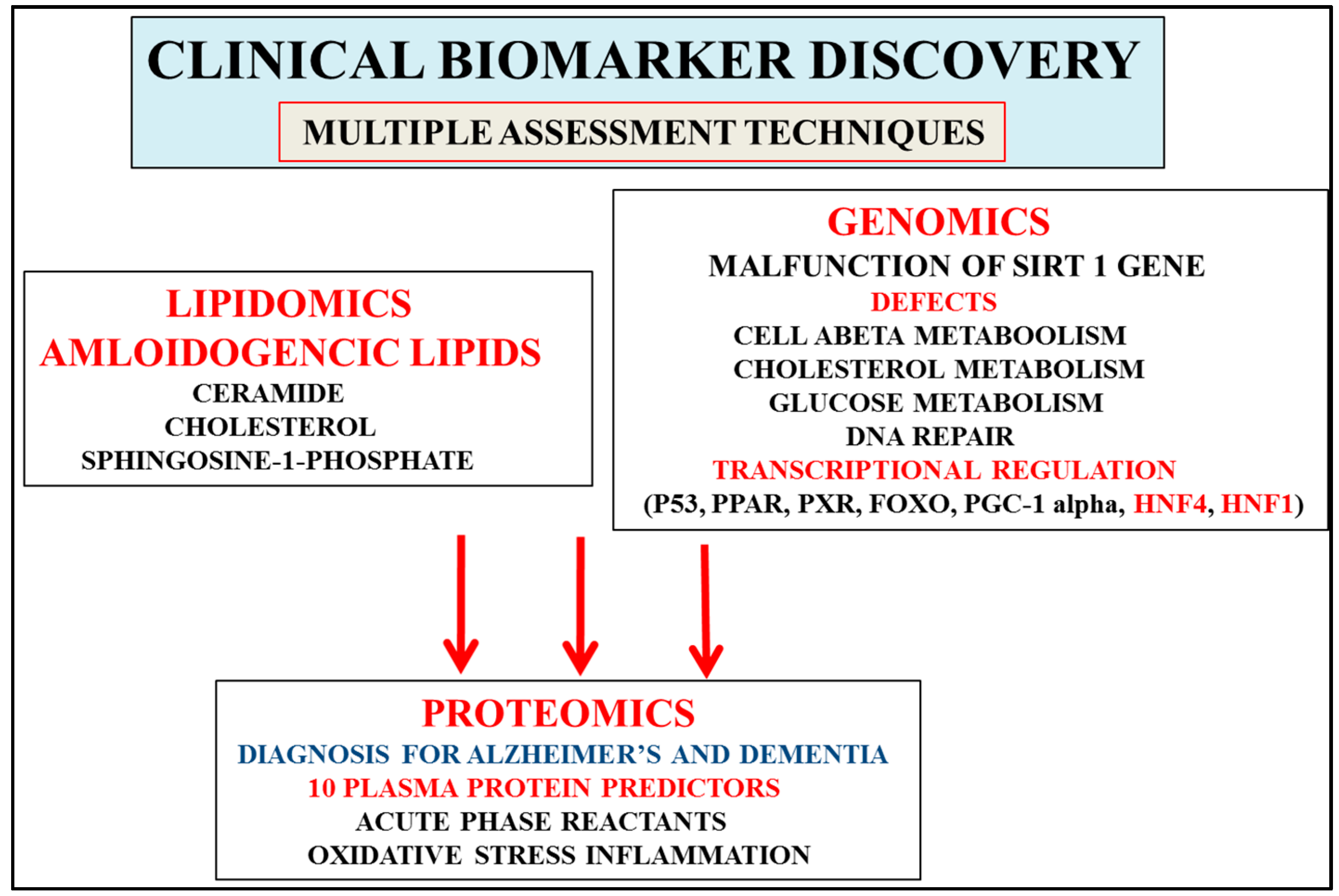
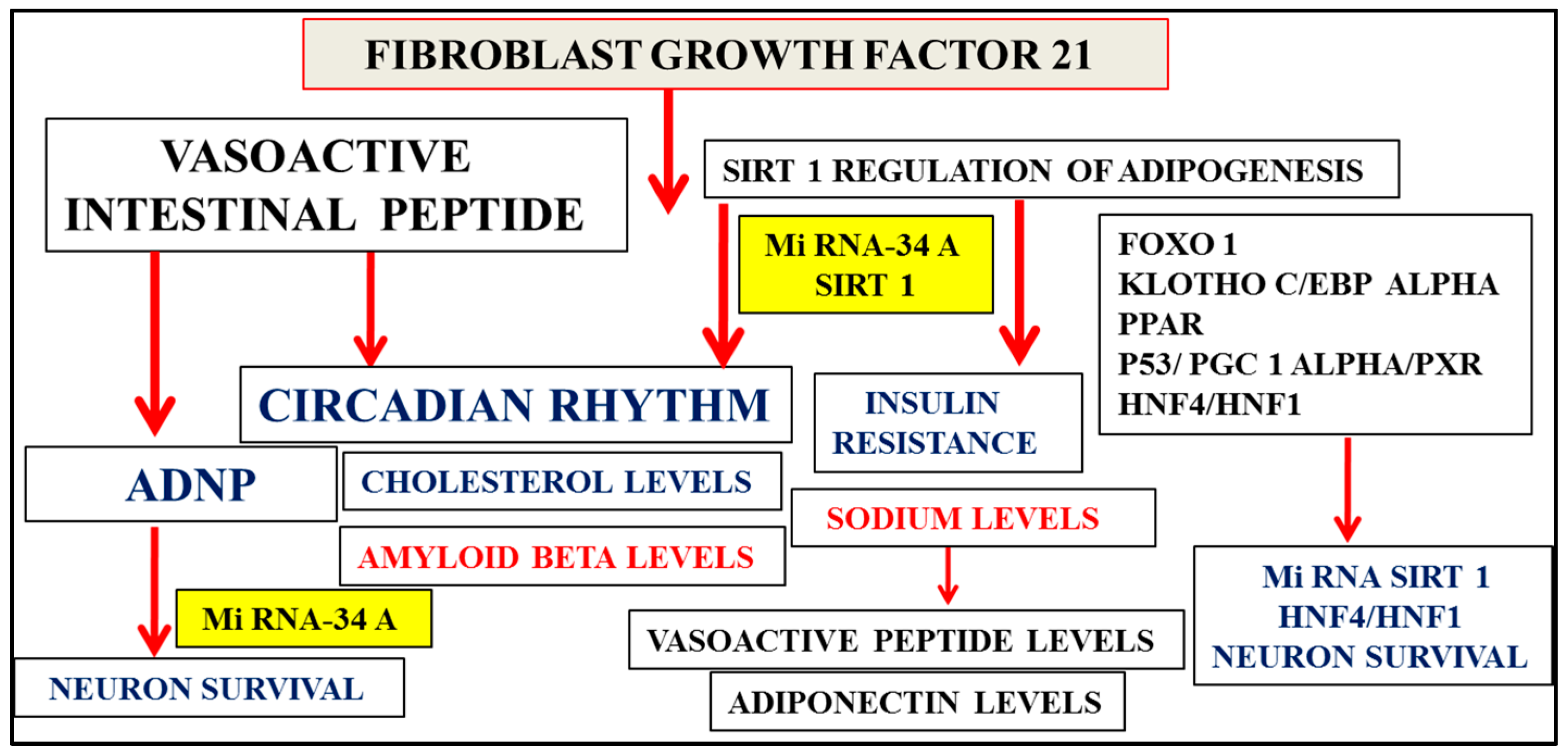
2. Multifactorial Nature of Alzheimer’s Disease Provides Important Links to Early Diagnosis
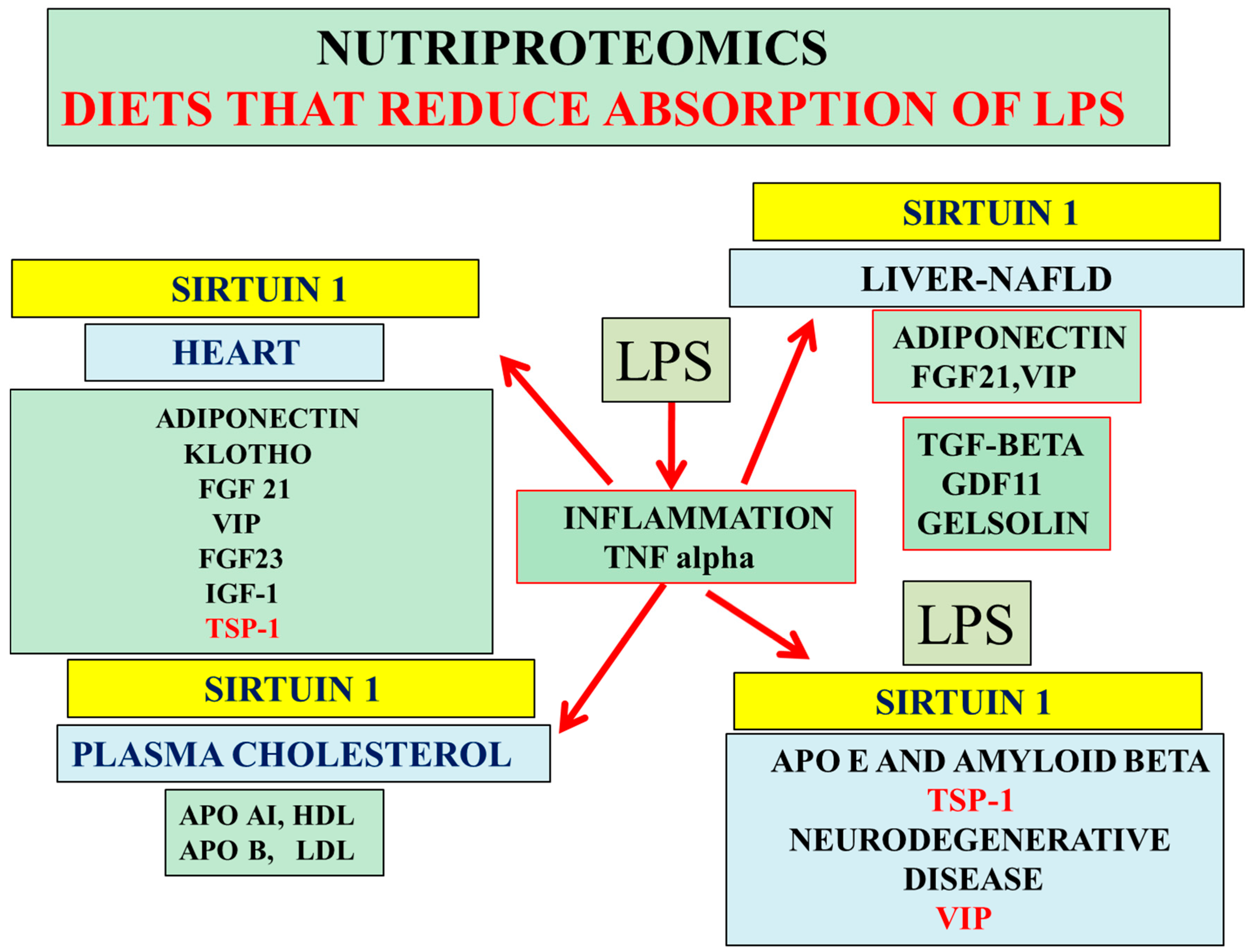
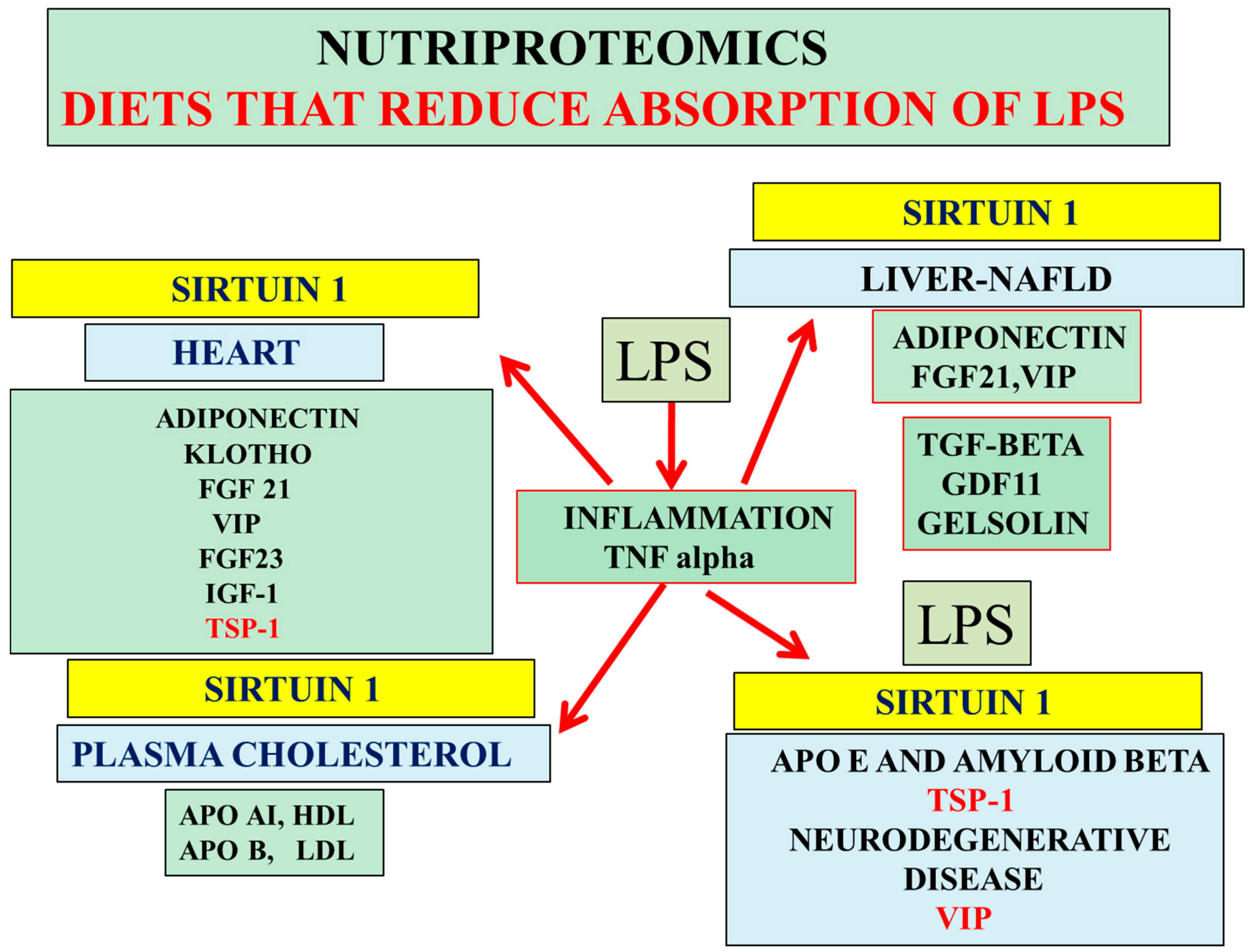
3. Nutriproteomic Diets Regulates Plasma Biomarkers and Reverses Neurodegeneration and Amyloidosis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Martins, I.J.; Creegan, R. Links between Insulin Resistance, Lipoprotein Metabolism and Amyloidosis in Alzheimer’s Disease. Health 2014, 6, 1549–1579. [Google Scholar] [CrossRef]
- Martins, I.J. Unhealthy Nutrigenomic Diets Accelerate NAFLD and Adiposity in Global communities. J. Mol. Genet. Med. 2015, 9, 1–11. [Google Scholar]
- Tamaoka, A.T.; Fukushima, N.; Sawamura, K.; Ishikawa, E.; Oguni, Y.; Komatsuzaki, Y.; Shoi, S. Amyloid beta protein in plasma from patients with sporadic Alzheimer’s disease. J. Neurol. Sci. 1996, 141, 65–68. [Google Scholar] [CrossRef]
- Vanderstichele, H.E.; van Kerschaver, C.; Hesse, P.; Davidsson, M.A.; Buyse, N.; Andreasen, N.; Minthon, L.; Wallin, A.; Blennow, K.; Vanmechelen, E. Standardization of measurement of β-amyloid(1–42) in cerebrospinal fluid and plasma. Amyloid 2000, 7, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R.; Tang, M.X.; Jacobs, D.M.; Manly, J.; Bell, K.; Merchant, C.; Small, S.A.; Stern, Y.; Wisniewski, H.M.; Mehta, P.D. Plasma amyloid β-peptide 1–42 and incipient Alzheimer’s disease. Ann. Neurol. 1999, 46, 412–416. [Google Scholar] [CrossRef]
- Martins, I.J.; Fernando, W. High Fibre Diets and Alzheimer’s Disease. Food Nutr. Sci. 2014, 5, 410–424. [Google Scholar] [CrossRef]
- Yu, C.; Alterman, M.; Dobrowsky, R.T. Ceramide displaces cholesterol from lipid rafts and decreases the association of the cholesterol binding protein caveolin-1. J. Lipid Res. 2005, 46, 1678–1691. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.R.; Cheng, K.H.; Huang, J. Ceramide drives cholesterol out of the ordered lipid bilayer phase into the crystal phase in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine/cholesterol/ceramide ternary mixtures. Biochemistry 2006, 45, 12629–12638. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.M.; Silva, L.C.; Fedorov, A.; de Almeida, R.F.; Prieto, M. Cholesterol-rich fluid membranes solubilize ceramide domains: Implications for the structure and dynamics of mammalian intracellular and plasma membranes. J. Biol. Chem. 2009, 284, 22978–22987. [Google Scholar] [CrossRef] [PubMed]
- Hye, A.; Riddoch-Contreras, J.; Baird, A.L.; Ashton, N.J.; Bazenet, C.; Leung, R.; Westman, E.; Simmons, A.; Dobson, R.; Sattlecker, M.; et al. Plasma proteins predict conversion to dementia from prodromal disease. Alzheimers Dement. 2014, 10, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Reisberg, B.; Ferris, S.H.; de Leon, M.J.; Crook, T. The global deterioration scale for assessment of primary degenerative dementia. Am. J. Psychiatry 1982, 139, 1136–1139. [Google Scholar] [PubMed]
- Mar, J.; Soto-Gordoa, M.; Arrospide, A.; Moreno-Izco, F.; Martínez-Lage, P. Fitting the epidemiology and neuropathology of the early stages of Alzheimer’s disease to prevent dementia. Alzheimers Res.Ther. 2015, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.F.L.; Martins, I.J.; Martins, R.N. Lipid metabolism and lipidomics: An emerging frontier in biology. J. Genet. Genom. 2014, 41, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Fonteh, A.N.; Fisher, R.D. Combining lipidomics and proteomics of human cerebrospinal fluids. Methods Mol. Biol. 2009, 579, 71–86. [Google Scholar] [PubMed]
- Colsch, B.; Seyer, A.; Boudah, S.; Junot, C. Lipidomic analysis of cerebrospinal fluid by mass spectrometry-based methods. J. Inherit. Metab. Dis. 2015, 38, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.J.; Creegan, R.; Lim, W.L.F.; Martins, R.N. Molecular insights into appetite control and neuroendocrine disease as risk factors for chronic diseases in Western countries. OJEMD 2013, 3, 11–33. [Google Scholar] [CrossRef]
- Martins, I.J. Increased Risk for Obesity and Diabetes with Neurodegeneration in Developing Countries. J. Mol. Genet. Med. 2013, S1, 1–8. [Google Scholar]
- Fiandaca, M.S.; Zhong, X.; Cheema, A.-K.; Orquiza, M.-H.; Chidambaram, S.; Tan, M.T.; Gresenz, C.; FitzGerald, K.T.; Nalls, M.A.; Singleton, A.B.; et al. Plasma 24-metabolite Panel Predicts Preclinical Transition to Clinical Stages of Alzheimer’s Disease. Front. Neurol. 2015, 6, 237. [Google Scholar] [CrossRef] [PubMed]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.J. Overnutrition Determines LPS Regulation of Mycotoxin Induced Neurotoxicity in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 29554–29573. [Google Scholar] [CrossRef] [PubMed]
- Deisenhammer, F.; Bartos, A.; Egg, R.; Gilhus, N.E.; Giovannoni, G.; Rauer, S.; Sellebjerg, F.; Tumani, H. Routine cerebrospinal fluid (CSF) analysis. In European Handbook of Neurological Management, 2nd ed.; Gilhus, N.E., Barnes, M.P., Brainin, M., Eds.; Blackwell Publishing Ltd: Oxford, UK, 2011; Volume 1. [Google Scholar]
- Martins, I.J.; Gupta, V.; Wilson, A.C.; Fuller, S.J.; Martins, R.N. Interactions between Apo E and Amyloid Beta and their Relationship to Nutriproteomics and Neurodegeneration. Curr. Proteom. 2014, 11, 173–183. [Google Scholar]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Stachowska, E.; Maciejewska, D.; Ossowski, P.; Drozd, A.; Ryterska, K.; Banaszczak, M.; Milkiewicz, M.; Raszeja-Wyszomirska, J.; Slebioda, M.; Milkiewicz, P.; et al. Apolipoprotein E4 allele is associated with substantial changes in the plasma lipids and hyaluronic acid content in patients with nonalcoholic fatty liver disease. J. Physiol. Pharmacol. 2013, 64, 711–717. [Google Scholar] [PubMed]
