

Advances in the Study of Protein Deamidation: Unveiling Its Influence on Aging, Disease Progression, Forensics and Therapeutic Efficacy


Abstract
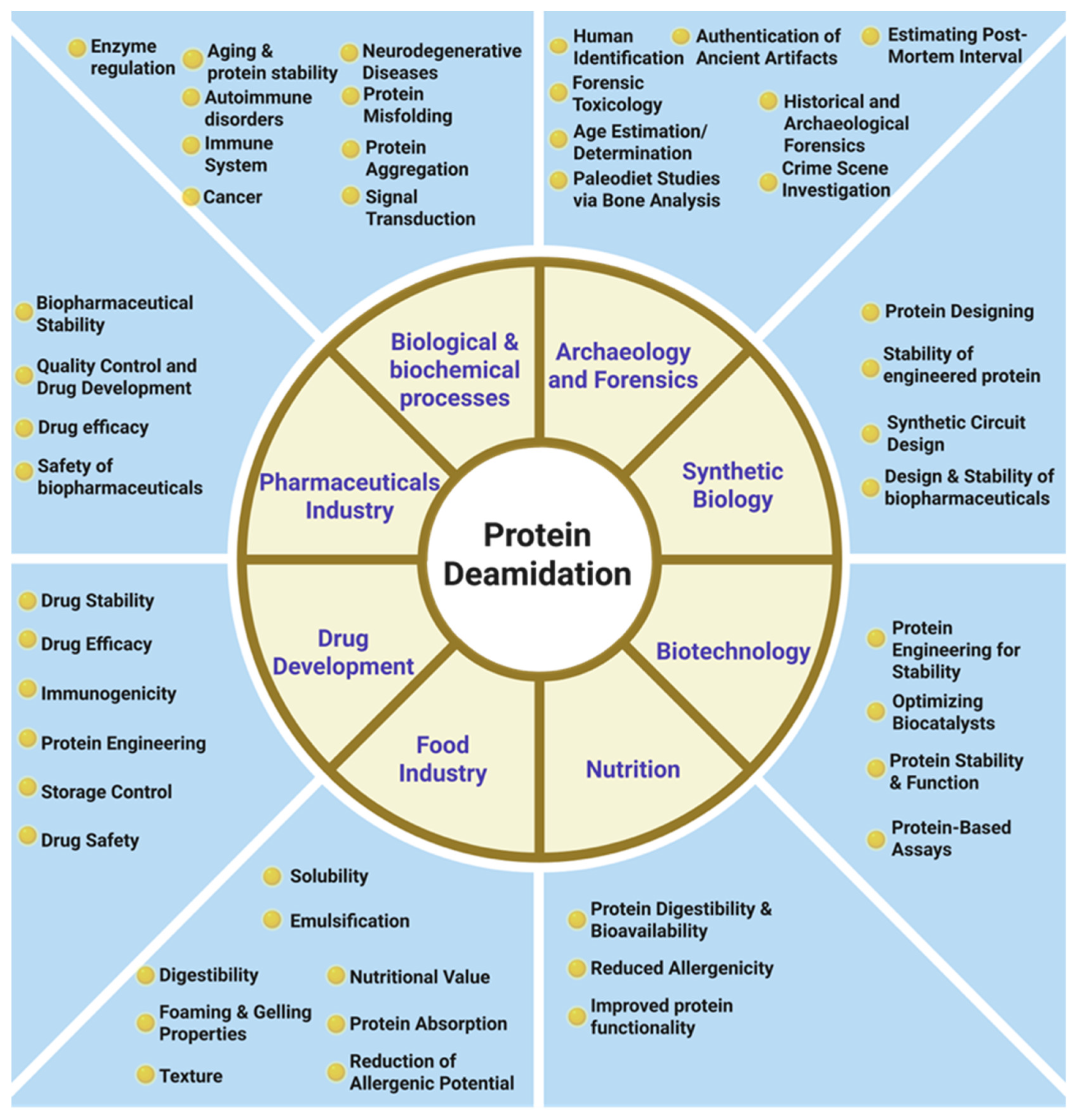
1. Introduction
2. Molecular Mechanisms of Protein Deamidation
3. Role of Deamidation in Protein Aging
3.1. Deamidation and Neurodegenerative Diseases
3.2. Deamidated Proteins as a Biomarker
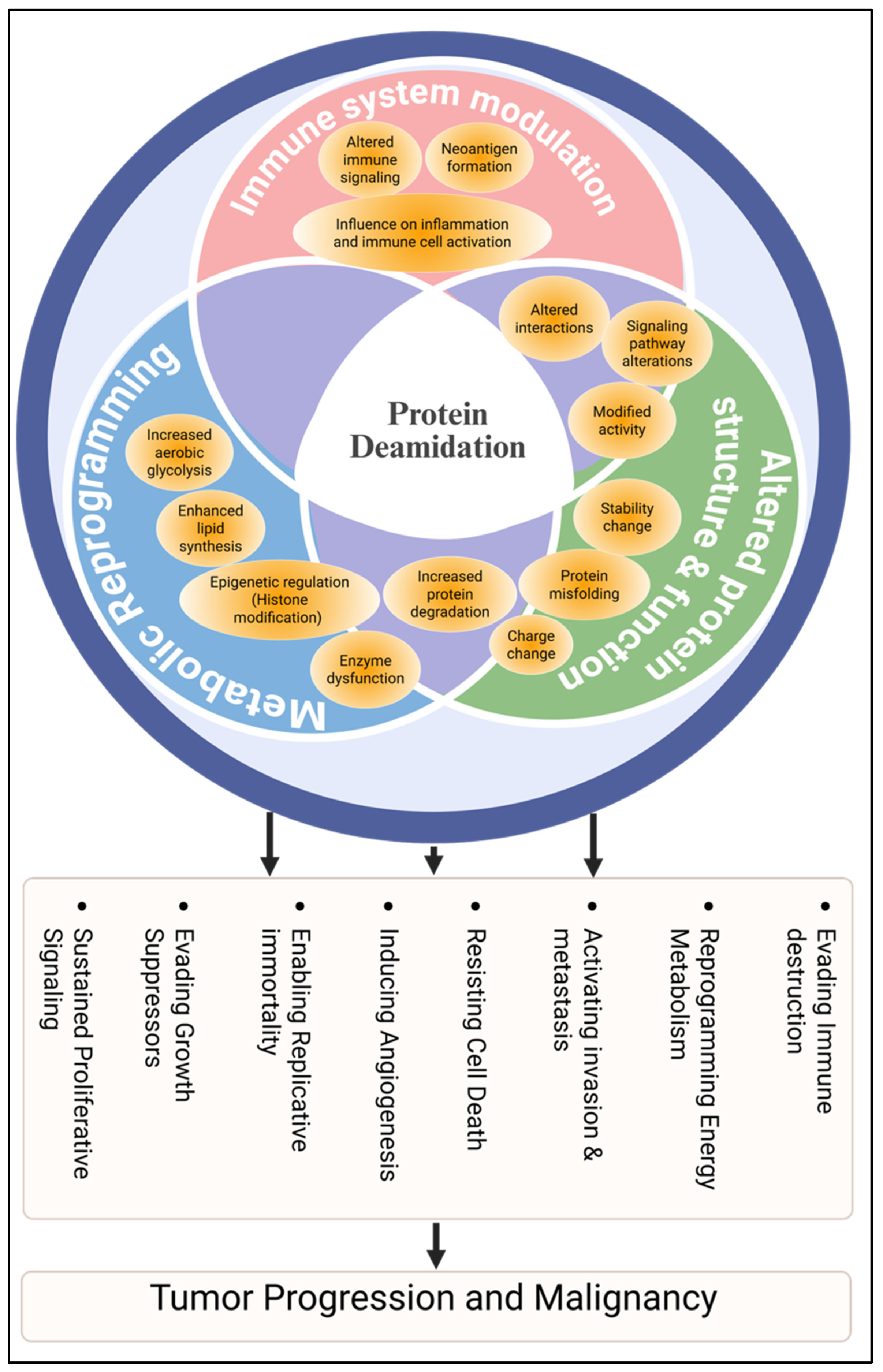
4. Protein Deamidation in Cancer
5. Protein Deamidation in Forensics and Archeology
6. Protein Deamidation in Biopharmaceuticals and Drug Development
7. Future Perspective
Funding
Data Availability Statement
Conflicts of Interest
References
- Adav, S.S.; Sze, S.K. Hypoxia-induced degenerative protein modifications associated with aging and age-associated disorders. Aging Dis. 2020, 11, 341–364. [Google Scholar] [CrossRef]
- Mycek, M.J.; Waelsch, H. The enzymatic deamidation of proteins. J. Biol. Chem. 1960, 235, 3513–3517. [Google Scholar] [CrossRef]
- Robinson, A.B.; Scotchler, J.W.; McKerrow, J.H. Rates of nonenzymatic deamidation of glutaminyl and asparaginyl residues in pentapeptides. J. Am. Chem. Soc. 1973, 95, 8156–8159. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E. Protein deamidation. Proc. Natl. Acad. Sci. USA 2002, 99, 5283–5288. [Google Scholar] [CrossRef]
- Robinson, N.E.; Robinson, A.B. Molecular clocks. Proc. Natl. Acad. Sci. USA 2001, 98, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, A.B. Prediction of protein deamidation rates from primary and three-dimensional structure. Proc. Natl. Acad. Sci. USA 2001, 98, 4367–4372. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, A.B. Deamidation of human proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 12409–12413. [Google Scholar] [CrossRef]
- Robinson, N.E.; Robinson, A.B.; Merrifield, R.B. Mass spectrometric evaluation of synthetic peptides as primary structure models for peptide and protein deamidation. J. Pept. Res. 2001, 57, 483–493. [Google Scholar] [CrossRef]
- Robinson, N.E.; Robinson, A.B. Amide molecular clocks in drosophila proteins: Potential regulators of aging and other processes. Mech. Ageing Dev. 2004, 125, 259–267. [Google Scholar] [CrossRef]
- Kalailingam, P.; Mohd-Kahliab, K.H.; Ngan, S.C.; Iyappan, R.; Melekh, E.; Lu, T.; Zien, G.W.; Sharma, B.; Guo, T.; MacNeil, A.J. Immunotherapy targeting isoDGR-protein damage extends lifespan in a mouse model of protein deamidation. EMBO Mol. Med. 2023, 15, e18526. [Google Scholar] [CrossRef]
- Kato, K.; Nakayoshi, T.; Kitamura, Y.; Kurimoto, E.; Oda, A.; Ishikawa, Y. Identification of the most impactful asparagine residues for γS-crystallin aggregation by deamidation. Biochemistry 2023, 62, 1679–1688. [Google Scholar] [CrossRef]
- Shimizu, T.; Watanabe, A.; Ogawara, M.; Mori, H.; Shirasawa, T. Isoaspartate formation and neurodegeneration in Alzheimer’s disease. Arch. Biochem. Biophys. 2000, 381, 225–234. [Google Scholar] [CrossRef]
- Robinson, N.E.; Robinson, M.L.; Schulze, S.E.; Lai, B.T.; Gray, H.B. Deamidation of alpha-synuclein. Protein Sci. 2009, 18, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Enríquez-Flores, S.; De la Mora-De la Mora, I.; García-Torres, I.; Flores-López, L.A.; Martínez-Pérez, Y.; López-Velázquez, G. Human triosephosphate isomerase is a potential target in cancer due to commonly occurring post-translational modifications. Molecules 2023, 28, 6163. [Google Scholar] [CrossRef]
- Zafar, S.; Fatima, S.I.; Schmitz, M.; Zerr, I. Current technologies unraveling the significance of post-translational modifications (PTMs) as crucial players in neurodegeneration. Biomolecules 2024, 14, 118. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Machiesky, L.A.; De Mel, N.; Du, Q.; Xu, W.; Washabaugh, M.; Jiang, X.-R.; Wang, J. Characterization of IgG1 Fc deamidation at asparagine 325 and its impact on antibody-dependent cell-mediated cytotoxicity and FcγRIIIa binding. Sci. Rep. 2020, 10, 383. [Google Scholar] [CrossRef]
- Giles, A.R.; Sims, J.J.; Turner, K.B.; Govindasamy, L.; Alvira, M.R.; Lock, M.; Wilson, J.M. Deamidation of amino acids on the surface of adeno-associated virus capsids leads to charge heterogeneity and altered vector function. Mol. Ther. 2018, 26, 2848–2862. [Google Scholar] [CrossRef] [PubMed]
- Narciso, J.O.; Gulzar, S.; Soliva-Fortuny, R.; Martín-Belloso, O. Emerging chemical, biochemical, and non-thermal physical treatments in the production of hypoallergenic plant protein ingredients. Foods 2024, 13, 2180. [Google Scholar] [CrossRef]
- Beaumatin, F.; El Dhaybi, M.; Bobo, C.; Verdier, M.; Priault, M. Bcl-xL deamidation and cancer: Charting the fame trajectories of legitimate child and hidden siblings. Biochim. Biophys. Acta 2017, 1864, 1734–1745. [Google Scholar] [CrossRef]
- Butreddy, A.; Janga, K.Y.; Ajjarapu, S.; Sarabu, S.; Dudhipala, N. Instability of therapeutic proteins—An overview of stresses, stabilization mechanisms and analytical techniques involved in Lyophilized proteins. Int. J. Biol. Macromol. 2021, 167, 309–325. [Google Scholar] [CrossRef]
- Wakankar, A.A.; Borchardt, R.T. Formulation considerations for proteins susceptible to asparagine deamidation and aspartate isomerization. J. Pharm. Sci. 2006, 95, 2321–2336. [Google Scholar] [CrossRef]
