
Identifying Endogenous Proteins of Perennial Ryegrass (Lolium perenne) with Ex Vivo Antioxidant Activity


 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Grass Cultivation and Wet Fractionation
2.2. Crude Protein Estimation and Dry Matter Analysis
2.3. One-Dimensional SDS-PAGE
2.4. Protein Fractionation by Size Exclusion Chromatography
Size Estimation of Protein Fractions
2.5. DPPH Radical Scavenging Activity Screening and Fraction Selection
2.6. Quantification of Antioxidant Properties in Selected Protein Fractions
2.6.1. Desalting and Protein Concentration Determination
2.6.2. Protein Concentration
2.6.3. DPPH Radical Scavenging Activity
2.6.4. Iron Chelation Activity
2.6.5. EC50 Calculations
2.7. Bottom-Up Proteomics by LC-MS/MS
2.7.1. In-Solution Digest of Selected SEC Fractions
2.7.2. Protein Extraction and In-Solution Digest of Crude Fractions
2.7.3. LC-MS/MS Analysis
2.7.4. LC-MS/MS Data Processing
2.7.5. Downstream Data Analysis of MaxQuant Data from SEC Fractions
Isoform Combination
Gene Ontology Analysis and Data Filtering
2.7.6. Downstream Data Analysis of Crude Fractions
2.8. Statistical Analysis
3. Results and Discussion
3.1. Protein Characterization of Wet Fractionation
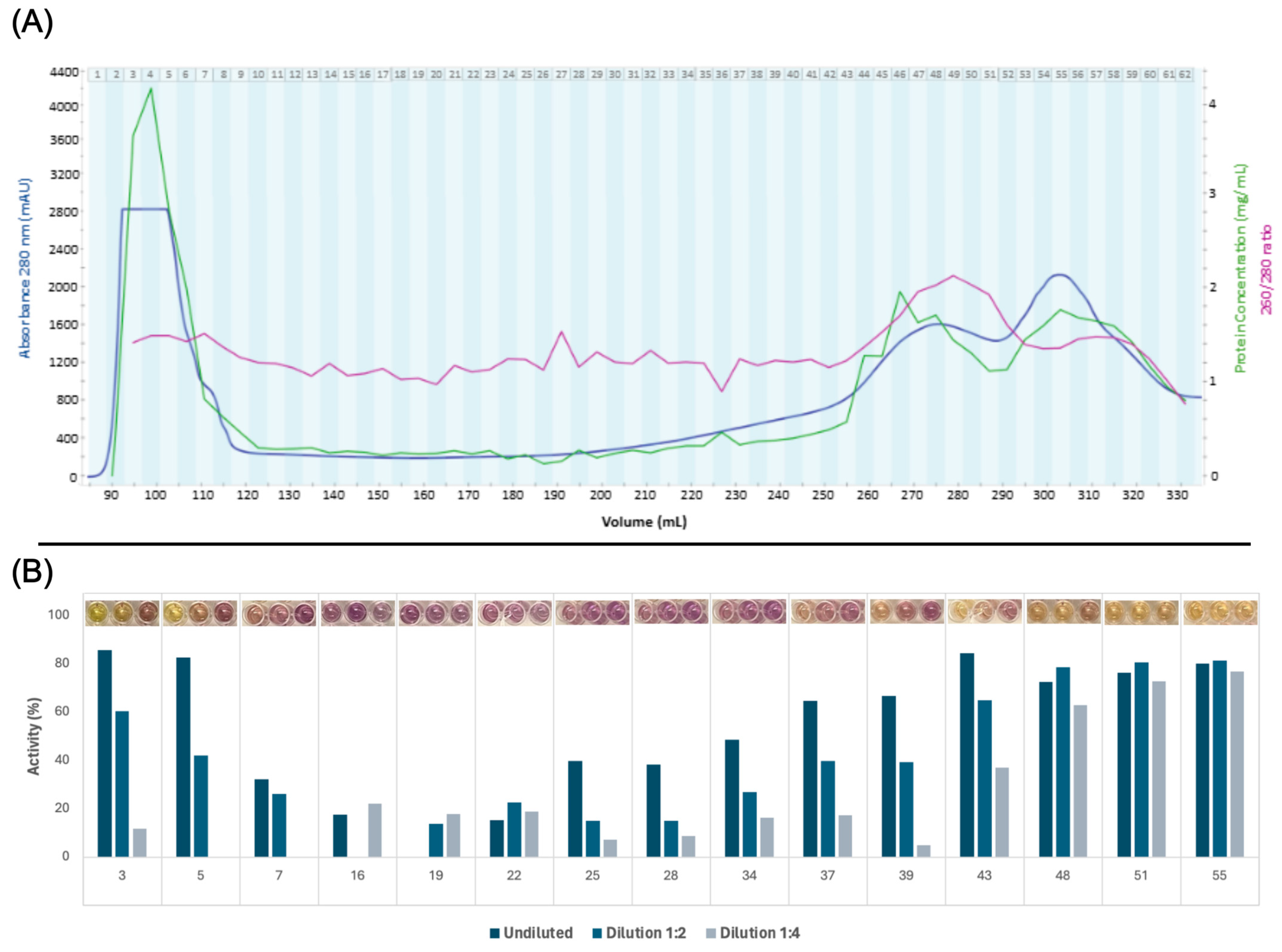
3.2. Protein Fractionation and Selection
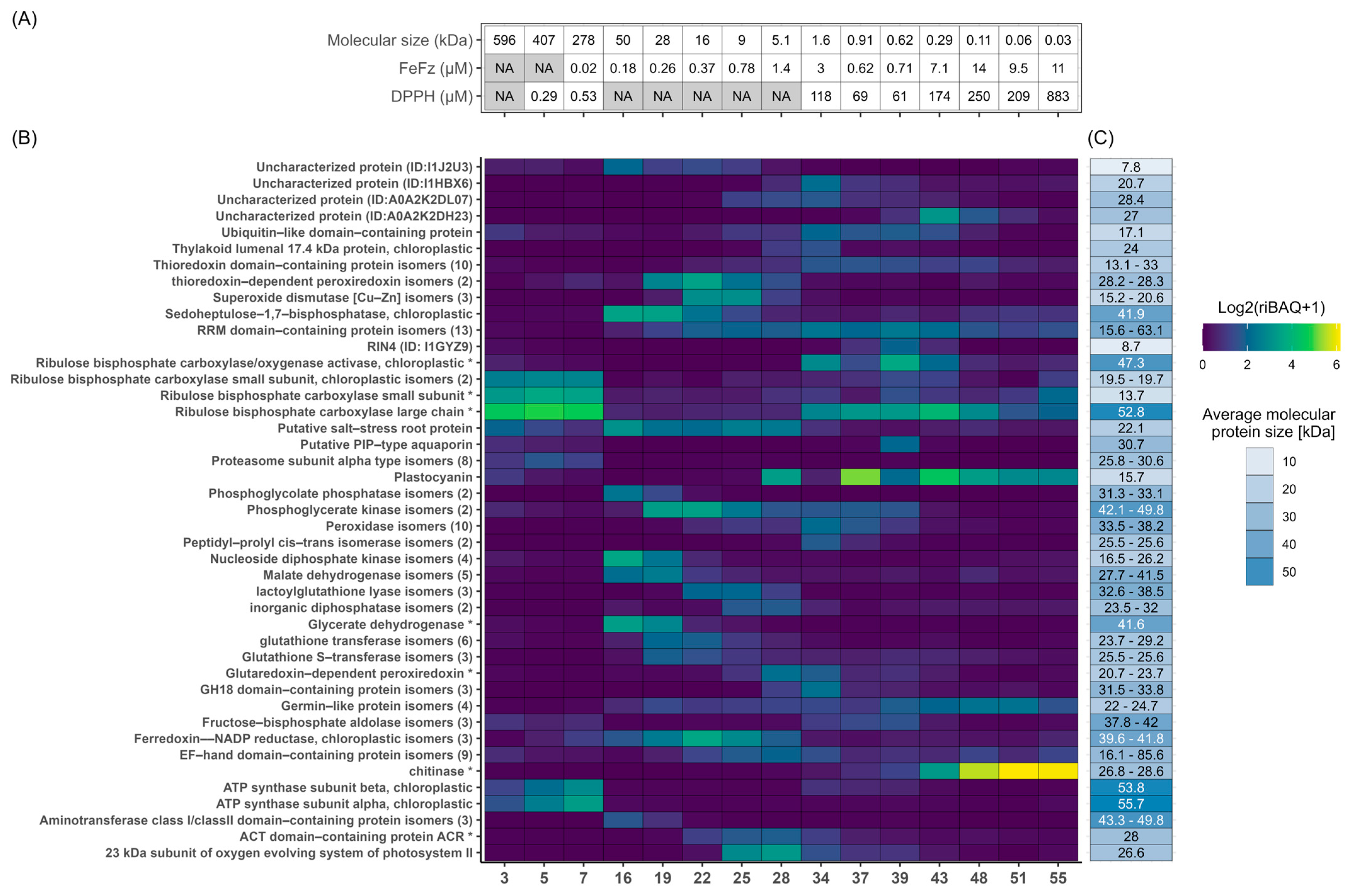
3.3. Ex Vivo Antioxidant Activity
3.4. Overview of Protein Composition and Most Abundant Proteins
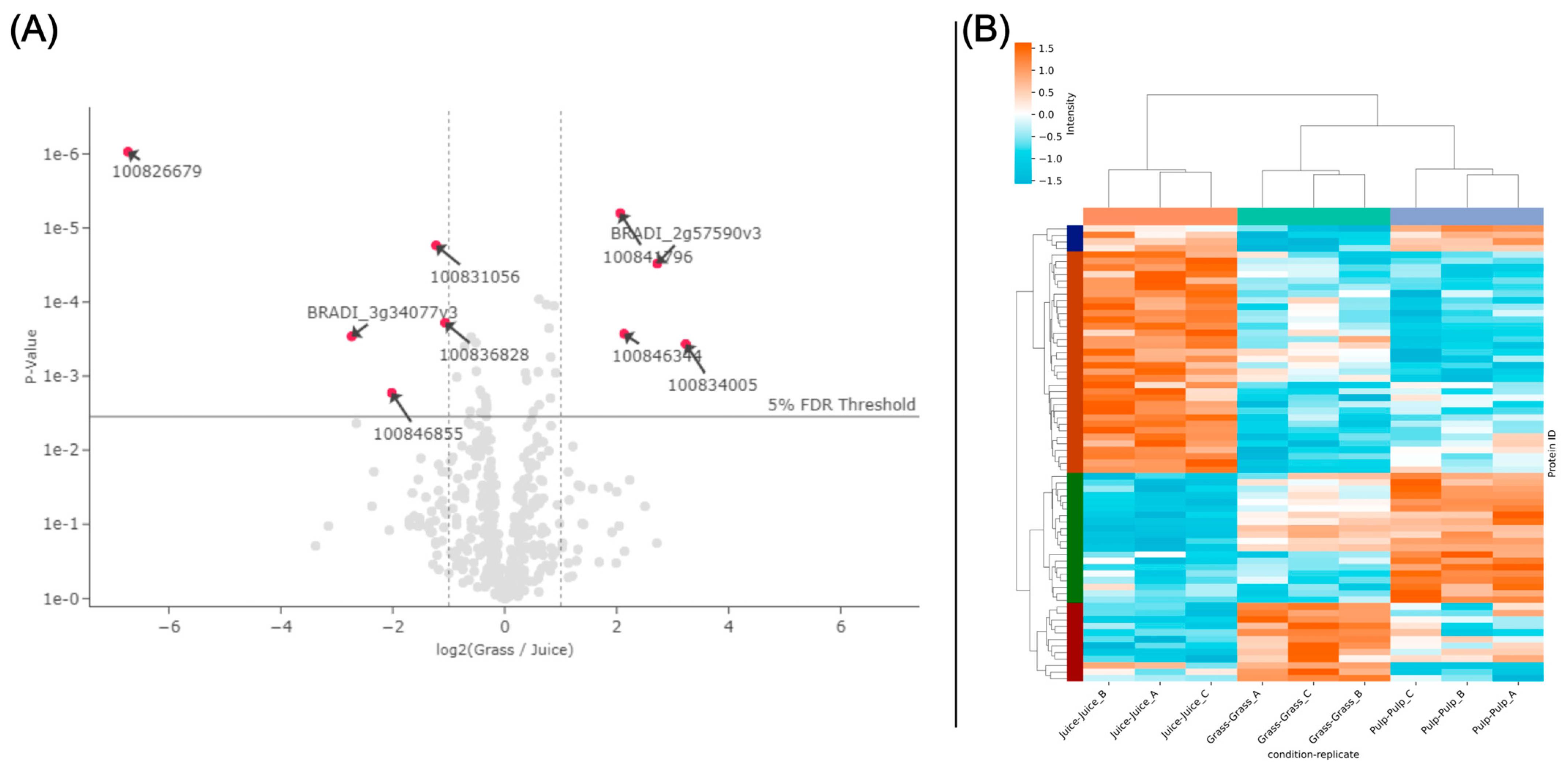
3.5. Prediction of Antioxidant Proteins Using GO-Term Analysis