- De Feo, E.; Cefalo, C.; Arzani, D.; Amore, R.; Landolfi, R.; Grieco, A.; Ricciardi, W.; Miele, L.; Boccia, S. A case-control study on the effects of the apolipoprotein E genotypes in nonalcoholic fatty liver disease. Mol. Biol. Rep. 2012, 39, 7381–7388. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.-W.; Sun, Q.-Q.; Zhang, B.-B.; Hu, A.-M.; Liu, H.-L.; Wang, Q.; Hou, Z.Z. Association between Apolipoprotein E Gene Polymorphism and the Risk of Coronary Artery Disease in Chinese Population: Evidence from a Meta-Analysis of 40 Studies. PLoS ONE 2013, 8, e66924. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Craft, S. The role of insulin resistance in the pathogenesis of Alzheimer’s disease: Implications for treatment. CNS Drugs 2003, 17, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Li, X. Sirtuin 1 in lipid metabolism and obesity. Ann. Med. 2011, 43, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Maizel, J.; Xavier, S.; Chen, J.; Lin, C.H.; Vasko, R.; Goligorsky, M.S. Sirtuin 1 ablation in endothelial cells is associated with impaired angiogenesis and diastolic dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1691–H1704. [Google Scholar] [CrossRef] [PubMed]
- Botti, C.; Caiafa, I.; Coppola, A.; Cuomo, F.; Miceli, M.; Altucci, L.; Cobellis, G. SIRT1 inhibition affects angiogenic properties of human MSCs. Biomed. Res. Int. 2014, 2014, 783459. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Ghaeni, L.; Baldessari, D.; Mostoslavsky, R.; Rossig, L.; Dequiedt, F.; Haendeler, J.; Mione, M.; Dejana, E.; Alt, F.W.; et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007, 21, 2644–2658. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.A.; Dominy, J.E.; Lee, Y.; Puigserver, P. The sirtuin family’s role in aging and age-associated pathologies. J. Clin. Invest. 2013, 123, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Lee, H.G.; Camins, A.; Pallàs, M.; Casadesus, G.; Smith, M.A.; Zhu, X. The sirtuin pathway in ageing and Alzheimer disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2011, 10, 275–279. [Google Scholar] [CrossRef]
- Martins, I.J. Nutritional and genotoxic stress contributes to diabetes and neurodegenerative diseases such as Parkinson’s and Alzheimer’s diseases. In Frontiers in Clinical Drug Research—CNS and Neurological Disorders; Atta-ur-Rahman, Ed.; Bentham Science: London, UK, 2015; Volume 3, pp. 158–192. [Google Scholar]
- Martins, I.J. Anti-Aging Genes Improve Appetite Regulation and Reverse Cell Senescence and Apoptosis in Global Populations. Adv. Aging Res. 2016, 5, 9–26. [Google Scholar] [CrossRef]
- Martins, I.J. Unhealthy diets determine benign or toxic amyloid beta states and promote brain amyloid beta aggregation. Austin J. Clin. Neurol. 2015, 2, 1060–1066. [Google Scholar]
- Martins, I.J. Diabetes and cholesterol dyshomeostasis involve abnormal α-synuclein and amyloid beta transport in neurodegenerative diseases. Austin Alzheimer’s J. Parkinson’s Dis. 2015, 2, 1020–1028. [Google Scholar]
- Ions, L.J.; Wakeling, L.A.; Bosomworth, H.J.; Hardyman, J.E.; Escolme, S.M.; Swan, D.C.; Valentine, R.A.; Mathers, J.C.; Ford, D. Effects of Sirt1 on DNA methylation and expression of genes affected by dietary restriction. Age (Dordr.) 2013, 35, 1835–1849. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qiao, L.; Shao, J. SIRT1 regulates adiponectin gene expression through Foxo1-C/enhancer-binding protein alpha transcriptional complex. J. Biol. Chem. 2006, 281, 39915–39924. [Google Scholar] [CrossRef] [PubMed]
- Chihara, Y.; Rakugi, H.; Ishikawa, K.; Ikushima, M.; Maekawa, Y.; Ohta, J.; Kida, I.; Ogihara, T. Klotho protein promotes adipocyte differentiation. Endocrinology 2006, 147, 3835–3842. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Zhang, F.; Yan, T.; Liu, Z.; Chunxi, W.; Ge, X.; Zhai, Q. C/EBPα regulates SIRT1 expression during adipogenesis. Cell Res. 2010, 20, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Alcendor, R.; Zhai, P.; Park, J.Y.; Shao, D.; Cho, J.; Yamamoto, T.; Tian, B.; Sadoshima, J. PPARα-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab. 2011, 14, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Westmacott, A.; Burke, Z.D.; Oliver, G.; Slack, J.M.; Tosh, D. C/EBPalpha and C/EBPbeta are markers of early liver development. Int. J Dev. Biol. 2006, 50, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Chau, M.D.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. PNAS 2010, 107, 12553–12558. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Uehara, Y.; Motomura-Matsuzaka, K.; Oki, J.; Koyama, Y.; Kimura, M.; Asada, M.; Komi-Kuramochi, A.; Oka, S.; Imamura, T. betaKlotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Mol. Endocrinol. 2008, 22, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Yie, J.; Wang, W.; Deng, L.; Tam, L.T.; Stevens, J.; Chen, M.M.; Li, Y.; Xu, J.; Lindberg, R.; Hecht, R.; et al. Understanding the physical interactions in the FGF21/FGFR/β-Klotho complex: Structural requirements and implications in FGF21 signaling. Chem. Biol. Drug Des. 2012, 79, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Wolf, I.; Levanon-Cohen, S.; Bose, S.; Ligumsky, H.; Sredni, B.; Kanety, H.; Kuro-o, M.; Karlan, B.; Kaufman, B.; Koeffler, H.P.; et al. Klotho: A tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 2008, 27, 7094–7105. [Google Scholar] [CrossRef] [PubMed]
- Piya, M.K.; Harte, A.L.; Chittari, M.V.; Tripathi, G.; Kumar, S.; McTernan, P.G. FGF21 action on human adipose tissue compromised by reduced βKlotho and FGFR1 expression in type 2 diabetes mellitus. Endocr. Abstr. 2013, 31, P179. [Google Scholar] [CrossRef]