- Liu, Y.D.; van Enk, J.Z.; Flynn, G.C. Human antibody Fc deamidation in vivo. Biologicals 2009, 37, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Liu, C.; Zhang, W.; Li, Y.; Liu, J. Improving water solubility of Ddocosahexaenoic acid with chickpea protein isolates deamidated by protein-glutaminase. ACS Food. Sci. Technol. 2024, 4, 2690–2698. [Google Scholar] [CrossRef]
- Zhao, M.; He, H.; Ma, A.; Hou, T. Sources, chemical synthesis, functional improvement and applications of food-derived protein/peptide-saccharide covalent conjugates: A review. Crit. Rev. Food Sci. Nutr. 2023, 63, 5985–6004. [Google Scholar] [CrossRef]
- Giuffrida, M.G.; Mazzoli, R.; Pessione, E. Back to the past: Deciphering cultural heritage secrets by protein identification. Appl. Microbiol. Biotechnol. 2018, 102, 5445–5455. [Google Scholar] [CrossRef] [PubMed]
- Leo, G.; Bonaduce, I.; Andreotti, A.; Marino, G.; Pucci, P.; Colombini, M.P.; Birolo, L. Deamidation at asparagine and glutamine as a major modification upon deterioration/aging of proteinaceous binders in mural paintings. AnaCh 2011, 83, 2056–2064. [Google Scholar] [CrossRef]
- Leo, G.; Cartechini, L.; Pucci, P.; Sgamellotti, A.; Marino, G.; Birolo, L. Proteomic strategies for the identification of proteinaceous binders in paintings. Anal. Bioanal. Chem. 2009, 395, 2269–2280. [Google Scholar] [CrossRef]
- Adav, S.S.; Qian, J.; Ang, Y.L.; Kalaria, R.N.; Lai, M.K.; Chen, C.P.; Sze, S.K. iTRAQ quantitative clinical proteomics revealed role of Na(+)K(+)-ATPase and its correlation with deamidation in vascular dementia. J. Proteome Res. 2014, 13, 4635–4646. [Google Scholar] [CrossRef]
- Adav, S.S.; Gallart-Palau, X.; Tan, K.H.; Lim, S.K.; Tam, J.P.; Sze, S.K. Dementia-linked amyloidosis is associated with brain protein deamidation as revealed by proteomic profiling of human brain tissues. Mol. Brain 2016, 9, 20. [Google Scholar] [CrossRef]
- Takata, T.; Oxford, J.T.; Brandon, T.R.; Lampi, K.J. Deamidation alters the structure and decreases the stability of human lens betaA3-crystallin. Biochemistry 2007, 46, 8861–8871. [Google Scholar] [CrossRef]
- Takata, T.; Oxford, J.T.; Demeler, B.; Lampi, K.J. Deamidation destabilizes and triggers aggregation of a lens protein, betaA3-crystallin. Protein Sci. 2008, 17, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Dutta, B.; Park, J.E.; Kumar, S.; Hao, P.; Gallart-Palau, X.; Serra, A.; Ren, Y.; Sorokin, V.; Lee, C.N.; Ho, H.H.; et al. Monocyte adhesion to atherosclerotic matrix proteins is enhanced by Asn-Gly-Arg deamidation. Sci. Rep. 2017, 7, 5765. [Google Scholar] [CrossRef]
- Hao, P.; Adav, S.S.; Gallart-Palau, X.; Sze, S.K. Recent advances in mass spectrometric analysis of protein deamidation. Mass. Spectrom. Rev. 2017, 36, 677–692. [Google Scholar] [CrossRef]
- Jin, Y.; Yi, Y.; Yeung, B. Mass spectrometric analysis of protein deamidation—A focus on top-down and middle-down mass spectrometry. Methods 2022, 200, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Lindner, H.; Helliger, W. Age-dependent deamidation of asparagine residues in proteins. Exp. Gerontol. 2001, 36, 1551–1563. [Google Scholar] [CrossRef]
- Catak, S.; Monard, G.; Aviyente, V.; Ruiz-López, M.F. Deamidation of asparagine residues: Direct hydrolysis versus succinimide-mediated deamidation mechanisms. J. Phys. Chem. A 2009, 113, 1111–1120. [Google Scholar] [CrossRef]
- Oliyai, C.; Borchardt, R.T. Chemical pathways of peptide degradation. IV. Pathways, kinetics, and mechanism of degradation of an aspartyl residue in a model hexapeptide. Pharm. Res. 1993, 10, 95–102. [Google Scholar] [CrossRef]
- Takahashi, O.; Kirikoshi, R.; Manabe, N. Racemization of the succinimide intermediate formed in proteins and peptides: A computational study of the mechanism catalyzed by dihydrogen phosphate ion. Int. J. Mol. Sci. 2016, 17, 1698. [Google Scholar] [CrossRef]
- Li, X.; Lin, C.; O’Connor, P.B. Glutamine deamidation: Differentiation of glutamic acid and gamma-glutamic acid in peptides by electron capture dissociation. Anal. Chem. 2010, 82, 3606–3615. [Google Scholar] [CrossRef]
- Hao, P.; Ren, Y.; Alpert, A.J.; Sze, S.K. Detection, evaluation and minimization of nonenzymatic deamidation in proteomic sample preparation. Mol. Cell. Proteomics 2011, 10, O111.009381. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Alves, J.; Patel, R.; Adams, M.; Nashine, V.; Goueli, S. A high-throughput bioluminescent assay to monitor the deamidation of asparagine and isomerization of aspartate residues in therapeutic proteins and antibodies. J. Pharm. Sci. 2017, 106, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.M.; Ozumerzifon, T.J.; Henry, C.S.; Manning, M.C. High throughput detection of deamidation using S-(5′-adenosyl)-l-homocysteine hydrolase and a fluorogenic reagent. J. Pharm. Biomed. Anal. 2018, 156, 323–327. [Google Scholar] [CrossRef]
- Sze, S.K.; JebaMercy, G.; Ngan, S.C. Profiling the ‘deamidome’ of complex biosamples using mixed-mode chromatography-coupled tandem mass spectrometry. Methods 2022, 200, 31–41. [Google Scholar] [CrossRef]
- Ying, Y.; Li, H. Recent progress in the analysis of protein deamidation using mass spectrometry. Methods 2022, 200, 42–57. [Google Scholar] [CrossRef]
- Viña, J.; Borrás, C.; Miquel, J. Theories of ageing. IUBMB Life 2007, 59, 249–254. [Google Scholar] [CrossRef]
- Jin, K. Modern biological theories of aging. Aging Dis. 2010, 1, 72–74. [Google Scholar] [PubMed]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef]