3.6. Correlating Protein Abundance and GO-Term Analysis with In Vitro Antioxidant Activity
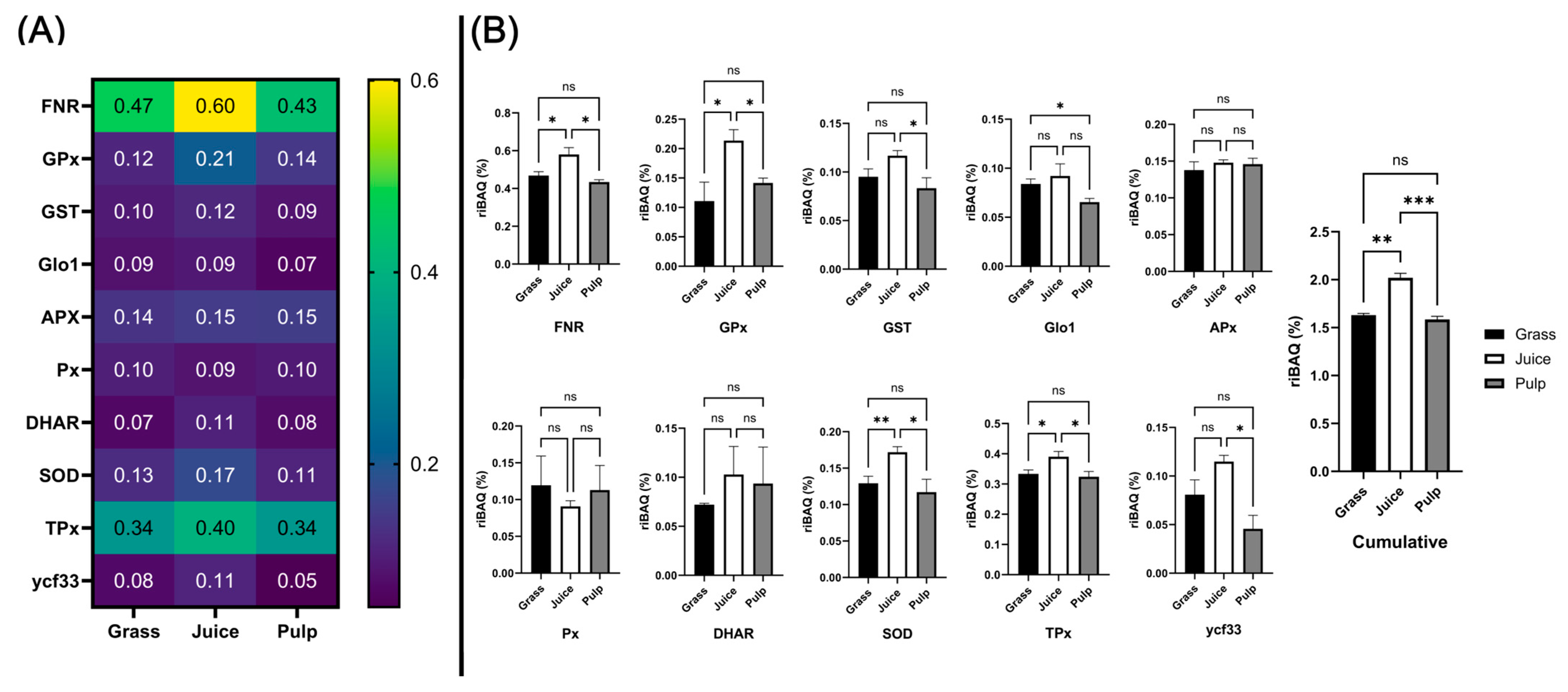
3.7. Abundance of Known Antioxidant Proteins in Crude Fractions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
Appendix C
Appendix D
References
- Lynch, H.; Johnston, C.; Wharton, C. Plant-Based Diets: Considerations for Environmental Impact, Protein Quality, and Exercise Performance. Nutrients 2018, 10, 1841. [Google Scholar] [CrossRef] [PubMed]
- Sá, A.G.A.; Moreno, Y.M.F.; Carciofi, B.A.M. Plant proteins as high-quality nutritional source for human diet. Trends Food Sci. Technol. 2020, 97, 170–184. [Google Scholar] [CrossRef]
- Kromus, S.; Kamm, B.; Kamm, M.; Fowler, P.; Narodoslawsky, M. Green Biorefineries: The Green Biorefinery Concept—Fundamentals and