- Bass, J. Forever (FGF) 21. Nat. Med. 2013, 19, 1090–1092. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, J.; Tselykh, T.V.; Maiorana, F.; Eriksson, O.; Do, H.T.; Mudò, G.; Korhonen, L.T.; Belluardo, N.; Lindholm, D. Fibroblast growth factor-21 enhances mitochondrial functions and increases the activity of PGC-1α in human dopaminergic neurons via Sirtuin-1. Springerplus 2014, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Lundåsen, T.; Hunt, M.C.; Nilsson, L.M.; Sanyal, S.; Angelin, B.; Alexson, S.E.; Rudling, M. PPARalpha is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 2007, 360, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Akbar, H.; Batistel, F.; Drackley, J.K.; Loor, J.J. Alterations in Hepatic FGF21, Co-Regulated Genes, and Upstream Metabolic Genes in Response to Nutrition, Ketosis and Inflammation in Peripartal Holstein Cows. PLoS ONE 2015, 10, e0139963. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wong, K.; Giles, A.; Jiang, J.; Lee, J.W.; Adams, A.C.; Kharitonenkov, A.; Yang, Q.; Gao, B.; Guarente, L.; et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 2014, 146, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Tian, H.; Lam, K.S.; Lin, S.; Hoo, R.C.; Konishi, M.; Itoh, N.; Wang, Y.; Bornstein, S.R.; Xu, A.; Li, X. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013, 17, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N. FGF21 as a Hepatokine, Adipokine, and Myokine in Metabolism and Diseases. Front. Endocrinol. 2014, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Zhong, L.; Zhang, J.; Wang, Y.; Bornstein, S.R.; Triggle, C.R.; Ding, H.; Lam, K.S.; Xu, A. FGF21 maintains glucose homeostasis by mediating the cross talk between liver and brain during prolonged fasting. Diabetes 2014, 63, 4064–4075. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Lin, V.Y.; Goetz, R.; Mohammadi, M.; Mangelsdorf, D.J.; Kliewer, S.A. Inhibition of Growth Hormone Signaling by the Fasting-Induced Hormone FGF21. Cell Metab. 2008, 8, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Adams, A.C.; Brozinick, J.T.; Bui, H.H.; Miyauchi, Y.; Kusminski, C.M.; Bauer, S.M.; Wade, M.; Singhal, E.; Cheng, C.C.; et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 2013, 17, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.K.; Hallschmid, M.; Adya, R.; Kern, W.; Lehnert, H.; Randeva, H.S. Fibroblast growth factor 21 (FGF21) in human cerebrospinal fluid: relationship with plasma FGF21 and body adiposity. Diabetes 2011, 60, 2758–2762. [Google Scholar] [CrossRef] [PubMed]
- Tao, C.; Sifuentes, A.; Holland, W.L. Regulation of glucose and lipid homeostasis by adiponectin: Effects on hepatocytes, pancreatic β cells and adipocytes. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Okua, H.; Matsuuraa, F.; Kosekia, M.; Sandovala, J.C.; Yuasa-Kawasea, M.; Tsubakio-Yamamotoa, K.; Masudab, D.; Maedab, N.; Ohamaa, T.; Ishigamic, M.; et al. Adiponectin deficiency suppresses ABCA1 expression and ApoA-I synthesis in the liver. FEBS Lett. 2007, 581, 5029–5033. [Google Scholar] [CrossRef] [PubMed]
- Dantas, K.C.; Bydlowski, S.P.; Novak, E.M. Study of activity transcription factors C/EBPα in region-53 to -33 of promoter apolipoprotein B gene. Rev. Bras. Ciências Farm. Braz. J. Pharm. Sci. 2006, 42, 405–411. [Google Scholar] [CrossRef]
- Bhalla, S.; Ozalp, C.; Fang, S.; Xiang, L.; Kemper, J.K. Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1alpha. Functional implications in hepatic cholesterol and glucose metabolism. J. Biol. Chem. 2004, 279, 45139–45147. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.; Ge, H.; Yang, W.; Fan, M.; Handschin, C.; Cooper, M.; Lin, J.; Li, C.; Spiegelman, B.M. Partnership of PGC-1alpha and HNF4alpha in the regulation of lipoprotein metabolism. J. Biol. Chem. 2006, 281, 14683–14690. [Google Scholar] [CrossRef] [PubMed]
- Mogilenko, D.A.; Dizhe, E.B.; Shavva, V.S.; Lapikov, I.A.; Orlov, S.V.; Perevozchikov, A.P. Role of the nuclear receptors HNF4 alpha, PPAR alpha, and LXRs in the TNF alpha-mediated inhibition of human apolipoprotein A-I gene expression in HepG2 cells. Biochemistry 2009, 48, 11950–11960. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Shimano, H.; Nakagawa, Y.; Ide, T.; Yahagi, N.; Matsuzaka, T.; Nakakuki, M.; Takahashi, A.; Suzuki, H.; Sone, H.; et al. SREBP-1 interacts with hepatocyte nuclear factor-4 alpha and interferes with PGC-1 recruitment to suppress hepatic gluconeogenic genes. J. Biol. Chem. 2004, 279, 12027–12035. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Jung-Hynes, B.; Ahmad, N. SIRT1 controls circadian clock circuitry and promotes cell survival: A connection with age-related neoplasms. FASEB J. 2009, 23, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Loh, D.H.; Dragich, J.M.; Kudo, T.; Schroeder, A.M.; Nakamura, T.J.; Waschek, J.A.; Block, G.D.; Colwell, C.S. Effects of vasoactive intestinal peptide genotype on circadian gene expression in the suprachiasmatic nucleus and peripheral organs. J. Biol. Rhythms 2011, 26, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Piggins, H.D.; Cutler, D.J. The roles of vasoactive intestinal polypeptide in the mammalian circadian clock. J. Endocrinol. 2003, 177, 7–15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aton, S.J.; Colwell, C.S.; Harmar, A.J.; Waschek, J.; Herzog, E.D. Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat. Neurosci. 2005, 8, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Nussdorfer, G.G.; Malendowicz, L.K. Role of VIP, PACAP, and related peptides in the regulation of the hypothalamo-pituitary-adrenal axis. Peptides 1998, 19, 1443–1467. [Google Scholar] [CrossRef]
- Gozes, I.; Zamostiano, R.; Pinhasov, A.; Bassan, M.; Giladi, E.; Steingart, R.A.; Brenneman, D.E. A novel VIP responsive gene. Activity dependent neuroprotective protein. Ann. N. Y. Acad. Sci. 2000, 921, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I.; Divinsky, I.; Pilzer, I.; Fridkin, M.; Brenneman, D.E.; Spier, A.D. From vasoactive intestinal peptide (VIP) through activity-dependent neuroprotective protein (ADNP) to NAP: A view of neuroprotection and cell division. J. Mol. Neurosci. 2003, 20, 315–322. [Google Scholar] [CrossRef]
- Gozes, I. Activity-dependent neuroprotective protein: From gene to drug candidate. Pharmacol. Ther. 2007, 114, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Yang, Y.H.; Lu, C.Y.; Jong, S.B.; Chen, L.J.; Lin, Y.F.; Wu, S.J.; Chu, P.Y.; Chung, T.W.; Tyan, Y.C. Activity-dependent neuroprotector homeobox protein: A candidate protein identified in serum as diagnostic biomarker for Alzheimer’s disease. J. Proteom. 2012, 75, 3617–3629. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I.; Bardea, A.; Reshef, A.; Zamostiano, R.; Zhukovsky, S.; Rubinraut, S.; Fridkin, M.; Brenneman, D.E. Neuroprotective strategy for Alzheimer disease: intranasal administration of a fatty neuropeptide. PNAS 1996, 96, 427–432. [Google Scholar] [CrossRef]
- White, C.M.; Ji, S.; Cai, H.; Maudsley, S.; Martin, B. Therapeutic potential of vasoactive intestinal peptide and its receptors in neurological disorders. CNS Neurol. Disord. Drug Targets 2010, 9, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Hawley, C.M.; Duggan, K.A.; Macdonald, G.J.; Shelley, S. Oral sodium regulates extrahepatic metabolism of vasoactive intestinal peptide. Clin. Sci. 1991, 81, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Shelley, S.; MacDonald, G.J.; Duggan, K.A. The effects of a high sodium diet on the metabolism and secretion of vasoactive intestinal peptide in the rabbit. J. Physiol. 1992, 451, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.J. The Global Obesity Epidemic is Related to Stroke, Dementia and Alzheimer’s disease. JSM Alzheimer’s Dis. Relat. Dement. 2014, 1, 1010–1018. [Google Scholar]
- Fuenmayor, N.; Moreira, E.; Cubeddu, L.X. Salt sensitivity is associated with insulin resistance in essential hypertension. Am. J. Hypertens. 1998, 11, 397–402. [Google Scholar] [CrossRef]
- Rocchini, A.P. The relationship of sodium sensitivity to insulin resistance. Am. J. Med. Sci. 1994, 307 (Suppl. 1), S75–S80. [Google Scholar] [PubMed]
- Rocchini, A.P.; Katch, V.; Kveselis, D.; Moorehead, C.; Martin, M.; Lampman, R.; Gregory, M. Insulin and renal sodium retention in obese adolescents. Hypertension 1989, 14, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Sawmiller, D.R.; Henning, R.J.; Cuevas, J.; Dehaven, W.I.; Vesely, D.L. Coronary vascular effects of vasoactive intestinal peptide in the isolated perfused rat heart. Neuropeptides 2004, 38, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Dogrukol-Ak, D.; Tore, F.; Tuncel, N. Passage of VIP/PACAP/secretin family across the blood-brain barrier: Therapeutic effects. Curr. Pharm. Des. 2004, 10, 1325–1340. [Google Scholar] [CrossRef] [PubMed]
- Dogrukol-Ak, D.; Banks, W.A.; Tuncel, N.; Tuncel, M. Passage of vasoactive intestinal peptide across the blood-brain barrier. Peptides 2009, 24, 437–444. [Google Scholar] [CrossRef]
- Ji, H.; Zhang, Y.; Liu, Y.; Shen, X.D.; Gao, F.; Nguyen, T.T.; Busuttil, R.W.; Waschek, J.A.; Kupiec-Weglinski, J.W. Vasoactive intestinal peptide attenuates liver ischemia/reperfusion injury in mice via the cyclic adenosine monophosphate-protein kinase a pathway. Liver Transplant. 2013, 19, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015, 6, 7466. [Google Scholar] [CrossRef] [PubMed]
- Morgado, A.L.; Xavier, J.M.; Dionísio, P.A.; Ribeiro, M.F.; Dias, R.B.; Sebastião, A.M.; Solá, S.; Rodrigues, C.M. MicroRNA-34a Modulates Neural Stem Cell Differentiation by Regulating Expression of Synaptic and Autophagic Proteins. Mol. Neurobiol. 2015, 5, 1168–1183. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cheng, H.; Li, X.; Lu, W.; Wang, K.; Wen, T. Regulation of neural stem cell differentiation by transcription factors HNF4-1 and MAZ-1. Mol. Neurobiol. 2013, 47, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Kullak-Ublick, G.A. Hepatocyte nuclear factor 1 alpha: A key mediator of the effect of bile acids on gene expression. Hepatology 2003, 37, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Satija, Y.K.; Das, S. PGC-1α, a Key Modulator of p53, Promotes Cell Survival upon Metabolic Stress. Mol. Cell 2011, 44, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Inoue, Y.; Gonzalez, F.J. Role of the hepatocyte nuclear factor 4alpha in control of the pregnane X receptor during fetal liver development. Hepatology 2003, 37, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- ADNP activity-dependent neuroprotector homeobox [Homo sapiens (human)] Gene ID: 23394. 2016. Available online: http://www.ncbi.nlm.nih.gov/gene/23394 (assessed on 17 March 2016).
- Lu, P.; Liu, J.; Melikishvili, M.; Fried, M.G.; Chi, Y.I. Crystallization of hepatocyte nuclear factor 4 alpha (HNF4 alpha) in complex with the HNF1 alpha promoter element. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2008, 64, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.A.; Brace, C.S.; Wang, T.; Stormo, G.D.; Imai, S. A nutrient-sensitive interaction between Sirt1 and HNF-1α regulates Crp expression. Aging Cell 2011, 10, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S. Hepatocyte nuclear factor 1 alpha (HNF-1 alpha) mutations in maturity-onset diabetes of the young. Hum. Mutat. 2000, 16, 377–385. [Google Scholar] [CrossRef]
- Maetzler, W.; Tian, Y.; Baur, S.M.; Gauger, T.; Odoj, B.; Schmid, B. Serum and Cerebrospinal Fluid Levels of Transthyretin in Lewy Body Disorders with and without Dementia. PLoS ONE 2012, 7, e48042. [Google Scholar] [CrossRef] [PubMed]
- Woodward, D.K.; Hatton, J.; Ensom, M.H.; Young, B.; Dempsey, R.; Clifton, G.D. Alpha1-acid glycoprotein concentrations and cerebrospinal fluid drug distribution after subarachnoid hemorrhage. Pharmacotherapy 1998, 18, 1062–1068. [Google Scholar] [PubMed]
- Mattsson, N.; Insel, P.; Nosheny, R.; Trojanowski, J.Q.; Shaw, L.M.; Jack, C.R.; Weiner, M. Effects of cerebrospinal fluid proteins on brain atrophy rates in cognitively healthy older adults. Neurobiol. Aging 2014, 35, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Kaynar, M.Y.; Tanriverdi, T.; Kafadar, A.M.; Kacira, T.; Uzun, H.; Aydin, S.; Gumustas, K.; Dirican, A.; Kuday, C. Detection of soluble intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in both cerebrospinal fluid and serum of patients after aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2004, 101, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Jongen, P.J.; Doesburg, W.H.; Ibrahim-Stappers, J.L.; Lemmens, W.A.; Hommes, O.R.; Lamers, K.J. Cerebrospinal fluid C3 and C4 indexes in immunological disorders of the central nervous system. Acta Neurol. Scand. 2000, 101, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Kuncl, R.W.; Bilak, M.M.; Bilak, S.R.; Corse, A.M.; Royal, W.; Becerra, S.P. Pigment epithelium-derived factor is elevated in CSF of patients with amyotrophic lateral sclerosis. J. Neurochem. 2002, 81, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Pearl, G.S.; Mullins, R.E. Alpha 1-antitrypsin in cerebrospinal fluid of patients with neurologic diseases. Arch. Neurol. 1985, 42, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Rentzos, M.; Nikolaou, C.; Rombos, A.; Boufidou, F.; Zoga, M.; Dimitrakopoulos, A.; Tsoutsou, A.; Vassilopoulos, D. RANTES levels are elevated in serum and cerebrospinal fluid in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2007, 8, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Moghekar, A.R.; Hu, J.; Sun, K.; Turner, R.; Ferrucci, L.; O’Brien, R. Klotho in the cerebrospinal fluid of adults with and without Alzheimer's disease. Neurosci. Lett. 2014, 558, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.J.; Rohlfing, F.; Anand, S.; Johnson, W.E.; Alvarez, M.T.; Cobell, J.; King, J.; Young, S.A.; Kauwe, J.S.; Graves, S.W. Discovery and Subsequent Confirmation of Novel Serum Biomarkers Diagnosing Alzheimer’s Disease. Alzheimers Dis. 2015, 49, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Bilic, E.; Bilic, E.; Rudan, I.; Kusec, V.; Zurak, N.; Delimar, D.; Zagar, M. Comparison of the growth hormone, IGF-1 and insulin in cerebrospinal fluid and serum between patients with motor neuron disease and healthy controls. Eur. J Neurol. 2006, 13, 1340–1345. [Google Scholar] [CrossRef] [PubMed]
- Kos, K.; Harte, A.L.; da Silva, N.F.; Tonchev, A.; Chaldakov, G.; James, S.; Snead, D.R.; Hoggart, B.; O’Hare, J.P.; McTernan, P.G.; et al. Adiponectin and resistin in human cerebrospinal fluid and expression of adiponectin receptors in the human hypothalamus. J. Clin. Endocrinol. Metab. 2007, 92, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Pirttilä, T.; Vanhatalo, S.; Turpeinen, U.; Riikonen, R. Cerebrospinal fluid insulin-like growth factor-1, insulin growth factor binding protein-2 or nitric oxide are not increased in MS or ALS. Acta Neurol. Scand. 2004, 109, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Zhao, X.; Hua, Z. Potential therapeutic implications of gelsolin in Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 13–25. [Google Scholar] [PubMed]
- Güntert, A.; Campbell, J.; Saleem, M.; O’Brien, D.P.; Thompson, A.J.; Byers, H.L.; Ward, M.A.; Lovestone, S. Plasma gelsolin is decreased and correlates with rate of decline in Alzheimer's disease. J. Alzheimers Dis. 2010, 21, 585–596. [Google Scholar] [PubMed]
- Restituto, P.; Colina, I.; Varo, J.J.; Varo, N. Adiponectin diminishes platelet aggregation and sCD40L release. Potential role in the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1072–E1077. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Q.; Zhang, H.F.; Gao, G.X.; Bai, Q.X.; Li, R.; Wang, X.M. Adiponectin inhibits hyperlipidemia-induced platelet aggregation via attenuating oxidative/nitrative stress. Physiol. Res. 2011, 60, 347–354. [Google Scholar] [PubMed]
- Carnevale, R.; Pastori, D.; Peruzzi, M.; de Falco, E.; Chimenti, I.; Biondi-Zoccai, G.; Greco, E.; Marullo, A.G.M.; Nocella, C.; Violi, F.; et al. Total Adiponectin Is Inversely Associated with Platelet Activation and CHA2DS2-VASc Score in Anticoagulated Patients with Atrial Fibrillation. Mediat. Inflamm. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Kobayashi, H.; Kihara, S.; Kumada, M.; Sato, K.; Inoue, T.; Funahashi, T.; Walsh, K. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J. Biol. Chem. 2004, 279, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, L.Y.; Lam, K.S.; Lu, G.; Cooper, G.J.; Xu, A. Proteomic characterization of human serum proteins associated with the fat-derived hormone adiponectin. Proteomics 2006, 6, 3862–3870. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Nam, D.W.; Cha, M.Y.; Kim, K.H.; Byun, J.; Ryu, H.; Mook-Jung, I. Thrombospondin-1 prevents amyloid beta-mediated synaptic pathology in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 3214–3227. [Google Scholar] [CrossRef] [PubMed]
- Rama Rao, K.V.; Curtis, K.M.; Johnstone, J.T.; Norenberg, M.D. Amyloid-β inhibits thrombospondin 1 release from cultured astrocytes: effects on synaptic protein expression. J. Neuropathol. Exp. Neurol. 2013, 72, 735–744. [Google Scholar] [PubMed]
- Liauw, J.; Hoang, S.; Choi, M.; Eroglu, C.; Choi, M.; Sun, G.H.; Percy, M.; Wildman-Tobriner, B.; Bliss, T.; Guzman, R.G.; et al. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J. Cereb. Blood Flow Metab. 2008, 8, 1722–1732. [Google Scholar] [CrossRef] [PubMed]
- Kounnas, M.Z.; Morris, R.E.; Thompson, M.R.; FitzGerald, D.J.; Strickland, D.K.; Saelinger, C.B. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J. Biol. Chem. 1992, 267, 12420–12423. [Google Scholar] [PubMed]
- Naganuma, H.; Satoh, E.; Asahara, T.; Amagasaki, K.; Watanabe, A.; Satoh, H.; Kuroda, K.; Zhang, L.; Nukui, H. Quantification of thrombospondin-1 secretion and expression of alphavbeta3 and alpha3beta1 integrins and syndecan-1 as cell-surface receptors for thrombospondin-1 in malignant glioma cells. J. Neurooncol. 2004, 70, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Resovi, A.; Pinessi, D.; Chiorino, G.; Taraboletti, G. Current understanding of the thrombospondin-1 interactome. Matrix Biol. 2014, 37, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Kim, D.B.; Kim, M.J.; Kwon, B.J.; Chang, S.Y.; Jang, S.W.; Cho, E.J.; Rho, T.H.; Kim, J.H. Higher plasma thrombospondin-1 levels in patients with coronary artery disease and diabetes mellitus. Korean Circ. J. 2012, 42, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.D.; Miller, T.W.; Rogers, N.M.; Yao, M.; Isenberg, J.S. The matricellular protein thrombospondin-1 globally regulates cardiovascular function and responses to stress via CD47. Matrix Biol. 2012, 31, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cavalera, M.; Frangogiannis, N.G. The role of thrombospondin (TSP)-1 in obesity and diabetes. Adipocyte 2014, 3, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Maier, K.G.; Han, X.; Sadowitz, B.; Gentile, K.L.; Middleton, F.A.; Gahtan, V. Thrombospondin-1: A proatherosclerotic protein augmented by hyperglycemia. J. Vasc. Surg. 2010, 51, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, J.S.; Romeo, M.J.; Yu, C.; Yu, C.K.; Nghiem, K.; Monsale, J.; Rick, M.E.; Wink, D.A.; Frazier, W.A.; Roberts, D.D. Thrombospondin-1 stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide/cGMP signaling. Blood 2008, 111, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Starlinger, P.; Moll, H.P.; Assinger, A.; Nemeth, C.; Hoetzenecker, K.; Gruenberger, B.; Gruenberger, T.; Kuehrer, I.; Schoppmann, S.F.; Gnant, M.; et al. Thrombospondin-1: A unique marker to identify in vitro platelet activation when monitoring in vivo processes. J. Thromb. Haemost. 2010, 8, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Lawler, P.R.; Lawler, J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb. Perspect. Med. 2012, 2, a006627. [Google Scholar] [CrossRef] [PubMed]
- Maimaitiyiming, H.; Clemons, K.; Zhou, Q.; Norman, H.; Wang, S. Thrombospondin1 Deficiency Attenuates Obesity-Associated Microvascular Complications in ApoE−/− Mice. PLoS ONE 2015, 10, e0121403. [Google Scholar]
- Dabrowska, K.; Lewandowski, E.; Skowronska-Jozwiak, A.; Brona, A.; Milewicz, A.; Lewinski, A. Serum concentrations of thrombospondin-1 and adiponectin in patients with hyperthyroidism before and after normalisation of thyroid function. Endocr. Abstr. 2012, 29, P1672. [Google Scholar]
- Daniel, C.; Schaub, K.; Amann, K.; Lawler, J.; Hugo, C. Thrombospondin-1 is an endogenous activator of TGF-beta in experimental diabetic nephropathy in vivo. Diabetes 2007, 56, 2982–2989. [Google Scholar] [CrossRef] [PubMed]
- Murphy-Ullrich, J.E.; Poczatek, M. Activation of latent TGF-beta by thrombospondin-1: Mechanisms and physiology. Cytokine Growth Factor Rev. 2000, 11, 59–69. [Google Scholar] [CrossRef]
- Ribeiro, S.M.; Poczatek, M.; Schultz-Cherry, S.; Villain, M.; Murphy-Ullrich, J.E. The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-beta. J. Biol. Chem. 1999, 274, 13586–13593. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Wiede, J.; Krutzsch, H.C.; Ribeiro, S.M.; Roberts, D.D.; Murphy-Ullrich, J.E.; Hugo, C. Thrombospondin-1 is a major activator of TGF-beta in fibrotic renal disease in the rat in vivo. Kidney Int. 2004, 65, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Sakai, K.; Baba, H.; Sakai, T. Thrombospondin-1 is a novel negative regulator of liver regeneration after partial hepatectomy via TGF-β1 activation in mice. Hepatology 2012, 55, 1562–1573. [Google Scholar] [CrossRef] [PubMed]
- Tesseur, I.; Zou, K.; Esposito, L.; Bard, F.; Berber, E.; Can, J.V.; Lin, A.H.; Crews, L.; Tremblay, P.; Mathews, P.; et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J. Clin. Invest. 2006, 116, 3060–3069. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Lin, C.; Yan, F.; Yu, G.Q.; Rohde, M.; McConlogue, L.; Masliah, E.; Mucke, L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat. Med. 2001, 7, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Golde, T. Dysfunction of TGF-beta signaling in Alzheimer’s disease. J. Clin. Invest. 2006, 116, 2855–2857. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Masliah, E.; Mallory, M.; McConlogue, L.; Johnson-Wood, K.; Lin, C.; Mucke, L. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer’s disease. Nature 1997, 389, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Roh, Y.S.; Song, J.; Zhang, B.; Liu, C.; Loomba, R.; Seki, E. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology 2014, 59, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; Ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Tian, Q.; Zhao, X.; Wang, X. Serum transforming growth factor beta 3 predicts future development of nonalcoholic fatty liver disease. Int. J. Clin. Exp. Med. 2015, 8, 4545–4550. [Google Scholar] [PubMed]
- Chao, C.C.; Hu, S.; Frey, W.H.; Ala, T.A.; Tourtellotte, W.W.; Peterson, P.K. Transforming growth factor beta in Alzheimer’s disease. Clin. Diagn. Lab. Immunol. 1994, 1, 109–110. [Google Scholar] [PubMed]
- Sinha, M.; Jang, Y.C.; Oh, J.; Khong, D.; Wu, E.Y.; Manohar, R.; Miller, C.; Regalado, S.G.; Loffredo, F.S.; Pancoast, J.R.; et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014, 344, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Ge, G.; Hopkins, D.R.; Ho, W.-B.; Greenspan, D.S. GDF11 Forms a Bone Morphogenetic Protein 1-Activated Latent Complex That Can Modulate Nerve Growth Factor-Induced Differentiation of PC12 Cells. Mol. Cell. Biol. 2005, 25, 5846–5858. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, V.; Hettiarachchy, N.S. Nutriproteomics: A promising tool to link diet and diseases in nutritional research. Biochim. Biophys. Acta 2012, 1824, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Sénéchal, S.; Kussmann, M. Nutriproteomics: Technologies and applications for identification and quantification of biomarkers and ingredients. Proc. Nutr. Soc. 2011, 70, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Sauer, S.; Luge, T. Nutriproteomics: Facts, concepts, and perspectives. Proteomics 2015, 15, 997–1013. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.T.; Chen-Plotkin, A.; Arnold, S.E.; Grossman, M.; Clark, C.M.; Shaw, L.M.; Pickering, E.; Kuhn, M.; Chen, Y.; McCluskey, L.; et al. Novel CSF biomarkers for Alzheimer’s disease and mild cognitive impairment. Acta Neuropathol. 2010, 119, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Neumeier, M.; Weigert, J.; Buettner, R.; Wanninger, J.; Schäffler, A.; Müller, A.M.; Killian, S.; Sauerbruch, S.; Schlachetzki, F.; Steinbrecher, A.; et al. Detection of adiponectin in cerebrospinal fluid in humans. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E965–E969. [Google Scholar] [CrossRef] [PubMed]
- Une, K.; Takei, Y.A.; Tomita, N.; Asamura, T.; Ohrui, T.; Furukawa, K.; Arai, H. Adiponectin in plasma and cerebrospinal fluid in MCI and Alzheimer’s disease. Eur. J. Neurol. 2011, 18, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Kusminski, C.M.; McTernan, P.G.; Schraw, T.; Kos, K.; O’Hare, J.P.; Ahima, R.; Kumar, S.; Scherer, P.E. Adiponectin complexes in human cerebrospinal fluid: Distinct complex distribution from serum. Diabetologia 2007, 50, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Choe, Y.G.; Jin, W.; Cho, Y.K.; Chung, W.G.; Kim, H.J.; Jeon, W.K.; Kim, B.I. Apolipoprotein B/AI ratio is independently associated with non-alcoholic fatty liver disease in nondiabetic subjects. J. Gastroenterol. Hepatol. 2013, 28, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.J. LPS Regulates Apolipoprotein E and Aβ Interactions with Effects on Acute Phase Proteins and Amyloidosis. Adv. Aging Res. 2015, 4, 69–77. [Google Scholar] [CrossRef]
- De Almeida, R.L.; Constancio, J.; Vendramini, R.C.; Fracasso, J.F.; Menani, J.V. Lipopolysaccharide reduces sodium intake and sodium excretion in dehydrated rats. Physiol. Behav. 2011, 102, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.J. Diabetes and organ dysfunction in the developing and developed. World Glob. J. Med. Res. F Dis. 2015, 15, 1–8. [Google Scholar]
- Affarah, H.B.; Hall, W.D.; Heymsfield, S.B.; Kutner, M.; Wells, J.O. High-carbohydrate diet: antinatriuretic and blood pressure response in normal men. Am. J. Clin. Nutr. 1986, 44, 341–348. [Google Scholar] [PubMed]
- Matsuura, F.; Oku, H.; Koseki, M.; Sandoval, J.C.; Yuasa-Kawase, M.; Tsubakio-Yamamoto, K.; Masuda, D.; Maeda, N.; Tsujii, K.; Ishigami, M.; et al. Adiponectin accelerates reverse cholesterol transport by increasing high density lipoprotein assembly in the liver. Biochem. Biophys. Res. Commun. 2007, 358, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Wang, X.; Guo, X.; Yang, Z.; Bai, R.; Liu, M.; Xiao, C.; Bian, Y. Adiponectin upregulates ABCA1 expression through liver X receptor alpha signaling pathway in RAW 264.7 macrophages. Int. J. Clin. Exp. Pathol. 2015, 8, 450–457. [Google Scholar] [PubMed]
- Inoue, M.; Jiang, Y.; Tokunaga, M.; Martinez-Santibañez, G.; Geletka, L.; Lumeng, C.N.; Buchner, D.A.; Chun, T.H. Thrombospondin 1 mediates high-fat diet-induced muscle fibrosis and insulin resistance in male mice. Endocrinology 2013, 154, 4548–4559. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Maimaitiyiming, H.; Qi, X.; Norman, H.; Wang, S. Thrombospondin 1 mediates renal dysfunction in a mouse model of high-fat diet-induced obesity. Am. J. Physiol. Renal Physiol. 2013, 305, F871–F880. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.J. Nutritional Diets Accelerate Amyloid Beta Metabolism and Prevent the Induction of Chronic Diseases and Alzheimer’s Disease, 1st ed.; Photon ebooks: Sherbrooke, QC, Canada, 2015; pp. 1–48. [Google Scholar]
- Naito, T.; Masaki, T.; Nikolic-Paterson, D.J.; Tanji, C.; Yorioka, N.; Kohno, N. Angiotensin II induces thrombospondin-1 production in human mesangial cells via p38 MAPK and JNK: A mechanism for activation of latent TGF-beta1. Am. J. Physiol. Renal Physiol. 2004, 286, F278–F287. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.C.; Hamdy, R.C.; Chua, B.H. Regulation of thrombospondin-1 production by angiotensin II in rat heart endothelial cells. Biochim. Biophys. Acta 1997, 1357, 209–214. [Google Scholar] [CrossRef]
- Lutz, J.; Huwiler, K.G.; Fedczyna, T.; Lechman, T.S.; Crawford, S.; Kinsella, T.R.; Pachman, L.M. Increased plasma thrombospondin-1 (TSP-1) levels are associated with the TNF alpha-308A allele in children with juvenile dermatomyositis. Clin. Immunol. 2002, 103, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Rege, T.A.; Stewart, J.; Dranka, B.; Benveniste, E.N.; Silverstein, R.L.; Gladson, C.L. Thrombospondin-1-induced apoptosis of brain microvascular endothelial cells can be mediated by TNF-R1. J. Cell. Physiol. 2009, 218, 94–103. [Google Scholar] [CrossRef] [PubMed]
- McMorrow, J.P.; Crean, D.; Gogarty, M.; Smyth, A.; Connolly, M.; Cummins, E.; Veale, D.; Fearon, U.; Tak, P.P.; Fitzgerald, O.; et al. Tumor necrosis factor inhibition modulates thrombospondin-1 expression in human inflammatory joint disease through altered NR4A2 activity. Am. J. Pathol. 2013, 183, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Belarbi, K.; Jopson, T.; Tweedie, D.; Arellano, C.; Luo, W.; Greig, N.H.; Rosi, S. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J. Neuroinflamm. 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E.; Gross, H.; Weinberger, A.; Cohen, H. TNF-α modulation for treatment of Alzheimer’s disease: A 6-month pilot study. MedGenMed 2006, 8, 25. [Google Scholar] [PubMed]
- Woo, Y.-C.; Tso, A.W.K.; Xu, A.; Law, L.S.C.; Fong, C.H.Y.; Lam, T.-H. Combined Use of Serum Adiponectin and Tumor Necrosis Factor-Alpha Receptor 2 Levels Was Comparable to 2-Hour Post-Load Glucose in Diabetes Prediction. PLoS ONE 2012, 7, e36868. [Google Scholar]
- Masaki, T.; Chiba, S.; Tatsukawa, H.; Yasuda, T.; Noguchi, H.; Seike, M.; Yoshimatsu, H. Adiponectin protects LPS-induced liver injury through modulation of TNF-α in KK-Ay obese mice. Hepatology 2004, 40, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, L.-M.; Wu, J. Cross-Talk between Apolipoprotein E and Cytokines. Mediat. Inflamm. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Laskowitz, D.T.; Goel, S.; Bennett, E.R.; Matthew, W.D. Apolipoprotein E suppresses glial cell secretion of TNF alpha. J. Neuroimmunol. 1997, 76, 70–74. [Google Scholar] [CrossRef]
- Song, H.; Saito, K.; Fujigaki, S.; Noma, A.; Ishiguro, H.; Nagatsu, T.; Seishima, M. IL-1 beta and TNF-alpha suppress apolipoprotein (apo) E secretion and apo A-I expression in HepG2 cells. Cytokine 1998, 10, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Delfín, J.; Hondares, E.; Iglesias, R.; Giralt, M.; Caelles, C.; Villarroya, F. TNF-α represses β-Klotho expression and impairs FGF21 action in adipose cells: involvement of JNK1 in the FGF21 pathway. Endocrinology 2012, 153, 4238–4245. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Grunfeld, C.; Heuer, J.G.; Gupta, A.; Cramer, M.; Zhang, T.; Shigenaga, J.K.; Patzek, S.M.; Chan, Z.W.; Moser, A.; et al. FGF21 is increased by inflammatory stimuli and protects leptin-deficient ob/ob mice from the toxicity of sepsis. Endocrinology 2012, 153, 2689–2700. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.C.; Xu, A.; Wang, Y.; Lam, K.S. Fibroblast growth factor 21 as an emerging metabolic regulator: Clinical perspectives. Clin. Endocrinol. 2013, 78, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.F.; Li, S.M.; Ren, G.P.; Zheng, W.; Lu, Y.J.; Yu, Y.H.; Xu, W.J.; Li, T.H.; Zhou, L.H.; Liu, Y.; et al. Recombinant murine fibroblast growth factor 21 ameliorates obesity-related inflammation in monosodium glutamate-induced obesity rats. Endocrine 2015, 49, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Kang, X.; Jiang, Y.; Song, Z.; Feng, W.; McClain, C.J.; Kang, Y.J. Preservation of hepatocyte nuclear factor-4alpha is associated with zinc protection against TNF-alpha hepatotoxicity in mice. Exp. Biol. Med. 2007, 232, 622–628. [Google Scholar]
- Von Bülow, V.; Dubben, S.; Engelhardt, G.; Hebel, S.; Plümäkers, B.; Heine, H.; Rink, L.; Haase, H. Zinc-dependent suppression of TNF-alpha production is mediated by protein kinase A-induced inhibition of Raf-1, I kappa B kinase beta, and NF-kappa B. J. Immunol. 2007, 179, 4180–4186. [Google Scholar] [CrossRef] [PubMed]
- Meerarani, P.; Ramadass, P.; Toborek, M.; Bauer, H.C.; Bauer, H.; Hennig, B. Zinc protects against apoptosis of endothelial cells induced by linoleic acid and tumor necrosis factor alpha. Am. J. Clin. Nutr. 2000, 71, 81–87. [Google Scholar] [PubMed]
- Fordham, J.B.; Hua, J.; Morwood, S.R.; Schewitz-Bowers, L.P.; Copland, D.A.; Dick, A.D.; Nicholson, L.B. Environmental conditioning in the control of macrophage thrombospondin-1 production. Sci. Rep. 2012, 2, 512. [Google Scholar] [CrossRef] [PubMed]
- Gokyu, M.; Kobayashi, H.; Nanbara, H.; Sudo, T.; Ikeda, Y.; Suda, T.; Izumi, Y. Thrombospondin-1 production is enhanced by Porphyromonas gingivalis lipopolysaccharide in THP-1 cells. PLoS ONE 2014, 9, e115107. [Google Scholar]
- Lu, W.; Jiang, J.P.; Hu, J.; Wang, J.; Zheng, M.Z. Curcumin protects against lipopolysaccharide-induced vasoconstriction dysfunction via inhibition of thrombospondin-1 and transforming growth factor-β1. Exp. Ther. Med. 2015, 9, 377–383. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Haas, C.J.; van Leeuwen, H.J.; Verhoef, J.; van Kessel, K.P.; van Strijp, J.A. Analysis of lipopolysaccharide (LPS)-binding characteristics of serum components using gel filtration of FITC-labeled LPS. J. Immunol. Methods 2000, 242, 79–89. [Google Scholar] [CrossRef]
- Wollenberg, G.K.; LaMarre, J.; Rosendal, S.; Gonias, S.L.; Hayes, M.A. Binding of tumor necrosis factor alpha to activated forms of human plasma alpha 2 macroglobulin. Am. J. Pathol. 1991, 138, 265–272. [Google Scholar] [PubMed]
- Webb, D.J.; Gonias, S.L. A modified human alpha 2-macroglobulin derivative that binds tumor necrosis factor-alpha and interleukin-1 beta with high affinity in vitro and reverses lipopolysaccharide toxicity in vivo in mice. Lab. Invest. 1998, 78, 939–948. [Google Scholar] [PubMed]
- Gourine, A.V.; Gourine, V.N.; Tesfaigzi, Y.; Caluwaerts, N.; Van Leuven, F.; Kluger, M.J. Role of alpha(2)-macroglobulin in fever and cytokine responses induced by lipopolysaccharide in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R218–R226. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.M.; Kim, H.V.; Lee, S.; Kim, H.Y.; Kim, W.; Kim, T.S.; Kim, D.J.; Kim, Y.S. Correlations of amyloid-β concentrations between CSF and plasma in acute Alzheimer mouse model. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Johanson, C.E.; Duncan, J.A.; Klinge, P.M.; Brinker, T.; Stopa, E.G.; Silverberg, G.D. Multiplicity of cerebrospinal fluid functions: New challenges in health and disease. Cerebrospinal Fluid Res. 2008, 5, 10. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, I.J. The Role of Clinical Proteomics, Lipidomics, and Genomics in the Diagnosis of Alzheimer’s Disease. Proteomes 2016, 4, 14. https://doi.org/10.3390/proteomes4020014
Martins IJ. The Role of Clinical Proteomics, Lipidomics, and Genomics in the Diagnosis of Alzheimer’s Disease. Proteomes. 2016; 4(2):14. https://doi.org/10.3390/proteomes4020014
Chicago/Turabian StyleMartins, Ian James. 2016. "The Role of Clinical Proteomics, Lipidomics, and Genomics in the Diagnosis of Alzheimer’s Disease" Proteomes 4, no. 2: 14. https://doi.org/10.3390/proteomes4020014
APA StyleMartins, I. J. (2016). The Role of Clinical Proteomics, Lipidomics, and Genomics in the Diagnosis of Alzheimer’s Disease. Proteomes, 4(2), 14. https://doi.org/10.3390/proteomes4020014