- Brunk, U.T.; Terman, A. The mitochondrial-lysosomal axis theory of aging: Accumulation of damaged mitochondria as a result of imperfect autophagocytosis. EJBio 2002, 269, 1996–2002. [Google Scholar] [CrossRef]
- Hipkiss, A.R. Energy metabolism and ageing regulation: Metabolically driven deamidation of triosephosphate isomerase may contribute to proteostatic dysfunction. Ageing Res. Rev. 2011, 10, 498–502. [Google Scholar] [CrossRef]
- Hipkiss, A.R. Accumulation of altered proteins and ageing: Causes and effects. Exp. Gerontol. 2006, 41, 464–473. [Google Scholar] [CrossRef]
- Moskalev, A.A.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Yanai, H.; Fraifeld, V.E. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev. 2013, 12, 661–684. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Aryal, B.; Jeong, J.; Rao, V.A. Doxorubicin-induced carbonylation and degradation of cardiac myosin binding protein C promote cardiotoxicity. Proc.Natl. Acad. Sci. USA 2014, 111, 2011–2016. [Google Scholar] [CrossRef]
- Watanabe, A.; Takio, K.; Ihara, Y. Deamidation and isoaspartate formation in smeared Tau in paired helical filaments: Unusual properties of the microtubule-binding domain of tau. J. Biol. Chem. 1999, 274, 7368–7378. [Google Scholar] [CrossRef]
- Joseph, D. The fundamental neurobiological mechanism of oxidative stress-related 4E-BP2 protein deamidation. Int. J. Mol. Sci. 2024, 25, 12268. [Google Scholar] [CrossRef]
- Flatmark, T.; Sletten, K. Multiple forms of cytochrome c in the rat: Precursor-product relationship between the main component cy i and the minor components cy ii and cy iii in vivo. J. Biol. Chem. 1968, 243, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.B.; McKerrow, J.H.; Legaz, M. Sequence dependent deamidation rates for model peptides of cytochrome c. Int. J. Pept. Protein Res. 1974, 6, 31–35. [Google Scholar] [CrossRef]
- McKerrow, J.H.; Robinson, A.B. Primary sequence dependence of the deamidation of rabbit muscle aldolase. Science 1974, 183, 85. [Google Scholar] [CrossRef]
- Sun, A.Q.; Yüksel, K.U.; Gracy, R.W. Terminal marking of triosephosphate isomerase: Consequences of deamidation. Arch. Biochem. Biophys. 1995, 322, 361–368. [Google Scholar] [CrossRef]
- Deverman, B.E.; Cook, B.L.; Manson, S.R.; Niederhoff, R.A.; Langer, E.M.; Rosová, I.; Kulans, L.A.; Fu, X.; Weinberg, J.S.; Heinecke, J.W.; et al. Bcl-xL deamidation is a critical switch in the regulation of the response to DNA damage. Cell 2002, 111, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Wilmarth, P.; Tanner, S.; Dasari, S.; Nagalla, S.; Riviere, M.; Bafna, V.; Pevzner, P.; David, L. Age-related changes in human crystallins determined from comparative analysis of post-translational modifications in young and aged lens: Does deamidation contribute to crystallin insolubility? J. Proteome Res. 2006, 5, 2554–2566. [Google Scholar] [CrossRef]
- Lampi, K.J.; Ma, Z.; Shih, M.; Shearer, T.R.; Smith, J.B.; Smith, D.L.; David, L.L. Sequence analysis of betaA3, betaB3, and betaA4 crystallins completes the identification of the major proteins in young human lens. J. Biol. Chem. 1997, 272, 2268–2275. [Google Scholar] [CrossRef]
- Robinson, A.B.; Rudd, C.J. Deamidation of glutaminyl and asparaginyl residues in peptides and proteins. Curr. Top. Cell. Regul. 1974, 8, 247–295. [Google Scholar] [PubMed]
- Lampi, K.J.; Oxford, J.T.; Bachinger, H.P.; Shearer, T.R.; David, L.L.; Kapfer, D.M. Deamidation of human beta B1 alters the elongated structure of the dimer. Exp. Eye Res. 2001, 72, 279–288. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kapfer, D.M.; Boekhorst, J.; Lubsen, N.H.; Bächinger, H.P.; Shearer, T.R.; David, L.L.; Feix, J.B.; Lampi, K.J. Deamidation, but not truncation, decreases the urea stability of a lens structural protein, betaB1-crystallin. Biochemistry 2002, 41, 14076–14084. [Google Scholar] [CrossRef]
- Creecy, A.; Brown, K.L.; Rose, K.L.; Voziyan, P.; Nyman, J.S. Post-translational modifications in collagen type I of bone in a mouse model of aging. Bone 2021, 143, 115763. [Google Scholar] [CrossRef] [PubMed]
- Lindner, H.; Sarg, B.; Grunicke, H.; Helliger, W. Age-dependent deamidation of H1(0) histones in chromatin of mammalian tissues. J. Cancer Res. Clin. Oncol. 1999, 125, 182–186. [Google Scholar] [CrossRef]
- Truscott, R.J. Macromolecular deterioration as the ultimate constraint on human lifespan. Ageing Res. Rev. 2011, 10, 397–403. [Google Scholar] [CrossRef]
- Joseph, D. The Unified Theory of Neurodegeneration Pathogenesis Based on Axon Deamidation. Int. J. Mol. Sci. 2025, 26, 4143. [Google Scholar] [CrossRef]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Dan, A.; Takahashi, M.; Masuda-Suzukake, M.; Kametani, F.; Nonaka, T.; Kondo, H.; Akiyama, H.; Arai, T.; Mann, D.M.A.; Saito, Y.; et al. Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2013, 1, 54. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Opare, S.K.; Xu, X.; Ganesan, A.; Rao, P.P. Post-translational modifications in tau and their roles in Alzheimer’s pathology. Curr. Alzheimer Res. 2024, 21, 24–49. [Google Scholar] [CrossRef]
- Miyasaka, T.; Watanabe, A.; Saito, Y.; Murayama, S.; Mann, D.M.A.; Yamazaki, M.; Ravid, R.; Morishima-Kawashima, M.; Nagashima, K.; Ihara, Y. Visualization of newly deposited tau in neurofibrillary tangles and neuropil threads. J. Neuropathol. Exp. Neurol. 2005, 64, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Tarutani, A.; Kametani, F.; Tahira, M.; Saito, Y.; Yoshida, M.; Robinson, A.C.; Mann, D.M.A.; Murayama, S.; Tomita, T.; Hasegawa, M. Distinct tau folds initiate templated seeding and alter the post-translational modification profile. Brain 2023, 146, 4988–4999. [Google Scholar] [CrossRef]
- Wang, J.; Guo, C.; Meng, Z.; Zwan, M.D.; Chen, X.; Seelow, S.; Lundström, S.L.; Rodin, S.; Teunissen, C.E.; Zubarev, R.A. Testing the link between isoaspartate and Alzheimer’s disease etiology. Alzheimer’s and Dement. 2023, 19, 1491–1502. [Google Scholar] [CrossRef]
- Lambeth, T.R.; Riggs, D.L.; Talbert, L.E.; Tang, J.; Coburn, E.; Kang, A.S.; Noll, J.; Augello, C.; Ford, B.D.; Julian, R.R. Spontaneous isomerization of long-lived proteins provides a molecular mechanism for the lysosomal failure observed in Alzheimer’s disease. ACS Cent. Sci. 2019, 5, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Barbariga, M.; Curnis, F.; Andolfo, A.; Zanardi, A.; Lazzaro, M.; Conti, A.; Magnani, G.; Volontè, M.A.; Ferrari, L.; Comi, G.; et al. Ceruloplasmin functional changes in Parkinson’s disease-cerebrospinal fluid. Mol. Neurodegener. 2015, 10, 59. [Google Scholar] [CrossRef]
- Zanardi, A.; Alessio, M. Ceruloplasmin deamidation in neurodegeneration: From loss to gain of function. Int. J. Mol. Sci. 2021, 22, 663. [Google Scholar] [CrossRef]
- Schilling, B.; Gafni, J.; Torcassi, C.; Cong, X.; Row, R.H.; LaFevre-Bernt, M.A.; Cusack, M.P.; Ratovitski, T.; Hirschhorn, R.; Ross, C.A.; et al. Huntingtin phosphorylation sites mapped by mass spectrometry. Modulation of cleavage and toxicity. J. Biol. Chem. 2006, 281, 23686–23697. [Google Scholar] [CrossRef]
- Gertsman, I.; Wuu, J.; McAlonis-Downes, M.; Ghassemian, M.; Ling, K.; Rigo, F.; Bennett, F.; Benatar, M.; Miller, T.M.; Da Cruz, S. An endogenous peptide marker differentiates SOD1 stability and facilitates pharmacodynamic monitoring in SOD1 amyotrophic lateral sclerosis. JCI insight 2019, 4, e122768. [Google Scholar] [CrossRef] [PubMed]
- Sandmeier, E.; Hunziker, P.; Kunz, B.; Sack, R.; Christen, P. Spontaneous deamidation and isomerization of Asn108 in Prion peptide 106–126 and in full-length Prion protein. BBRC 1999, 261, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Tahira, M.; Takao, M.; Matsubara, T.; Hasegawa, K.; Yoshida, M.; Saito, Y.; Murayama, S.; Hasegawa, M. Analysis and comparison of post-translational modifications of α-synuclein filaments in multiple system atrophy and dementia with Lewy bodies. Sci. Rep. 2024, 14, 22892. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, W.; Yang, Y.; Murzin, A.G.; Falcon, B.; Kotecha, A.; van Beers, M.; Tarutani, A.; Kametani, F.; Garringer, H.J. Structure-based classification of tauopathies. Nature 2021, 598, 359–363. [Google Scholar] [CrossRef]
- Shi, Y.; Rhodes, N.R.; Abdolvahabi, A.; Kohn, T.; Cook, N.P.; Marti, A.A.; Shaw, B.F. Deamidation of asparagine to aspartate destabilizes Cu, Zn superoxide dismutase, accelerates fibrillization, and Mmirrors ALS-linked mutations. J. Am. Chem. Soc. 2013, 135, 15897–15908. [Google Scholar] [CrossRef]
- Sanford, A.M. Lewy body dementia. Clin. Geriatr. Med. 2018, 34, 603–615. [Google Scholar] [CrossRef]
- Gallart-Palau, X.; Serra, A.; Qian, J.; Chen, C.P.; Kalaria, R.N.; Sze, S.K. Temporal lobe proteins implicated in synaptic failure exhibit differential expression and deamidation in vascular dementia. Neurochem. Int. 2015, 80, 87–98. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukuda, H.; Murayama, S.; Izumiyama, N.; Shirasawa, T. Isoaspartate formation at position 23 of amyloid beta peptide enhanced fibril formation and deposited onto senile plaques and vascular amyloids in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 451–461. [Google Scholar] [CrossRef]
- Tomidokoro, Y.; Rostagno, A.; Neubert, T.A.; Lu, Y.; Rebeck, G.W.; Frangione, B.; Greenberg, S.M.; Ghiso, J. Iowa variant of familial Alzheimer’s disease: Accumulation of posttranslationally modified AβD23N in parenchymal and cerebrovascular amyloid deposits. Am. J. Pathol. 2010, 176, 1841–1854. [Google Scholar] [CrossRef]
- Pareek, S.; Suter, U.; Snipes, G.J.; Welcher, A.A.; Shooter, E.M.; Murphy, R.A. Detection and processing of peripheral myelin protein PMP22 in cultured Schwann cells. J. Biol. Chem. 1993, 268, 10372–10379. [Google Scholar] [CrossRef]
- Sleat, D.E.; Wiseman, J.A.; Sohar, I.; El-Banna, M.; Zheng, H.; Moore, D.F.; Lobel, P. Proteomic analysis of mouse models of Niemann-Pick C disease reveals alterations in the steady-state levels of lysosomal proteins within the brain. Proteomics 2012, 12, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberger, E.B.; Buchanan, L.E.; Marek, P.; Cao, P.; Raleigh, D.P.; Zanni, M.T. Deamidation accelerates amyloid formation and alters amylin fiber structure. J. Am. Chem. Soc. 2012, 134, 12658–12667. [Google Scholar] [CrossRef]
- Nilsson, M.R.; Driscoll, M.; Raleigh, D.P. Low levels of asparagine deamidation can have a dramatic effect on aggregation of amyloidogenic peptides: Implications for the study of amyloid formation. Protein Sci. 2002, 11, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Tochio, N.; Murata, T.; Utsunomiya-Tate, N. Effect of site-specific amino acid D-isomerization on β-sheet transition and fibril formation profiles of Tau microtubule-binding repeat peptides. BBRC 2019, 508, 184–190. [Google Scholar] [CrossRef]