Potential. In Biorefineries-Industrial Processes and Products: Status Quo and Future Directions; John Wiley & Sons: Hoboken, NJ, USA, 2008; Volume 1, pp. 253–294. [Google Scholar] [CrossRef]
- Domokos-Szabolcsy, É.; Yavuz, S.R.; Picoli, E.; Fári, M.G.; Kovács, Z.; Tóth, C.; Kaszás, L.; Alshaal, T.; Elhawat, N. Green Biomass-Based Protein for Sustainable Feed and Food Supply: An Overview of Current and Future Prospective. Life 2023, 13, 307. [Google Scholar] [CrossRef] [PubMed]
- Gaffey, J.; Rajauria, G.; McMahon, H.; Ravindran, R.; Dominguez, C.; Ambye-Jensen, M.; Souza, M.F.; Meers, E.; Aragonés, M.M.; Skunca, D.; et al. Green Biorefinery systems for the production of climate-smart sustainable products from grasses, legumes and green crop residues. Biotechnol. Adv. 2023, 66, 108168. [Google Scholar] [CrossRef]
- Santamaría-Fernández, M.; Lübeck, M. Production of leaf protein concentrates in green biorefineries as alternative feed for monogastric animals. Anim. Feed Sci. Technol. 2020, 268, 114605. [Google Scholar] [CrossRef]
- Damborg, V.K.; Jensen, S.K.; Weisbjerg, M.R.; Adamsen, A.P.; Stødkilde, L. Screw-pressed fractions from green forages as animal feed: Chemical composition and mass balances. Anim. Feed Sci. Technol. 2020, 261, 114401. [Google Scholar] [CrossRef]
- Hu, L.; Li, H.; Pang, H.; Fu, J. Responses of antioxidant gene, protein and enzymes to salinity stress in two genotypes of perennial ryegrass (Lolium perenne) differing in salt tolerance. J. Plant Physiol. 2012, 169, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Solati, