
Mass Spectrometric Immunoassays in Characterization of Clinically Significant Proteoforms

Abstract
:1. Introduction
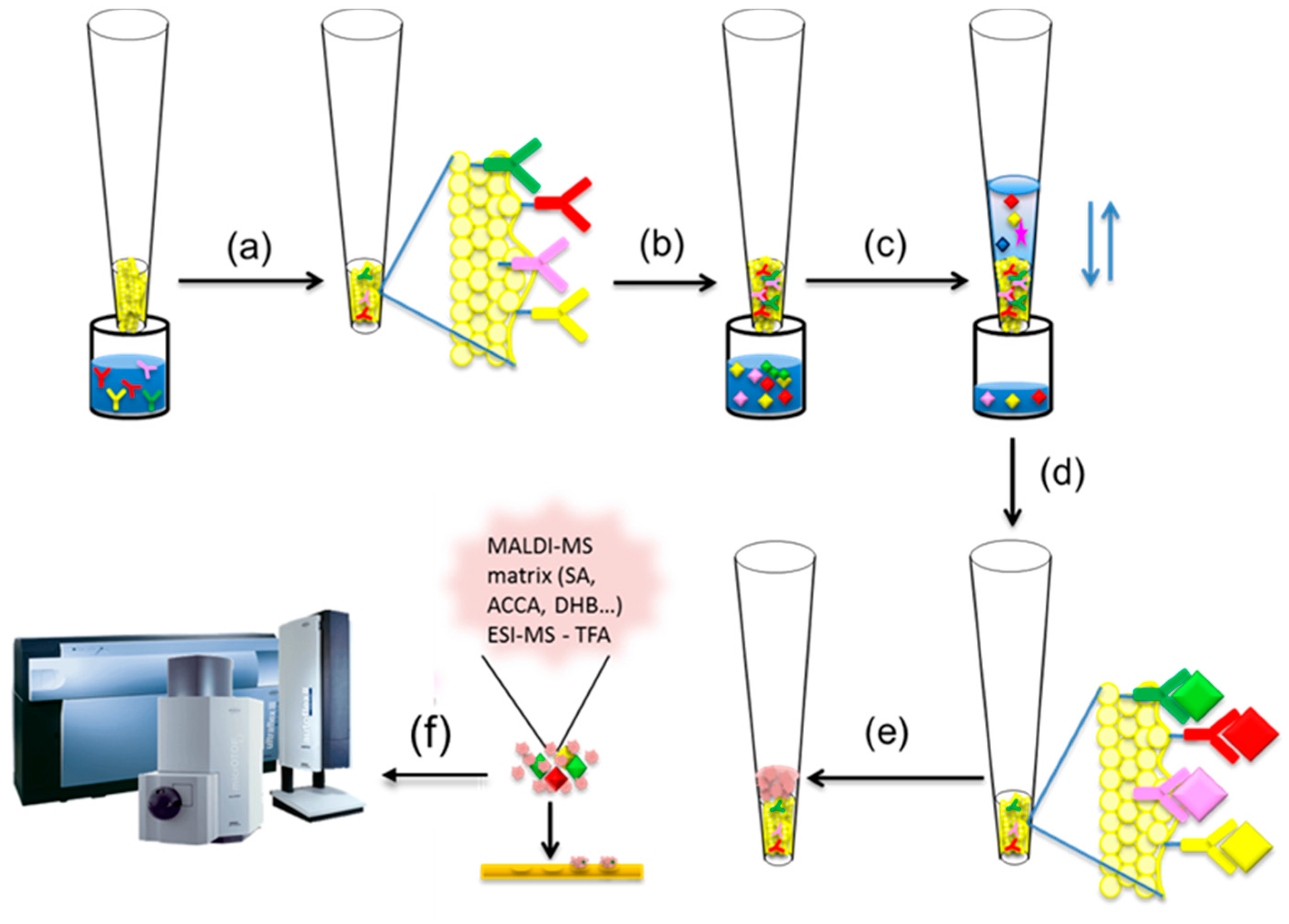
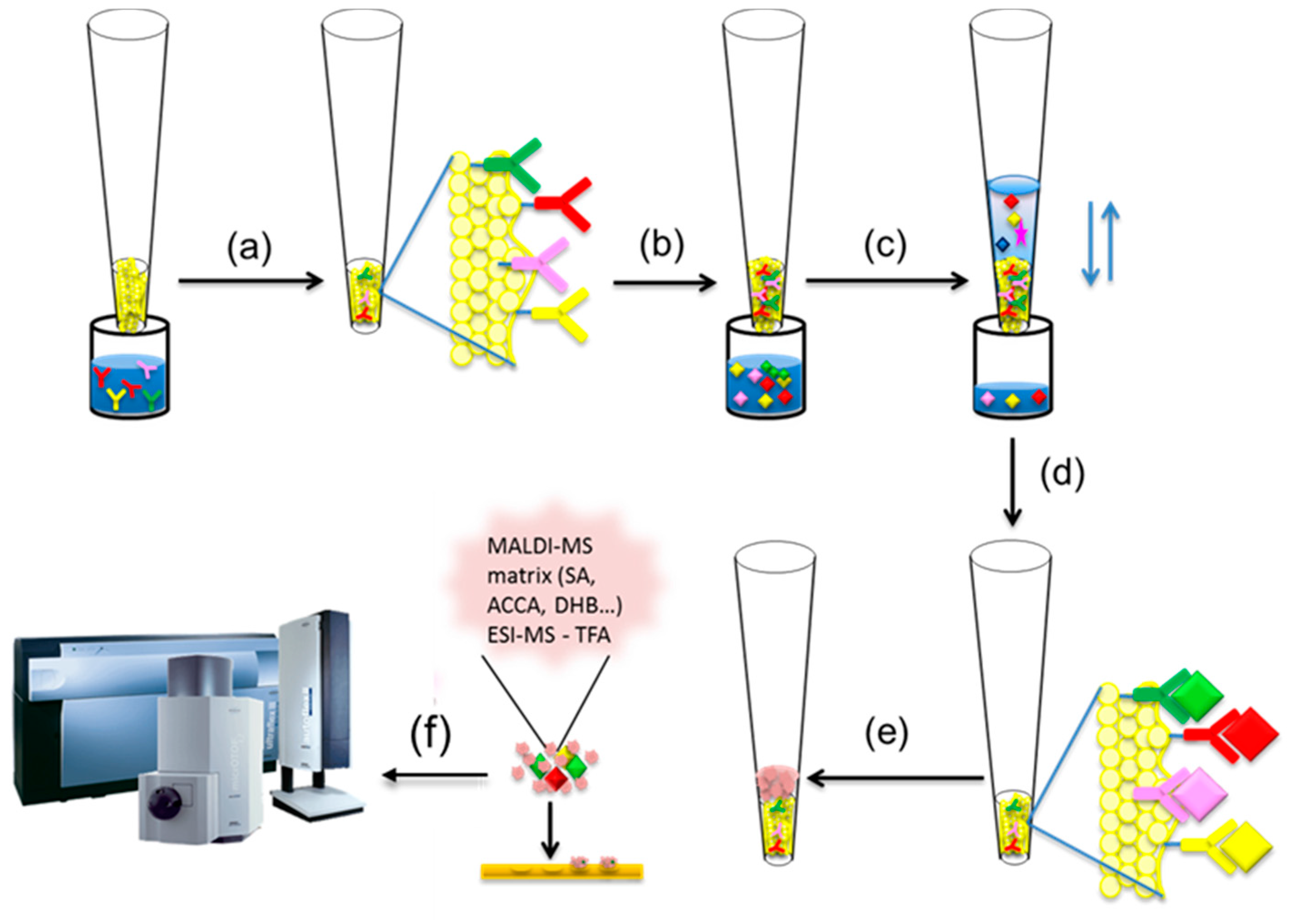
2. Mass Spectrometric Immunoassay Principle
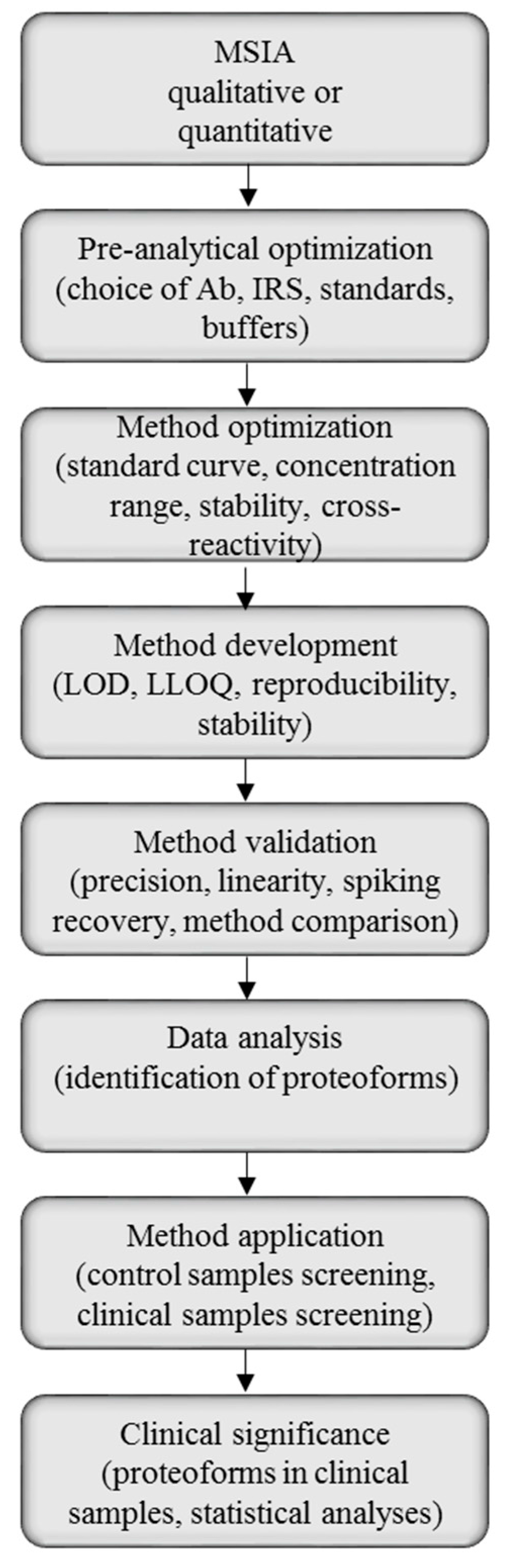
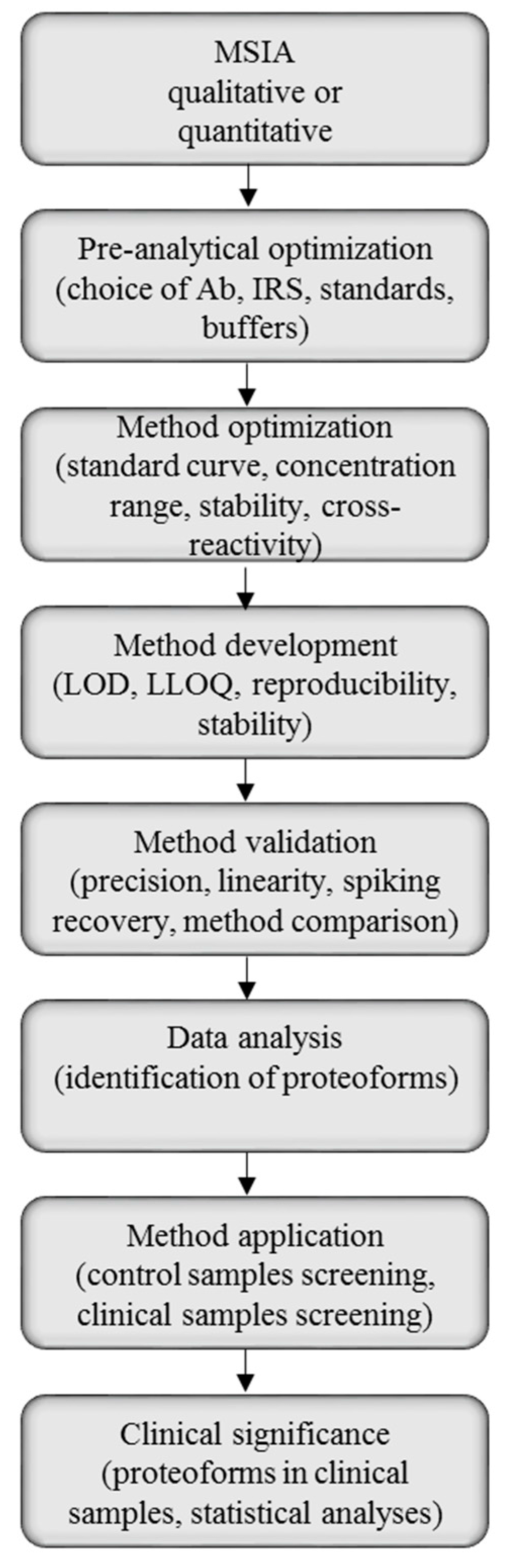
2.1. Method Development with Mass Spectrometric Immunoassay
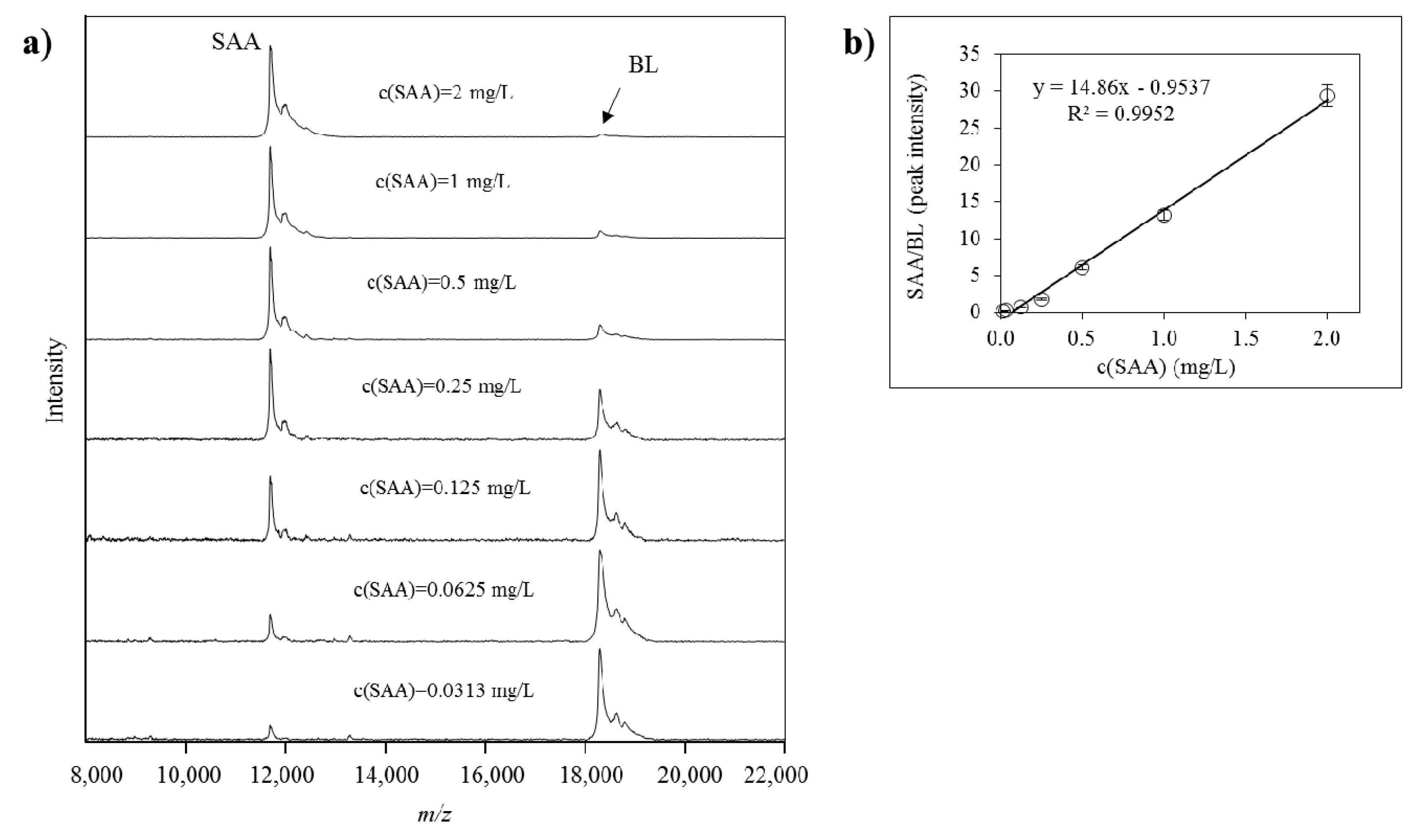
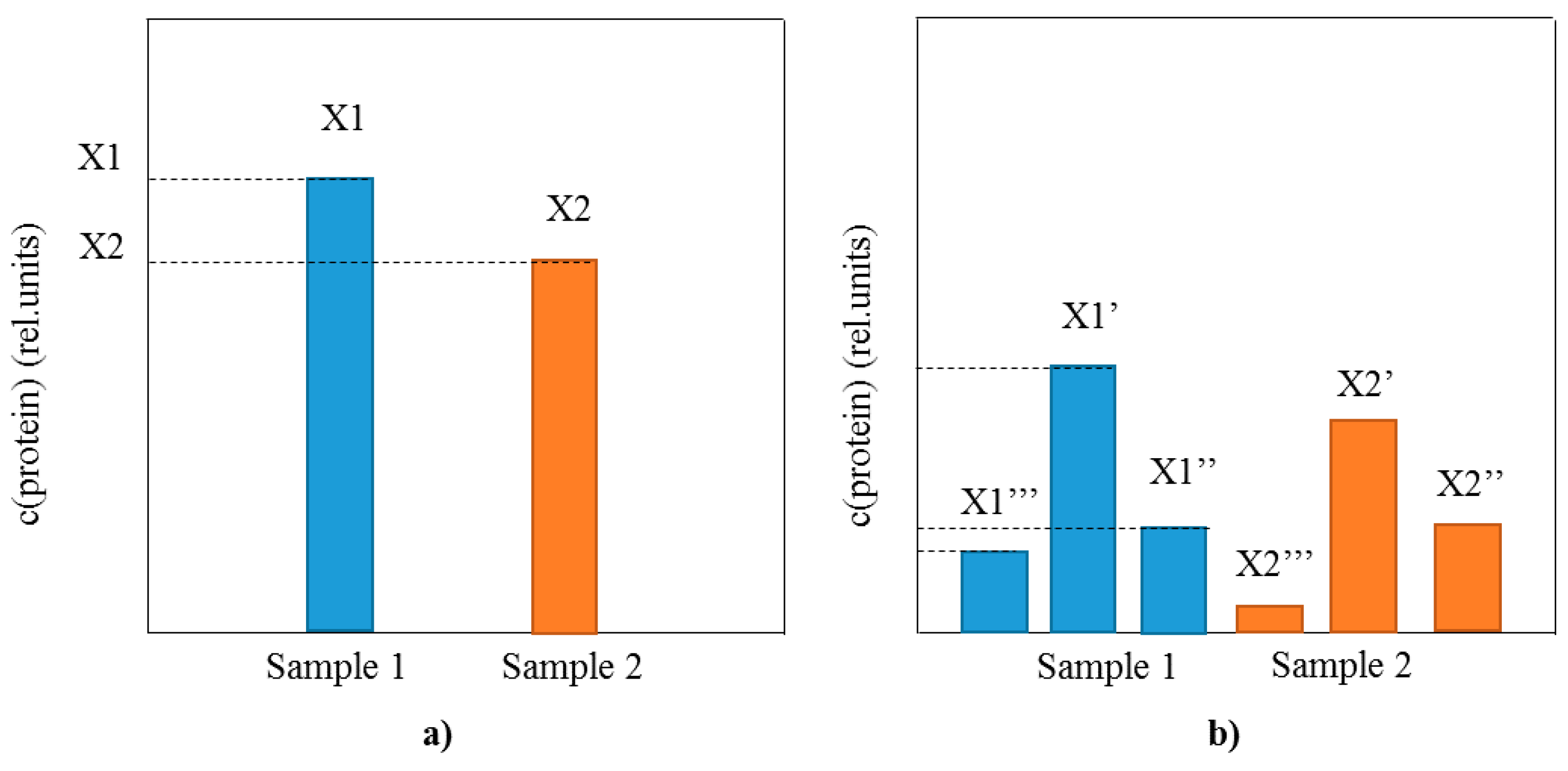
2.2. Method Validation for Quantitative Mass Spectrometric Immunoassay
2.3. Challenges and Limitations of Mass Spectrometric Immunoassay
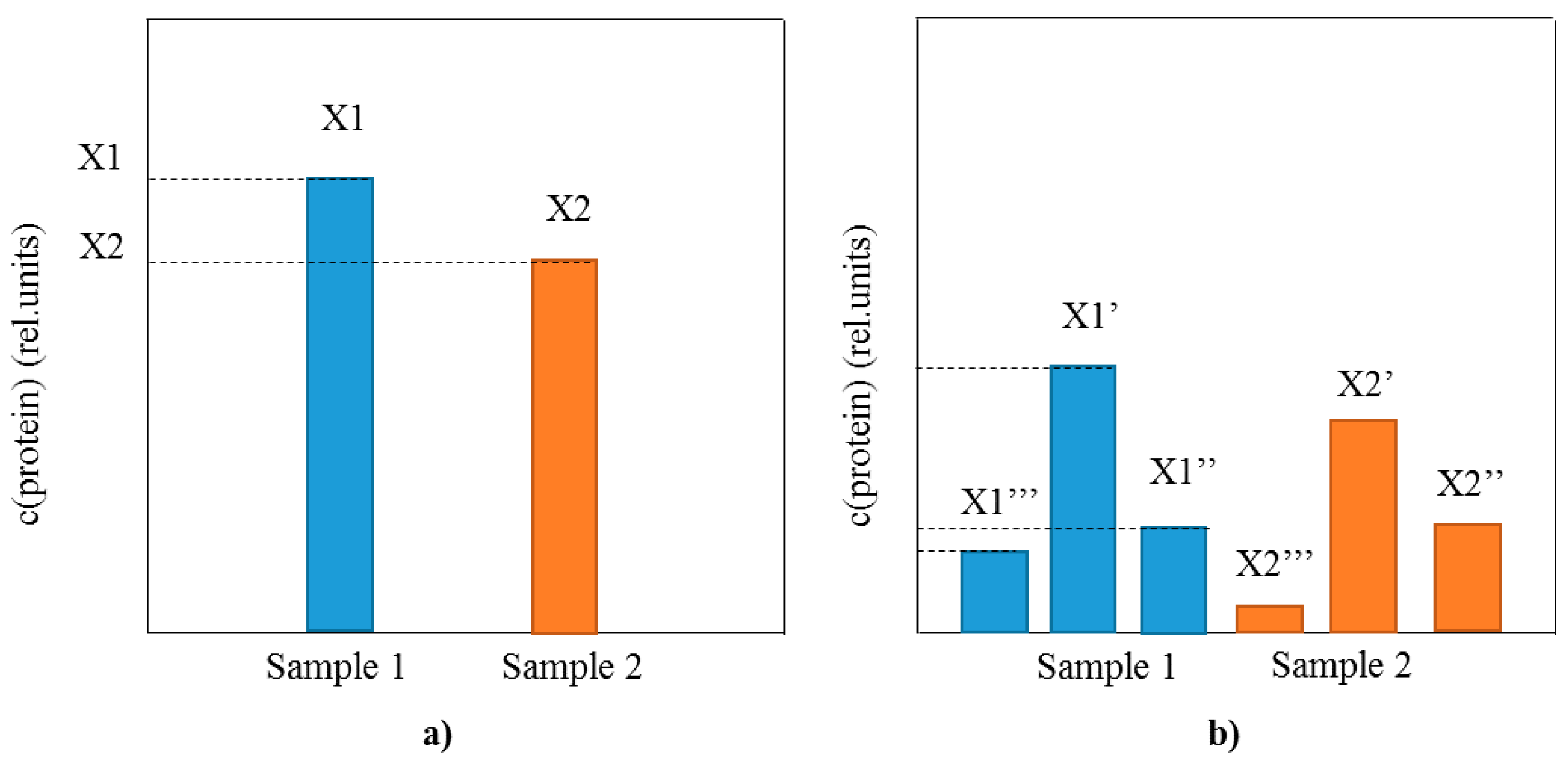
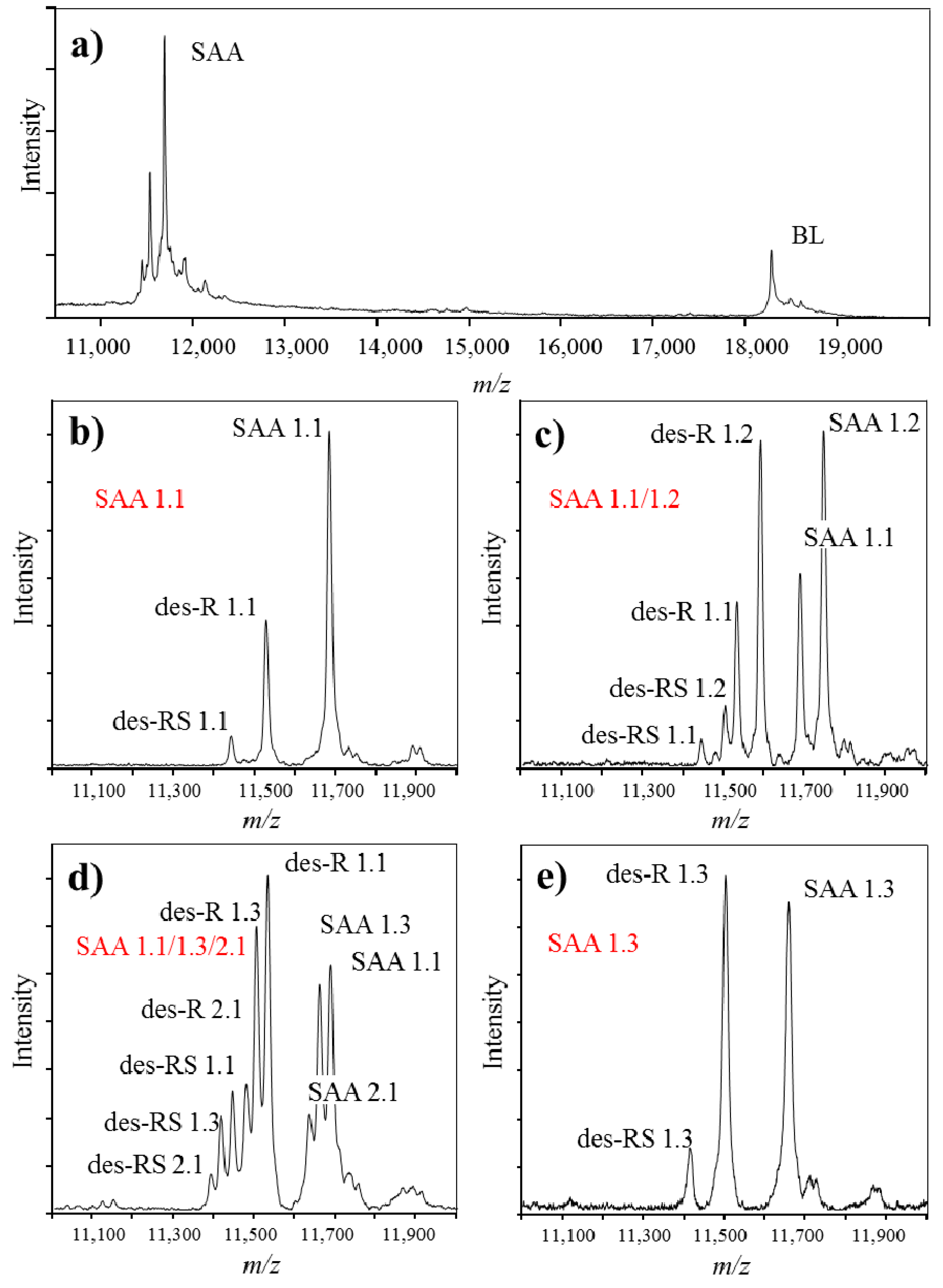
3. Identification and Analysis of Proteoforms with Mass Spectrometric Immunoassay
4. Mass Spectrometry Immunoassay for Clinically Significant Proteoforms
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| apoA-I | Apolipoprotein A-I |
| apoA-II | Apolipoprotein A-II |
| apoE | Apolipoprotein E |
| apoC-I | Apolipoprotein C-I |
| apoC-II | Apolipoprotein C-II |
| apoC-III | Apolipoprotein C-III |
| B2m | Beta 2-microglobulin |
| BNP | Brain natriuretic peptide |
| C-peptide | C-peptide |
| CRP | C-reactive protein |
| cysC | Cystatin C |
| GcG | Vitamin D-binding protein |
| IGF1 | Insulin-like growth factor 1 |
| IGF2 | Insulin-like growth factor 2 |
| MIF | Macrophage migration inhibitory factor |
| MYO | Myoglobin |
| PTH | Parathyroid hormone-related protein |
| RANTES | Regulated on Activation, Normal T Cell Expressed and Secreted |
| RBP | Retinol-binding protein |
| SAA | Serum amyloid A |
| TRFE | Transferrin |
| TTR | Transthyretin |
References
- Anderson, N.L. The roles of multiple proteomic platforms in a pipeline for new diagnostics. Mol. Cell. Proteom. 2005, 4, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Rifai, N.; Gillette, M.A.; Carr, S.A. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 24, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.; Agoram, B. Analytical assay platforms for soluble target engagement biomarkers: Old favorites and emerging technologies. Bioanalysis 2013, 5, 2919–2931. [Google Scholar] [CrossRef] [PubMed]
- Tighe, P.J.; Ryder, R.R.; Todd, I.; Fairclough, L.C. ELISA in the multiplex era: Potentials and pitfalls. Proteom. Clin. Appl. 2015, 9, 406–422. [Google Scholar] [CrossRef] [PubMed]
- Flodin, M.; Jonsson, A.S.; Hansson, L.O.; Danielsson, L.A.; Larsson, A. Evaluation of gentian cystatin c reagent on Abbott Ci8200 and calculation of glomerular filtration rate expressed in mL/min/1.73 m2 from the cystatin c values in mg/L. Scand. J. Clin. Lab. Investig. 2007, 67, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Farley, A.R.; Link, A.J. Identification and quantification of protein posttranslational modifications. Methods Enzymol. 2009, 463, 725–763. [Google Scholar] [PubMed]
- Wu, J.R.; Zeng, R. Molecular basis for population variation: From SNPS to SAPS. FEBS Lett. 2012, 586, 2841–2845. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L. Consortium for Top-Down Proteomics. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jensen, O.N. Modification-specific proteomics: Strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 2009, 9, 4632–4641. [Google Scholar] [CrossRef] [PubMed]
- Walsh, G.; Jefferis, R. Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol 2006, 24, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Committee, I.E. International Expert Committee report on the role of the A1C assay in the diagnosis of diabetes. Diabetes Care 2009, 32, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L. The clinical plasma proteome: A survey of clinical assays for proteins in plasma and serum. Clin. Chem. 2010, 56, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Meri, S. Techniques for studying protein heterogeneity and post-translational modifications. Expert Rev. Proteom. 2004, 1, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Chait, B.T. Chemistry. Mass spectrometry: Bottom-up or top-down? Science 2006, 314, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Tabb, D.L.; Yates, J.R. Large-scale protein identification using mass spectrometry. Biochim. Biophys. Acta 2003, 1646, 1–10. [Google Scholar] [CrossRef]
- Anderson, N.L.; Anderson, N.G.; Haines, L.R.; Hardie, D.B.; Olafson, R.W.; Pearson, T.W. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by Anti-peptide Antibodies (SISCAPA). J. Proteome Res. 2004, 3, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Razavi, M.; Frick, L.E.; LaMarr, W.A.; Pope, M.E.; Miller, C.A.; Anderson, N.L.; Pearson, T.W. High-throughput SISCAPA quantitation of peptides from human plasma digests by ultrafast, liquid chromatography-free mass spectrometry. J. Proteome Res. 2012, 11, 5642–5649. [Google Scholar] [CrossRef] [PubMed]
- Mann, M. Functional and quantitative proteomics using silac. Nat. Rev. Mol. Cell Biol. 2006, 7, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Hennrich, M.L.; Gavin, A.C. Quantitative mass spectrometry of posttranslational modifications: Keys to confidence. Sci. Signal. 2015, 8, re5. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Altelaar, A.F.; Frese, C.K.; Preisinger, C.; Hennrich, M.L.; Schram, A.W.; Timmers, H.T.; Heck, A.J.; Mohammed, S. Benchmarking stable isotope labeling based quantitative proteomics. J. Proteom. 2013, 88, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Fíla, J.; Honys, D. Enrichment techniques employed in phosphoproteomics. Amino Acids 2012, 43, 1025–1047. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Vitorino, R.; Domingues, M.R.; Spickett, C.M.; Domingues, P. Post-translational modifications and mass spectrometry detection. Free Radic. Biol. Med. 2013, 65, 925–941. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A. What does the future hold for Top Down mass spectrometry? J. Am. Soc. Mass Spectrom. 2010, 21, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ge, Y. Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ. Cardiovasc. Genet. 2011, 4, 711. [Google Scholar] [CrossRef] [PubMed]
- McLafferty, F.W.; Breuker, K.; Jin, M.; Han, X.; Infusini, G.; Jiang, H.; Kong, X.; Begley, T.P. Top-down MS, a powerful complement to the high capabilities of proteolysis proteomics. FEBS J 2007, 274, 6256–6268. [Google Scholar] [CrossRef] [PubMed]
- Siuti, N.; Kelleher, N.L. Decoding protein modifications using top-down mass spectrometry. Nat. Methods 2007, 4, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.J.; Håkansson, K.; Marshall, A.G. The role of electron capture dissociation in biomolecular analysis. Mass Spectrom. Rev. 2005, 24, 201–222. [Google Scholar] [CrossRef] [PubMed]
- Sarbu, M.; Ghiulai, R.M.; Zamfir, A.D. Recent developments and applications of electron transfer dissociation mass spectrometry in proteomics. Amino Acids 2014, 46, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, R.A. Electron-capture dissociation tandem mass spectrometry. Curr. Opin. Biotechnol. 2004, 15, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.; Pires, E.; Almeida, A.M.; Santos, R.; Gomes, R.; Koči, K.; Franco, C.F.; Coelho, A.V. Tandem mass spectrometry of peptides. In Tandem Mass Spectrometry—Applications and Principles; Prasain, J.K., Ed.; InTech: Rijeka, Croatia, 2012; Chapter 2; pp. 35–36. [Google Scholar]
- Elviri, L. ETD and ECD mass spectrometry fragmentation for the characterization of protein post translational modifications. In Tandem Mass Spectrometry—Applications and Principles; InTech: Rijeka, Croatia, 2012; Chapter 7; pp. 161–178. [Google Scholar]