- Hallinan, G.I.; Ozcan, K.A.; Hoq, M.R.; Cracco, L.; Vago, F.S.; Bharath, S.R.; Li, D.; Jacobsen, M.; Doud, E.H.; Mosley, A.L.; et al. Cryo-EM structures of prion protein filaments from Gerstmann–Sträussler–Scheinker disease. Acta Neuropathol. 2022, 144, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.B.; Murabito, J.M. The epidemiology of longevity and exceptional survival. Epidemiol. Rev. 2013, 35, 181–197. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef]
- Di Battista, A.M.; Heinsinger, N.M.; Rebeck, G.W. Alzheimer’s disease genetic risk fFactor APOE-ε4 also affects normal brain function. Curr. Alzheimer Res. 2016, 13, 1200–1207. [Google Scholar] [CrossRef]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef]
- Guo, L.; Jiao, B.; Liao, X.; Xiao, X.; Zhang, W.; Yuan, Z.; Liu, X.; Zhou, L.; Wang, X.; Zhu, Y.; et al. The role of NOTCH3 variants in Alzheimer’s disease and subcortical vascular dementia in the Chinese population. CNS Neurosci. Ther. 2021, 27, 930–940. [Google Scholar] [CrossRef]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.D.; Diaz-Cintra, S.; et al. Alzheimer’s disease: An updated overview of its genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Cheng, D.; Wang, J.; Duong, D.M.; Losik, T.G.; Gearing, M.; Rees, H.D.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J. Biol. Chem. 2004, 279, 37061–37068. [Google Scholar] [CrossRef]
- Lutz, D.; Loers, G.; Kleene, R.; Oezen, I.; Kataria, H.; Katagihallimath, N.; Braren, I.; Harauz, G.; Schachner, M. Myelin basic protein cleaves cell adhesion molecule L1 and promotes neuritogenesis and cell survival. J. Biol. Chem. 2014, 289, 13503–13518. [Google Scholar] [CrossRef]
- Zhang, C.; Walker, A.K.; Zand, R.; Moscarello, M.A.; Yan, J.M.; Andrews, P.C. Myelin basic protein undergoes a broader range of modifications in mammals than in lower vertebrates. J. Proteome Res. 2012, 11, 4791–4802. [Google Scholar] [CrossRef]
- Gallart-Palau, X.; Lee, B.S.T.; Adav, S.S.; Qian, J.; Serra, A.; Park, J.E.; Lai, M.K.P.; Chen, C.P.; Kalaria, R.N.; Sze, S.K. Gender differences in white matter pathology and mitochondrial dysfunction in Alzheimer’s disease with cerebrovascular disease. Mol. Brain 2016, 9, 27. [Google Scholar] [CrossRef]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.N.; David, S. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J. Biol. Chem. 1997, 272, 20185–20190. [Google Scholar] [CrossRef]
- Curnis, F.; Longhi, R.; Crippa, L.; Cattaneo, A.; Dondossola, E.; Bachi, A.; Corti, A. Spontaneous formation of L-isoaspartate and gain of function in fibronectin. J. Biol. Chem. 2006, 281, 36466–36476. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, J.; Xu, S.; Feng, P. Emerging roles of protein deamidation in innate immune signaling. J. Virol. 2016, 90, 4262–4268. [Google Scholar] [CrossRef]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef]
- Sekimori, T.; Fukunaga, K.; Finkelstein, D.I.; Kawahata, I. Advances in blood biomarkers and diagnosis approaches for neurodegenerative dementias and related diseases. JIN 2024, 23, 188. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Britschgi, M.; Herbert, C.; Takeda-Uchimura, Y.; Boxer, A.; Blennow, K.; Friedman, L.F.; Galasko, D.R.; Jutel, M.; Karydas, A.; et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat. Med. 2007, 13, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lyutvinskiy, Y.; Herukka, S.K.; Soininen, H.; Rutishauser, D.; Zubarev, R.A. Prognostic polypeptide blood plasma biomarkers of Alzheimer’s disease progression. J. Alzheimer’s Dis. 2014, 40, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Gaiottino, J.; Norgren, N.; Dobson, R.; Topping, J.; Nissim, A.; Malaspina, A.; Bestwick, J.P.; Monsch, A.U.; Regeniter, A.; Lindberg, R.L. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS ONE 2013, 8, e75091. [Google Scholar] [CrossRef]
- Paraskevaidi, M.; Morais, C.L.; Lima, K.M.; Snowden, J.S.; Saxon, J.A.; Richardson, A.M.; Jones, M.; Mann, D.M.; Allsop, D.; Martin-Hirsch, P.L. Differential diagnosis of Alzheimer’s disease using spectrochemical analysis of blood. Proc. Natl. Acad. Sci. USA 2017, 114, E7929–E7938. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.-R.; Shen, X.-N.; Han, J.; Cui, M.; Tan, L.; Dong, Q.; Zubarev, R.A.; Yu, J.-T. Deamidation-related blood biomarkers show promise for early diagnostics of neurodegeneration. Biomark. Res. 2022, 10, 91. [Google Scholar] [CrossRef]
- Doye, A.; Mettouchi, A.; Bossis, G.; Clément, R.; Buisson-Touati, C.; Flatau, G.; Gagnoux, L.; Piechaczyk, M.; Boquet, P.; Lemichez, E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002, 111, 553–564. [Google Scholar] [CrossRef]
- Desrosiers, R.R.; Fanelus, I. Damaged proteins bearing L-isoaspartyl residues and aging: A dynamic equilibrium between generation of isomerized forms and repair by PIMT. Curr. Aging Sci. 2011, 4, 8–18. [Google Scholar] [CrossRef]
- Johnson, B.A.; Langmack, E.; Aswad, D. Partial repair of deamidation-damaged calmodulin by protein carboxyl methyltransferase. J. Biol. Chem. 1987, 262, 12283–12287. [Google Scholar] [CrossRef]
- de la Mora-de la Mora, I.; Torres-Larios, A.; Enriquez-Flores, S.; Mendez, S.-T.; Castillo-Villanueva, A.; Gomez-Manzo, S.; Lopez-Velazquez, G.; Marcial-Quino, J.; Torres-Arroyo, A.; Garcia-Torres, I. Structural effects of protein aging: Terminal marking by deamidation in human triosephosphate isomerase. PLoS ONE 2015, 10, e0123379. [Google Scholar]
- Keibler, M.A.; Wasylenko, T.M.; Kelleher, J.K.; Iliopoulos, O.; Vander Heiden, M.G.; Stephanopoulos, G. Metabolic requirements for cancer cell proliferation. Cancer Metab. 2016, 4, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Enríquez-Flores, S.; Flores-López, L.A.; García-Torres, I.; de la Mora-de la Mora, I.; Cabrera, N.; Gutierrez-Castrellon, P.; Martinez-Perez, Y.; Lopez-Velazquez, G. Deamidated human triosephosphate isomerase is a promising druggable target. Biomolecules 2020, 10, 1050. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Tian, M.; Zhang, S.; Delfarah, A.; Gao, R.; Rao, Y.; Savas, A.C.; Lu, A.; Bubb, L.; Lei, X.; et al. Deamidation shunts RelA from mediating inflammation to aerobic glycolysis. Cell Metab. 2020, 31, 937.E7–955.E7. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Tanriver, G.; Monard, G.; Catak, S. Impact of deamidation on the structure and function of antiapoptotic Bcl-xL. J. Chem. Inf. Model. 2022, 62, 102–115. [Google Scholar] [CrossRef]
- Zhao, R.; Follows, G.A.; Beer, P.A.; Scott, L.M.; Huntly, B.J.; Green, A.R.; Alexander, D.R. Inhibition of the Bcl-xL deamidation pathway in myeloproliferative disorders. N. Engl. J. Med. 2008, 359, 2778–2789. [Google Scholar] [CrossRef]
- Takehara, T.; Takahashi, H. Suppression of Bcl-xL deamidation in human hepatocellular carcinomas. Cancer Res. 2003, 63, 3054–3057. [Google Scholar]
- Zhu, Y.; Yang, Y.; Bu, H.; Huang, H.; Chen, H.; Ran, J.; Qin, L.; Ni, Y.; Yao, M.; Song, T.; et al. Apelin-mediated deamidation of HMGA1 promotes tumorigenesis by enhancing SREBP1 activity and lipid synthesis. Cancer Sci. 2022, 113, 3722–3734. [Google Scholar] [CrossRef]
- Chang, C.Y.; Lin, Y.M.; Lee, W.P.; Hsu, H.H.; Chen, E.I.T. Involvement of Bcl-XL deamidation in E1A-mediated cisplatin sensitization of ovarian cancer cells. Oncogene 2006, 25, 2656–2665. [Google Scholar] [CrossRef]
- Deng, L.; Yao, P.; Li, L.; Ji, F.; Zhao, S.; Xu, C.; Lan, X.; Jiang, P. p53-mediated control of aspartate-asparagine homeostasis dictates LKB1 activity and modulates cell survival. Nat. Commun. 2020, 11, 1755. [Google Scholar] [CrossRef]
- Yuan, Q.; Yin, L.; He, J.; Zeng, Q.; Liang, Y.; Shen, Y.; Zu, X. Metabolism of asparagine in the physiological state and cancer. Cell Commun. Signal 2024, 22, 163. [Google Scholar] [CrossRef] [PubMed]
- Schotsmans, E.M.; Márquez-Grant, N.; Forbes, S.L. Taphonomy of Human Remains: Rorensic Analysis of the Dead and the Depositional Environment; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Procopio, N.; Williams, A.; Chamberlain, A.T.; Buckley, M. Forensic proteomics for the evaluation of the post-mortem decay in bones. J. Proteom. 2018, 177, 21–30. [Google Scholar] [CrossRef]
- Procopio, N.; Chamberlain, A.T.; Buckley, M. Intra- and interskeletal proteome variations in fresh and buried bones. J. Proteome Res. 2017, 16, 2016–2029. [Google Scholar] [CrossRef] [PubMed]
- Parisuthiman, D.; Mochida, Y.; Duarte, W.R.; Yamauchi, M. Biglycan modulates osteoblast differentiation and matrix mineralization. J. Bone Miner. Res. 2005, 20, 1878–1886. [Google Scholar] [CrossRef]
- Di Lullo, G.A.; Sweeney, S.M.; Korkko, J.; Ala-Kokko, L.; San Antonio, J.D. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. J. Biol. Chem. 2002, 277, 4223–4231. [Google Scholar] [CrossRef]
- Wadsworth, C.; Buckley, M. Proteome degradation in fossils: Investigating the longevity of protein survival in ancient bone. Rapid Commun. Mass. Spectrom. 2014, 28, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Orlando, L.; Ginolhac, A.; Zhang, G.; Froese, D.; Albrechtsen, A.; Stiller, M.; Schubert, M.; Cappellini, E.; Petersen, B.; Moltke, I. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 2013, 499, 74–78. [Google Scholar] [CrossRef]
- Chowdhury, M.P.; Buckley, M. Trends in deamidation across archaeological bones, ceramics and dental calculus. Methods 2022, 200, 67–79. [Google Scholar] [CrossRef]
- Smith, C.I.; Chamberlain, A.T.; Riley, M.S.; Stringer, C.; Collins, M.J. The thermal history of human fossils and the likelihood of successful DNA amplification. J. Hum. Evol. 2003, 45, 203–217. [Google Scholar] [CrossRef]
- Parker, G.J.; Leppert, T.; Anex, D.S.; Hilmer, J.K.; Matsunami, N.; Baird, L.; Stevens, J.; Parsawar, K.; Durbin-Johnson, B.P.; Rocke, D.M.; et al. Demonstration of protein-based human identification using the hair shaft proteome. PLoS ONE 2016, 11, e0160653. [Google Scholar] [CrossRef]
- Plott, T.J.; Karim, N.; Durbin-Johnson, B.P.; Swift, D.P.; Scott Youngquist, R.; Salemi, M.; Phinney, B.S.; Rocke, D.M.; Davis, M.G.; Parker, G.J.; et al. Age-related changes in hair shaft protein profiling and genetically variant peptides. Forensic Sci. Int. Genet. 2020, 47, 102309. [Google Scholar] [CrossRef] [PubMed]
- Adav, S.S.; Leung, C.Y.; Ng, K.W. Profiling of hair proteome revealed individual demographics. Forensic Sci. Int. Genet. 2023, 66, 102914. [Google Scholar] [CrossRef]
- Adav, S.S.; Wu, A.R.Y.L.; Ng, K.W. Insights into structural and proteomic alterations related to pH-induced changes and protein deamidation in hair. Int. J. Cosmetic Sci. 2024, 47, 281–296. [Google Scholar] [CrossRef]
- Adav, S.S.; Subbaiaih, R.S.; Kerk, S.K.; Lee, A.Y.; Lai, H.Y.; Ng, K.W.; Sze, S.K.; Schmidtchen, A. Studies on the proteome of human hair-identification of histones and deamidated keratins. Sci. Rep. 2018, 8, 1599. [Google Scholar] [CrossRef]
- Araki, N.; Moini, M. Age estimation of museum wool textiles from Ovis aries using deamidation rates utilizing matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass. Spectrom. 2011, 25, 3396–3400. [Google Scholar] [CrossRef]
- Solazzo, C.; Wilson, J.; Dyer, J.M.; Clerens, S.; Plowman, J.E.; von Holstein, I.; Walton Rogers, P.; Peacock, E.E.; Collins, M.J. Modeling deamidation in sheep α-keratin peptides and application to archeological wool textiles. AnaCh 2014, 86, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Hendy, J.; Colonese, A.C.; Franz, I.; Fernandes, R.; Fischer, R.; Orton, D.; Lucquin, A.; Spindler, L.; Anvari, J.; Stroud, E. Ancient proteins from ceramic vessels at Çatalhöyük West reveal the hidden cuisine of early farmers. Nat. Commun. 2018, 9, 4064. [Google Scholar] [CrossRef] [PubMed]
- Gamble, J.A.; Spicer, V.; Hunter, M.; Lao, Y.; Hoppa, R.D.; Pedersen, D.D.; Wilkins, J.A.; Zahedi, R.P. Advancing sex estimation from amelogenin: Applications to archaeological, deciduous, and fragmentary dental enamel. J. Archaeol. Sci. Rep. 2024, 54, 104430. [Google Scholar] [CrossRef]
- Shevchenko, A.; Schuhmann, A.; Thomas, H.; Wetzel, G. Fine Endmesolithic fish caviar meal discovered by proteomics in foodcrusts from archaeological site Friesack 4 (Brandenburg, Germany). PLoS ONE 2018, 13, e0206483. [Google Scholar] [CrossRef]
- Tokarski, C.; Martin, E.; Rolando, C.; Cren-Olivé, C. Identification of proteins in renaissance paintings by proteomics. AnaCh 2006, 78, 1494–1502. [Google Scholar] [CrossRef]
- Sharma, P.; Joshi, R.V.; Pritchard, R.; Xu, K.; Eicher, M.A. Therapeutic antibodies in medicine. Molecules 2023, 28, 6438. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. In Proceedings of the MAbs, Istanbul, Turkey, 4–5 May 2015; pp. 9–14. [Google Scholar]
- Krause, M.E.; Sahin, E. Chemical and physical instabilities in manufacturing and storage of therapeutic proteins. Curr. Opin. Biotechnol. 2019, 60, 159–167. [Google Scholar] [CrossRef]
- Perkins, M.; Theiler, R.; Lunte, S.; Jeschke, M. Determination of the origin of charge heterogeneity in a murine monoclonal antibody. Pharm. Res. 2000, 17, 1110–1117. [Google Scholar] [CrossRef]
- Dengl, S.; Wehmer, M.; Hesse, F.; Lipsmeier, F.; Popp, O.; Lang, K. Aggregation and chemical modification of monoclonal antibodies under upstream processing conditions. Pharm. Res. 2013, 30, 1380–1399. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Sato, R.; Aoyagi, H. Changes in the quality of antibodies produced by Chinese hamster ovary cells during the death phase of cell culture. J. Biosci. Bioeng. 2010, 109, 281–287. [Google Scholar] [CrossRef]
- Lu, X.; Nobrega, R.P.; Lynaugh, H.; Jain, T.; Barlow, K.; Boland, T.; Sivasubramanian, A.; Vásquez, M.; Xu, Y. Deamidation and isomerization liability analysis of 131 clinical-stage antibodies. MAbs 2019, 11, 45–57. [Google Scholar] [CrossRef]
- Gupta, R.; Srivastava, O.P. Deamidation affects structural and functional properties of human alphaA-crystallin and its oligomerization with alphaB-crystallin. J. Biol. Chem. 2004, 279, 44258–44269. [Google Scholar] [CrossRef] [PubMed]
- Gervais, D. Protein deamidation in biopharmaceutical manufacture: Understanding, control and impact. J. Chem. Technol. Biotechnol. 2016, 91, 569–575. [Google Scholar] [CrossRef]
- Schmid, I.; Bonnington, L.; Gerl, M.; Bomans, K.; Thaller, A.L.; Wagner, K.; Schlothauer, T.; Falkenstein, R.; Zimmermann, B.; Kopitz, J. Assessment of susceptible chemical modification sites of trastuzumab and endogenous human immunoglobulins at physiological conditions. Commun. Biol. 2018, 1, 28. [Google Scholar] [CrossRef]
- Strader, M.B.; Jana, S.; Meng, F.; Heaven, M.R.; Shet, A.S.; Thein, S.L.; Alayash, A.I. Post-translational modification as a response to cellular stress induced by hemoglobin oxidation in sickle cell disease. Sci. Rep. 2020, 10, 14218. [Google Scholar] [CrossRef]
- Robinson, N.; Robinson, Z.; Robinson, B.; Robinson, A.; Robinson, J.; Robinson, M.; Robinson, A.B. Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J. Pep. Res. 2004, 63, 426–436. [Google Scholar] [CrossRef]
- Alam, M.E.; Barnett, G.V.; Slaney, T.R.; Starr, C.G.; Das, T.K.; Tessier, P.M. Deamidation can compromise antibody colloidal stability and enhance aggregation in a pH-dependent manner. Mol. Pharm. 2019, 16, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Pace, A.L.; Wong, R.L.; Zhang, Y.T.; Kao, Y.-H.; Wang, Y.J. Asparagine deamidation dependence on buffer type, pH, and temperature. J. Pharm. Sci. 2013, 102, 1712–1723. [Google Scholar] [CrossRef]
- Robinson, N.E.; Robinson, A.B. Prediction of primary structure deamidation rates of asparaginyl and glutaminyl peptides through steric and catalytic effects. J. Pept. Res. 2004, 63, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.R.; Alonso, L.G.; Sánchez, I.E. Prediction of spontaneous protein deamidation from sequence-derived secondary structure and intrinsic disorder. PLoS ONE 2015, 10, e0145186. [Google Scholar] [CrossRef] [PubMed]
- Sydow, J.F.; Lipsmeier, F.; Larraillet, V.; Hilger, M.; Mautz, B.; Mølhøj, M.; Kuentzer, J.; Klostermann, S.; Schoch, J.; Voelger, H.R.; et al. Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PLoS ONE 2014, 9, e100736. [Google Scholar] [CrossRef]
- Yan, Q.; Huang, M.; Lewis, M.J.; Hu, P. Structure based prediction of asparagine deamidation propensity in monoclonal antibodies. MAbs 2018, 10, 901–912. [Google Scholar] [CrossRef]
- Plotnikov, N.V.; Singh, S.K.; Rouse, J.C.; Kumar, S. Quantifying the risks of asparagine deamidation and aspartate isomerization in biopharmaceuticals by computing reaction free-energy surfaces. J. Phys. Chem. B 2017, 121, 719–730. [Google Scholar] [CrossRef]
- Irudayanathan, F.J.; Jonathan, Z.; Jasper, L.; Izadi, S. Deciphering deamidation and isomerization in therapeutic proteins: Effect of neighboring residue. MAbs 2022, 14, 2143006. [Google Scholar] [CrossRef]
- Bults, P.; van der Voort, A.; Meijer, C.; Sonke, G.S.; Bischoff, R.; van de Merbel, N.C. Analytical and pharmacological consequences of the in vivo deamidation of trastuzumab and pertuzumab. Anal. Bioanal. Chem. 2022, 414, 1513–1524. [Google Scholar] [CrossRef]
- Spanov, B.; Olaleye, O.; Mesurado, T.; Govorukhina, N.; Jungbauer, A.; van de Merbel, N.C.; Lingg, N.; Bischoff, R. Pertuzumab charge Vvariant analysis and complementarity-determining region stability assessment to deamidation. AnaCh 2023, 95, 3951–3958. [Google Scholar] [CrossRef]
- Lynce, F.; Swain, S.M. Pertuzumab for the Treatment of Breast Cancer. Cancer Investig. 2014, 32, 430–438. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Sr No | Neurodegenerative Diseases | Deamidated Protein | Functional Consequences | References |
|---|---|---|---|---|
| 1. | Alzheimer’s disease | Tau | Increased aggregation, reduced microtubule binding. | [72,73,74,75] |
| Human serum albumin, neurofilament light protein (NfL), glial fibrillary acidic protein (GFAP) | Deamidation protein biomarkers for detection of neurodegeneration. | [76] | ||
| Amyloid-beta (Aβ), Tau, Protein S100A9, 4E-BP2 Protein, Na+/K+-ATPase, Ion-Channel Proteins | Structural changes, functional inactivation, and enhanced aggregation. Impacts neurons and axons. | [12,28,29,56,77] | ||
| 2. | Parkinson’s disease | α-Synuclein, 4E-BP2 Protein | Aggregation, Impact neurons and axons. | [13,56] |
| Ceruloplasmin | Ceruloplasmin in the CSF of PD patients undergoes conformational changes and NGR-motif deamidation, which promote the gain of integrin-binding function. Leads to loss of enzymatic activity, also confers gain of function to Cp. | [78,79] | ||
| 3. | Huntington’s disease | Huntingtin (HTT) | Abnormal conformation. | [80] |
| 4. | Amyotrophic lateral sclerosis | Superoxide dismutase | Structure destabilization, protein aggregation, toxic oligomer formation. | [81] |
| 5. | Prion diseases, Creutzfeldt-Jakob disease | Prion protein (PrP) | Change in conformation, misfolding. | [82] |
| 6. | Frontotemporal dementia | Tau, TDP-43 | Neuronal loss. | [74] |
| 7. | Spinocerebellar ataxia | Ataxin (varies by type) | Alter the protein’s stability, folding, and interactions. | |
| 8. | Multiple system atrophy | α-Synuclein | Misfolding and protein aggregation. | [83] |
| 9. | Progressive supranuclear palsy | Tau | Misfolding, aggregation, reduced microtubule binding, acceleration of NFT formation and disease progression. | [75,84] |
| 10. | Corticobasal degeneration | Tau | Misfolding, aggregation, reduced microtubule binding. | [75,84] |
| 11. | Cataract | α-Crystallin, β-crystallin, γ-crystallin | Altered structure, dimer formation, protein aggregation. | [31,62,85] |
| 12. | Lewy body dementia | α-Synuclein | Aggregation. | [13,86] |
| 13. | Vascular dementia | amyloid β peptides, Synapsin1, α-tubulin 1B (TUBA1B) and β-tubulin 2A (TUBB2A) proteins, human serum albumin, Na+/K+-ATPase, ion-channel proteins | Functional impairment and synaptic impairment. | [3,76,77,87,88] |
| 14. | Familial Alzheimer’s disease | Amyloid-beta (Aβ), Tau | Oligomerization/fibrillization, amyloid-related neurodegeneration. | [77,89] |
| 15 | Charcot–Marie–Tooth disease | Peripheral myelin proteins (PMP22) | Intracellular aggregation. | [90] |
| 16. | Niemann-Pick disease | Sphingomyelinase | Alterations in the function of the lysosomal system. | [91] |
| 17 | Diabetes (type 2 diabetes) | Amylin, islet amyloid polypeptide | Accelerates amyloid formation. | [92,93] |
| 18 | Alzheimer’s disease with tauopathy | Tau | Increased rates of β-sheet transition and fibril formation. | [94] |
| 19 | Gerstmann–Sträussler–Scheinker syndrome | Prion protein (PrP) | Aggregation and formation of PrP amyloid. Misfolding and pathogenicity of prion proteins. | [82,95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adav, S.S. Advances in the Study of Protein Deamidation: Unveiling Its Influence on Aging, Disease Progression, Forensics and Therapeutic Efficacy. Proteomes 2025, 13, 24. https://doi.org/10.3390/proteomes13020024
Adav SS. Advances in the Study of Protein Deamidation: Unveiling Its Influence on Aging, Disease Progression, Forensics and Therapeutic Efficacy. Proteomes. 2025; 13(2):24. https://doi.org/10.3390/proteomes13020024
Chicago/Turabian StyleAdav, Sunil S. 2025. "Advances in the Study of Protein Deamidation: Unveiling Its Influence on Aging, Disease Progression, Forensics and Therapeutic Efficacy" Proteomes 13, no. 2: 24. https://doi.org/10.3390/proteomes13020024
APA StyleAdav, S. S. (2025). Advances in the Study of Protein Deamidation: Unveiling Its Influence on Aging, Disease Progression, Forensics and Therapeutic Efficacy. Proteomes, 13(2), 24. https://doi.org/10.3390/proteomes13020024