- Na, S.; Paek, E. Software eyes for protein post-translational modifications. Mass Spectrom. Rev. 2015, 34, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Caulfield, M.P.; McPhaul, M.J.; Reitz, R.E.; Taylor, S.W.; Clarke, N.J. Quantitative insulin analysis using liquid chromatography-tandem mass spectrometry in a high-throughput clinical laboratory. Clin. Chem. 2013, 59, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Keshishian, H.; Addona, T.; Burgess, M.; Kuhn, E.; Carr, S.A. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics 2007, 6, 2212–2229. [Google Scholar] [CrossRef] [PubMed]
- Guthals, A.; Bandeira, N. Peptide identification by tandem mass spectrometry with alternate fragmentation modes. Mol Cell Proteomics 2012, 11, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Rauh, M. LC-MS/MS for protein and peptide quantification in clinical chemistry. J. Chromatogr. B 2012, 883–884, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Messana, I.; Cabras, T.; Iavarone, F.; Vincenzoni, F.; Urbani, A.; Castagnola, M. Unraveling the different proteomic platforms. J. Sep. Sci. 2013, 36, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Weiß, F.; van den Berg, B.H.; Planatscher, H.; Pynn, C.J.; Joos, T.O.; Poetz, O. Catch and measure-mass spectrometry-based immunoassays in biomarker research. Biochim. Biophys. Acta 2014, 1844, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Angel, T.E.; Aryal, U.K.; Hengel, S.M.; Baker, E.S.; Kelly, R.T.; Robinson, E.W.; Smith, R.D. Mass spectrometry-based proteomics: Existing capabilities and future directions. Chem. Soc. Rev. 2012, 41, 3912–3928. [Google Scholar] [CrossRef] [PubMed]
- Lacey, J.M.; Bergen, H.R.; Magera, M.J.; Naylor, S.; O’Brien, J.F. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin. Chem. 2001, 47, 513–518. [Google Scholar] [PubMed]
- Bystrom, C.; Sheng, S.; Zhang, K.; Caulfield, M.; Clarke, N.J.; Reitz, R. Clinical utility of insulin-like growth factor 1 and 2; determination by high resolution mass spectrometry. PLoS ONE 2012, 7, e43457. [Google Scholar] [CrossRef] [PubMed]
- Bystrom, C.E.; Sheng, S.; Clarke, N.J. Narrow mass extraction of time-of-flight data for quantitative analysis of proteins: Determination of insulin-like growth factor-1. Anal. Chem. 2011, 83, 9005–9010. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.W.; Krone, J.R.; Bieber, A.L.; Williams, P. Mass spectrometric immunoassay. Anal. Chem. 1995, 67, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.W.; Borges, C.R. Mass spectrometric immunoassay revisited. J. Am. Soc. Mass Spectrom. 2011, 22, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, K.A.; Nedelkov, D.; Nelson, R.W. Detection and quantification of β-2-microglobulin using mass spectrometric immunoassay. Anal. Biochem. 2001, 289, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Sherma, N.D.; Borges, C.R.; Trenchevska, O.; Jarvis, J.W.; Rehder, D.S.; Oran, P.E.; Nelson, R.W.; Nedelkov, D. Mass spectrometric immunoassay for the qualitative and quantitative analysis of the cytokine macrophage migration inhibitory factor (MIF). Proteome Sci. 2014, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Trenchevska, O.; Sherma, N.D.; Oran, P.E.; Reaven, P.D.; Nelson, R.W.; Nedelkov, D. Quantitative mass spectrometric immunoassay for the chemokine rantes and its variants. J. Proteom. 2015, 116, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Oran, P.E.; Trenchevska, O.; Nedelkov, D.; Borges, C.R.; Schaab, M.R.; Rehder, D.S.; Jarvis, J.W.; Sherma, N.D.; Shen, L.; Krastins, B.; et al. Parallel workflow for high-throughput (>1000 samples/day) quantitative analysis of human insulin-like growth factor 1 using mass spectrometric immunoassay. PLoS ONE 2014, 9, e92801. [Google Scholar] [CrossRef] [PubMed]
- Rehder, D.S.; Gundberg, C.M.; Booth, S.L.; Borges, C.R. Gamma-carboxylation and fragmentation of osteocalcin in human serum defined by mass spectrometry. Mol. Cell. Proteom. 2015, 14, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Rehder, D.S.; Nelson, R.W.; Borges, C.R. Glycosylation status of vitamin d binding protein in cancer patients. Protein Sci. 2009, 18, 2036–2042. [Google Scholar] [CrossRef] [PubMed]
- Borges, C.R.; Rehder, D.S.; Jarvis, J.W.; Schaab, M.R.; Oran, P.E.; Nelson, R.W. Full-length characterization of proteins in human populations. Clin. Chem. 2010, 56, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Strathmann, F.G.; Hoofnagle, A.N. Current and future applications of mass spectrometry to the clinical laboratory. Am. J. Clin. Pathol. 2011, 136, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.J.; Hoofnagle, A.N. Mass spectrometry continues its march into the clinical laboratory. Clin. Lab. Med. 2011, 31, IX–XI. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.P.; Hoofnagle, A.N. From lost in translation to paradise found: Enabling protein biomarker method transfer by mass spectrometry. Clin. Chem. 2014, 60, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Carr, S.A.; Abbatiello, S.E.; Ackermann, B.L.; Borchers, C.; Domon, B.; Deutsch, E.W.; Grant, R.P.; Hoofnagle, A.N.; Hüttenhain, R.; Koomen, J.M.; et al. Targeted peptide measurements in biology and medicine: Best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell. Proteom. 2014, 13, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, D.A.; Pry, T. Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev. 2008, 29 (Suppl. S1), S49–S52. [Google Scholar] [PubMed]
- Bland, J.M.; Altman, D.G. Measuring agreement in method comparison studies. Stat. Methods Med. Res. 1999, 8, 135–160. [Google Scholar] [CrossRef] [PubMed]
- Passing, H.; Bablok, W. A new biometrical procedure for testing the equality of measurements from two different analytical methods. Application of linear regression procedures for method comparison studies in clinical chemistry, part I. J. Clin. Chem. Clin. Biochem. 1983, 21, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Stapels, M.D.; Barofsky, D.F. Complementary use of MALDI and esi for the HPLC-MS/MS analysis of DNA-binding proteins. Anal. Chem 2004, 76, 5423–5430. [Google Scholar] [CrossRef] [PubMed]
- Dreisewerd, K. The desorption process in maldi. Chem. Rev. 2003, 103, 395–426. [Google Scholar] [CrossRef] [PubMed]
- Albrethsen, J. Reproducibility in protein profiling by maldi-tof mass spectrometry. Clin. Chem. 2007, 53, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Yassine, H.; Borges, C.R.; Schaab, M.R.; Billheimer, D.; Stump, C.; Reaven, P.; Lau, S.S.; Nelson, R. Mass spectrometric immunoassay and MRM as targeted MS-based quantitative approaches in biomarker development: Potential applications to cardiovascular disease and diabetes. Proteom. Clin. Appl. 2013, 7, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Nedelkov, D.; Shaik, S.; Trenchevska, O.; Aleksovski, V.; Mitrevski, A.; Stojanoski, K. Targeted mass spectrometric immunoassay for detection of cystatin c isoforms in cerebrospinal fluid. Open Proteom. J. 2008, 1, 54–58. [Google Scholar] [CrossRef]
- Meyer, K.; Ueland, P.M. Targeted quantification of c-reactive protein and cystatin c and its variants by immuno-MALDI-MS. Anal. Chem. 2014, 86, 5807–5814. [Google Scholar] [CrossRef] [PubMed]
- Végvári, A.; Sjödin, K.; Rezeli, M.; Malm, J.; Lilja, H.; Laurell, T.; Marko-Varga, G. Identification of a novel proteoform of prostate specific antigen (SNP-L132I) in clinical samples by multiple reaction monitoring. Mol. Cell. Proteom. 2013, 12, 2761–2773. [Google Scholar] [CrossRef] [PubMed]
- Henze, M.; Patrick, S.E.; Hinken, A.; Scruggs, S.B.; Goldspink, P.; de Tombe, P.P.; Kobayashi, M.; Ping, P.; Kobayashi, T.; Solaro, R.J. New insights into the functional significance of the acidic region of the unique N-terminal extension of cardiac troponin i. Biochim. Biophys. Acta 2013, 1833, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; McCully, M.E.; Luo, Z.; McMichael, J.; Tu, A.Y.; Daggett, V.; Regnier, M. Structural and functional consequences of cardiac troponin C l57Q and I61Q Ca2+-desensitizing variants. Arch. Biochem. Biophys. 2013, 535, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Steinkasserer, A.; Weiss, E.H.; Schwaeble, W.; Linke, R.P. Heterogeneity of human serum amyloid A protein. Five different variants from one individual demonstrated by cdna sequence analysis. Biochem. J. 1990, 268, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Malle, E.; Steinmetz, A.; Raynes, J.G. Serum amyloid A (SAA): An acute phase protein and apolipoprotein. Atherosclerosis 1993, 102, 131–146. [Google Scholar] [CrossRef]
- Raynes, J.G.; McAdam, K.P. Serum amyloid A isoforms in inflammation. Scand. J. Immunol 1991, 33, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Ducret, A.; Bruun, C.F.; Bures, E.J.; Marhaug, G.; Husby, G.; Aebersold, R. Characterization of human serum amyloid A protein isoforms separated by two-dimensional electrophoresis by liquid chromatography/electrospray ionization tandem mass spectrometry. Electrophoresis 1996, 17, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Farwig, Z.N.; McNeal, C.J.; Little, D.; Baisden, C.E.; Macfarlane, R.D. Novel truncated isoforms of constitutive serum amyloid A detected by MALDI mass spectrometry. Biochem. Biophys. Res. Commun. 2005, 332, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Uhlar, C.M.; Whitehead, A.S. Serum amyloid A, the major vertebrate acute-phase reactant. Eur. J. Biochem. 1999, 265, 501–523. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, A.I.; Rothenberg, L.S.; DeBeer, F.C.; Cantor, R.M.; Rotter, J.I.; Lusis, A.J. Association between serum amyloid A proteins and coronary artery disease: Evidence from two distinct arteriosclerotic processes. Circulation 1997, 96, 2914–2919. [Google Scholar] [CrossRef] [PubMed]
- Ristori, G.; Laurenti, F.; Stacchini, P.; Gasperini, C.; Buttinelli, C.; Pozzilli, C.; Salvetti, M. Serum amyloid A protein is elevated in relapsing-remitting multiple sclerosis. J. Neuroimmunol. 1998, 88, 9–12. [Google Scholar] [CrossRef]
- Artl, A.; Marsche, G.; Lestavel, S.; Sattler, W.; Malle, E. Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Wada, A.; Itoh, Y.; Itoh, K. Serum amyloid A1 alleles and plasma concentrations of serum amyloid A. Amyloid 1999, 6, 199–204. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, A.J.; Gallimore, J.R.; Spector, T.D.; Pepys, M.B. Genetic effects on baseline values of c-reactive protein and serum amyloid A protein: A comparison of monozygotic and dizygotic twins. Clin. Chem. 2004, 50, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, U.A.; Tubbs, K.A.; Nedelkov, D.; Niederkofler, E.E.; Nelson, R.W. Detection of novel truncated forms of human serum amyloid A protein in human plasma. FEBS Lett. 2003, 537, 166–170. [Google Scholar] [CrossRef]
- Sipe, J. Revised nomenclature for serum amyloid A (SAA). Nomenclature committee of the international society of amyloidosis. Part 2. Amyloid 1999, 6, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Trenchevska, O.; Kamcheva, E.; Nedelkov, D. Mass spectrometric immunoassay for quantitative determination of protein biomarker isoforms. J. Proteome Res. 2010, 9, 5969–5973. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, U.A.; Phillips, D.A.; Trenchevska, O.; Nedelkov, D. Quantitative mass spectrometry evaluation of human retinol binding protein 4 and related variants. PLoS ONE 2011, 6, e17282. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, U.A.; Tubbs, K.A.; Gruber, K.; Nedelkov, D.; Niederkofler, E.E.; Williams, P.; Nelson, R.W. High-throughput protein characterization using mass spectrometric immunoassay. Anal. Biochem. 2002, 301, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.W.; Nedelkov, D.; Tubbs, K.A.; Kiernan, U.A. Quantitative mass spectrometric immunoasay of insulin like growth factor 1. J. Proteome Res. 2004, 3, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Borges, C.R.; Oran, P.E.; Buddi, S.; Jarvis, J.W.; Schaab, M.R.; Rehder, D.S.; Rogers, S.P.; Taylor, T.; Nelson, R.W. Building multidimensional biomarker views of type 2 diabetes on the basis of protein microheterogeneity. Clin. Chem. 2011, 57, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Yassine, H.N.; Jackson, A.M.; Reaven, P.D.; Nedelkov, D.; Nelson, R.W.; Lau, S.S.; Borchers, C.H. The application of multiple reaction monitoring to assess ApoA-I methionine oxidations in diabetes and cardiovascular disease. Transl. Proteom. 2014, 4–5, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Yassine, H.N.; Trenchevska, O.; Ramrakhiani, A.; Parekh, A.; Koska, J.; Walker, R.W.; Billheimer, D.; Reaven, P.D.; Yen, F.T.; Nelson, R.W.; et al. The association of human apolipoprotein C-III sialylation proteoforms with plasma triglycerides. PLoS ONE 2015, 10, e0144138. [Google Scholar] [CrossRef] [PubMed]
- Niederkofler, E.E.; Tubbs, K.A.; Gruber, K.; Nedelkov, D.; Kiernan, U.A.; Williams, P.; Nelson, R.W. Determination of β-2 microglobulin levels in plasma using a high-throughput mass spectrometric immunoassay system. Anal. Chem. 2001, 73, 3294–3299. [Google Scholar] [CrossRef] [PubMed]
- Niederkofler, E.E.; Kiernan, U.A.; O’Rear, J.; Menon, S.; Saghir, S.; Protter, A.A.; Nelson, R.W.; Schellenberger, U. Detection of endogenous b-type natriuretic peptide at very low concentrations in patients with heart failure. Circ. Heart Fail. 2008, 1, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Oran, P.E.; Jarvis, J.W.; Borges, C.R.; Nelson, R.W. C-peptide microheterogeneity in type 2 diabetes populations. Proteom. Clin. Appl. 2010, 4, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, U.A.; Addobbati, R.; Nedelkov, D.; Nelson, R.W. Quantitative multiplexed c-reactive protein mass spectrometric immunoassay. J. Proteome Res. 2006, 5, 1682–1687. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, U.A.; Nedelkov, D.; Niederkofler, E.E.; Tubbs, K.A.; Nelson, R.W. High-throughput affinity mass spectrometry. Methods Mol. Biol. 2006, 328, 141–150. [Google Scholar] [PubMed]
- Trenchevska, O.; Nedelkov, D. Targeted quantitative mass spectrometric immunoassay for human protein variants. Proteome Sci. 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Borges, C.R.; Jarvis, J.W.; Oran, P.E.; Nelson, R.W. Population studies of vitamin d binding protein microheterogeneity by mass spectrometry lead to characterization of its genotype-dependent O-glycosylation patterns. J. Proteome Res. 2008, 7, 4143–4153. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, K.A.; Kiernan, U.A.; Niederkofler, E.E.; Nedelkov, D.; Bieber, A.L.; Nelson, R.W. High-throughput MS-based protein phenotyping: Application to haptoglobin. Proteomics 2005, 5, 5002–5007. [Google Scholar] [CrossRef] [PubMed]
- Oran, P.E.; Jarvis, J.W.; Borges, C.R.; Sherma, N.D.; Nelson, R.W. Mass spectrometric immunoassay of intact insulin and related variants for population proteomics studies. Proteom. Clin. Appl. 2011, 5, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.F.; Rezai, T.; Sarracino, D.A.; Prakash, A.; Krastins, B.; Athanas, M.; Singh, R.J.; Barnidge, D.R.; Oran, P.; Borges, C.; et al. Selected reaction monitoring-mass spectrometric immunoassay responsive to parathyroid hormone and related variants. Clin. Chem. 2010, 56, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Oran, P.E.; Sherma, N.D.; Borges, C.R.; Jarvis, J.W.; Nelson, R.W. Intrapersonal and populational heterogeneity of the chemokine rantes. Clin. Chem. 2010, 56, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, U.A.; Tubbs, K.A.; Nedelkov, D.; Niederkofler, E.E.; Nelson, R.W. Comparative phenotypic analyses of human plasma and urinary retinol binding protein using mass spectrometric immunoassay. Biochem. Biophys. Res. Commun. 2002, 297, 401–405. [Google Scholar] [CrossRef]
- Yassine, H.N.; Trenchevska, O.; He, H.; Borges, C.R.; Nedelkov, D.; Mack, W.; Kono, N.; Koska, J.; Reaven, P.D.; Nelson, R.W. Serum amyloid A truncations in type 2 diabetes mellitus. PLoS ONE 2015, 10, e0115320. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, U.A.; Nedelkov, D.; Tubbs, K.A.; Niederkofler, E.E.; Nelson, R.W. Proteomic characterization of novel serum amyloid p component variants from human plasma and urine. Proteomics 2004, 4, 1825–1829. [Google Scholar] [CrossRef] [PubMed]
- Trenchevska, O.; Kamcheva, E.; Nedelkov, D. Mass spectrometric immunoassay for quantitative determination of transthyretin and its variants. Proteomics 2011, 11, 3633–3641. [Google Scholar] [CrossRef] [PubMed]
- Trenchevska, O.; Schaab, M.R.; Nelson, R.W.; Nedelkov, D. Development of multiplex mass spectrometric immunoassay for detection and quantification of apolipoproteins C-I, C-II, C-III and their proteoforms. Methods 2015, 81, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Niederkofler, E.E.; Tubbs, K.A.; Kiernan, U.A.; Nedelkov, D.; Nelson, R.W. Novel mass spectrometric immunoassays for the rapid structural characterization of plasma apolipoproteins. J. Lipid Res. 2003, 44, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Nedelkov, D.; Nelson, R.W.; Kiernan, U.A.; Niederkofler, E.E.; Tubbs, K.A. Detection of bound and free IGF-1 and IGF-2 in human plasma via biomolecular interaction analysis mass spectrometry. FEBS Lett. 2003, 536, 130–134. [Google Scholar] [CrossRef]
- Kiernan, U.A.; Nedelkov, D.; Nelson, R.W. Multiplexed mass spectrometric immunoassay in biomarker research: A novel approach to the determination of a myocardial infarct. J. Proteome Res. 2006, 5, 2928–2934. [Google Scholar] [CrossRef] [PubMed]
- Nedelkov, D.; Tubbs, K.A.; Niederkofler, E.E.; Kiernan, U.A.; Nelson, R.W. High-throughput comprehensive analysis of human plasma proteins: A step toward population proteomics. Anal. Chem. 2004, 76, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Trenchevska, O.; Phillips, D.A.; Nelson, R.W.; Nedelkov, D. Delineation of concentration ranges and longitudinal changes of human plasma protein variants. PLoS ONE 2014, 9, e100713. [Google Scholar] [CrossRef] [PubMed]
- Krastins, B.; Prakash, A.; Sarracino, D.A.; Nedelkov, D.; Niederkofler, E.E.; Kiernan, U.A.; Nelson, R.; Vogelsang, M.S.; Vadali, G.; Garces, A.; et al. Rapid development of sensitive, high-throughput, quantitative and highly selective mass spectrometric targeted immunoassays for clinically important proteins in human plasma and serum. Clin. Biochem. 2013, 46, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Nedelkov, D. Population proteomics: Investigation of protein diversity in human populations. Proteomics 2008, 8, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Nedelkov, D.; Phillips, D.A.; Tubbs, K.A.; Nelson, R.W. Investigation of human protein variants and their frequency in the general population. Mol. Cell. Proteom. 2007, 6, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Nedelkov, D.; Kiernan, U.A.; Niederkofler, E.E.; Tubbs, K.A.; Nelson, R.W. Investigating diversity in human plasma proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 10852–10857. [Google Scholar] [CrossRef] [PubMed]
- Nedelkov, D. Population proteomics: Addressing protein diversity in humans. Expert Rev. Proteom. 2005, 2, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Nedelkov, D.; Kiernan, U.A.; Niederkofler, E.E.; Tubbs, K.A.; Nelson, R.W. Population proteomics: The concept, attributes, and potential for cancer biomarker research. Mol. Cell. Proteom. 2006, 5, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Jong, M.C.; Hofker, M.H.; Havekes, L.M. Role of ApoCs in lipoprotein metabolism: Functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Bondarenko, P.V.; Cockrill, S.L.; Watkins, L.K.; Cruzado, I.D.; Macfarlane, R.D. Mass spectral study of polymorphism of the apolipoproteins of very low density lipoprotein. J. Lipid Res. 1999, 40, 543–555. [Google Scholar] [PubMed]
- Surinova, S.; Schiess, R.; Hüttenhain, R.; Cerciello, F.; Wollscheid, B.; Aebersold, R. On the development of plasma protein biomarkers. J. Proteome Res. 2011, 10, 5–16. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
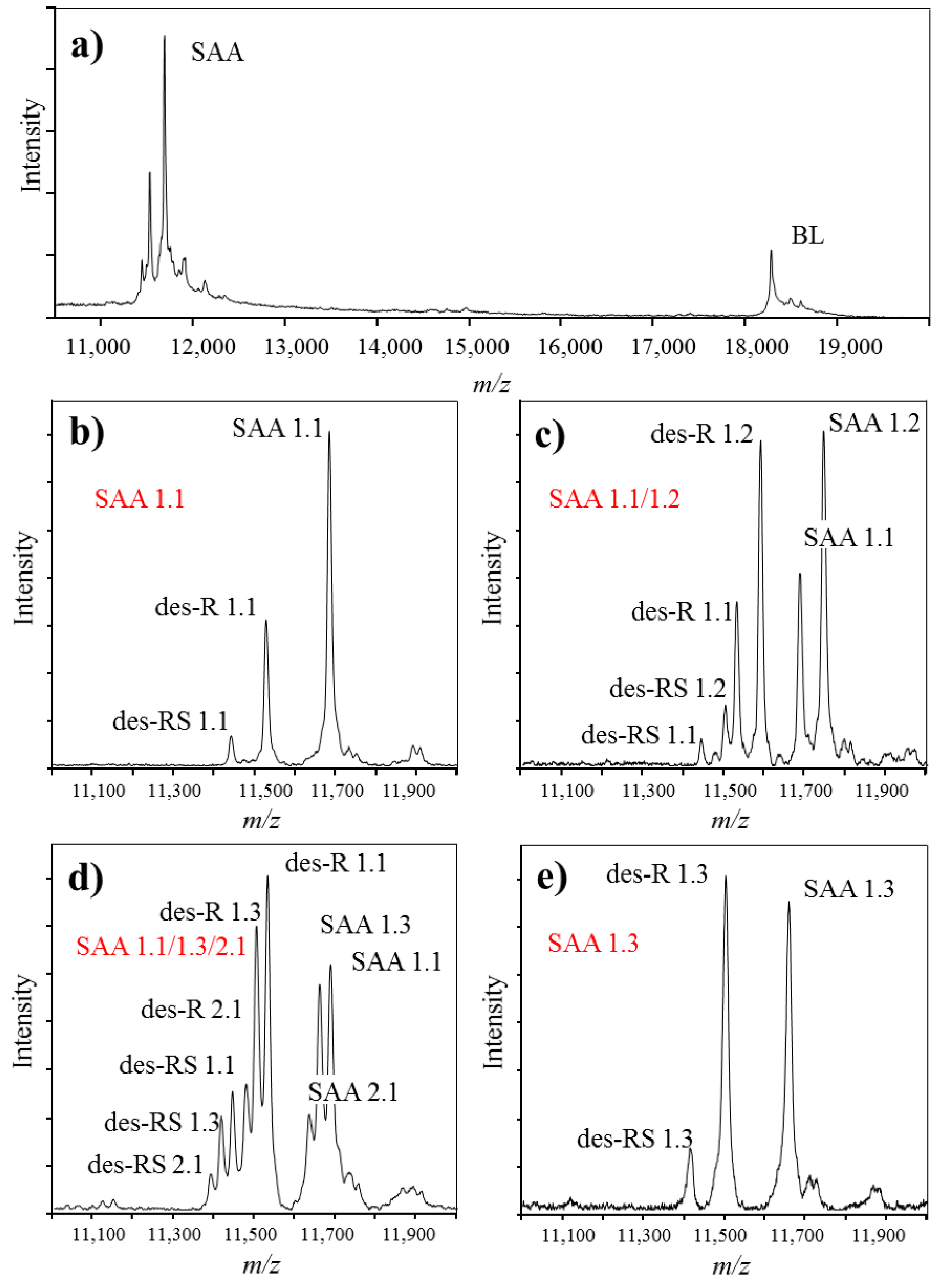{kind=link}
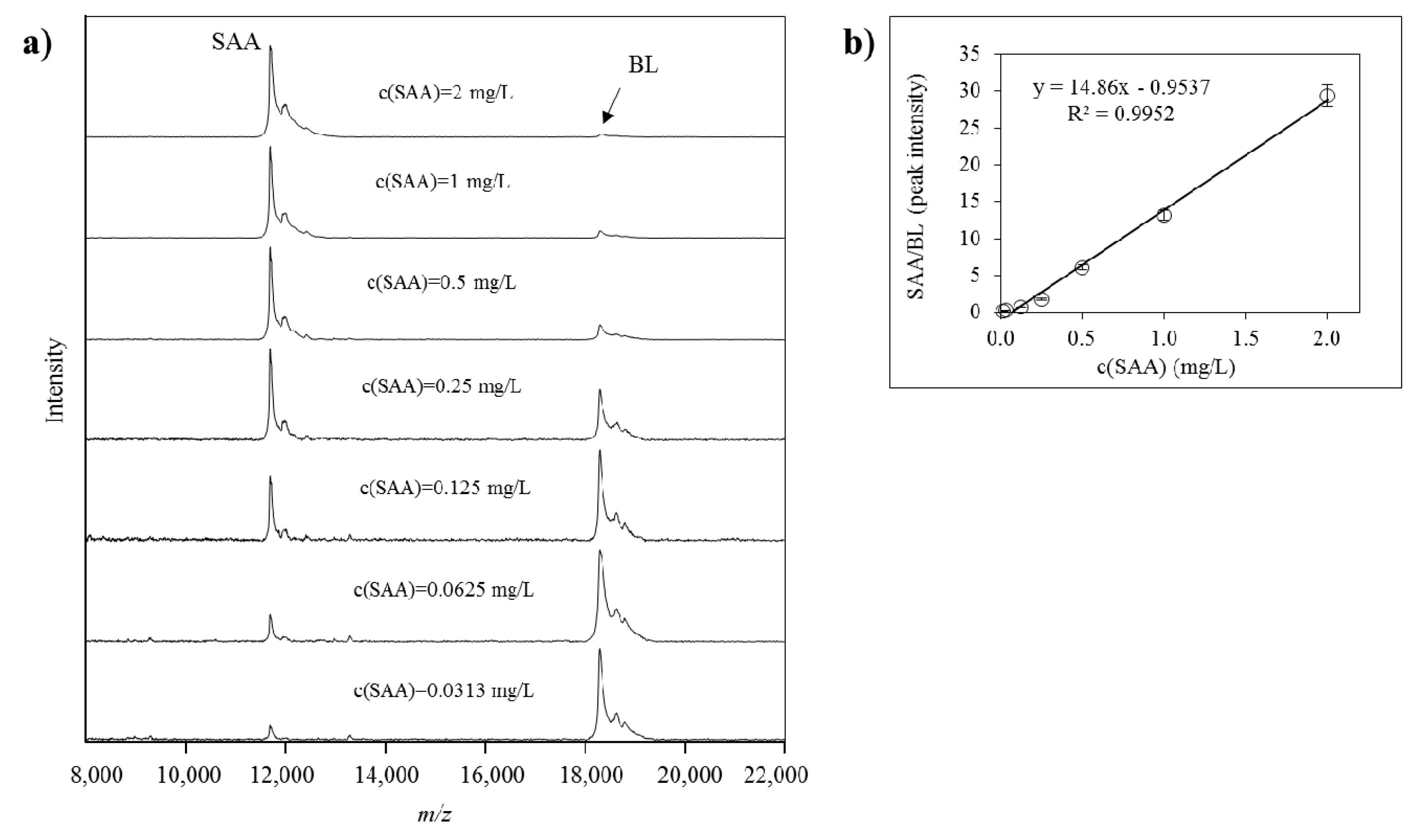{kind=link}
| Protein Target(s) * | MSIA Approach | Study | Reference |
|---|---|---|---|
| Single protein assays | |||
| Apolipoprotein A-I | Qualitative | Clinical application | [ 90] |
| Apolipoprotein C-III | Quantitative | Clinical application | [ 91] |
| Beta 2-microglobulin | Qualitative
Quantitative | Method development and application Method development | [
92] [85] |
| Brain natriuretic peptide | Quantitative | Method development and application | [ 93] |
| C-peptide | Qualitative | Method development and application | [ 94] |
| C-reactive protein | Quantitative | Method development and application | [ 95] |
| Cystatin C | Qualitative
Quantitative | Population proteomics Method development and application | [
96] [97] |
| Vitamin D-binding protein | Qualitative | Clinical proteomics | [ 54,55,98] |
| Haptoglobin | Qualitative | Method development and application | [ 99] |
| Insulin-like growth factor 1 | Quantitative | Method development Population proteomics | [
88] [52] |
| Insulin | Qualitative
Quantitative | Method development and application | [ 100] |
| Macrophage migration inhibitory factor | Quantitative | Method development | [ 50] |
| Osteocalcin | Qualitative | Method development and application | [ 53] |
| Parathyroid hormone-related protein | Qualitative | Method development and application | [ 101] |
| Regulated on activation, normal T cell expressed and secreted | Qualitative
Quantitative | Clinical proteomics Method development | [
102] [51] |
| Retinol-binding protein | Qualitative
Quantitative | Method development and application Method development | [
103] [86] |
| Serum amyloid A | Qualitative
Quantitative | Method development and application Clinical application | [
83] [104] |
| Serum amyloid P | Qualitative | Method development | [ 105] |
| Transthyretin | Quantitative | Method development | [ 106] |
| Multiplexed assays ** | |||
| Apolipoprotein C-I Apolipoprotein C-II Apolipoprotein C-III | Qualitative
Quantitative | Method development Method development | [ 107] |
| Apolipoprotein A-I, Apolipoprotein A-II, Apolipoprotein E | Qualitative | Method development and application | [ 108] |
| Insulin-like growth factor 1, Insulin-like growth factor 2 | Qualitative | Method development | [ 88,109] |
| Serum amyloid A, Transthyretin, Myoglobin | Qualitative | Method development and application | [ 110] |
| Multiple protein targets *** | |||
| Transthyretin, Transferrin | Qualitative | Method application, population proteomics | [ 111] |
| Transthyretin, Retinol-binding protein | Qualitative | Method development and application | [ 87] |
| Multiple targets | Quantitative | Population proteomics | [ 112,113,114,115,116,117] |
| Multiple targets | Qualitative | Clinical proteomics | [ 89] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trenchevska, O.; Nelson, R.W.; Nedelkov, D. Mass Spectrometric Immunoassays in Characterization of Clinically Significant Proteoforms. Proteomes 2016, 4, 13. https://doi.org/10.3390/proteomes4010013
Trenchevska O, Nelson RW, Nedelkov D. Mass Spectrometric Immunoassays in Characterization of Clinically Significant Proteoforms. Proteomes. 2016; 4(1):13. https://doi.org/10.3390/proteomes4010013
Chicago/Turabian StyleTrenchevska, Olgica, Randall W. Nelson, and Dobrin Nedelkov. 2016. "Mass Spectrometric Immunoassays in Characterization of Clinically Significant Proteoforms" Proteomes 4, no. 1: 13. https://doi.org/10.3390/proteomes4010013
APA StyleTrenchevska, O., Nelson, R. W., & Nedelkov, D. (2016). Mass Spectrometric Immunoassays in Characterization of Clinically Significant Proteoforms. Proteomes, 4(1), 13. https://doi.org/10.3390/proteomes4010013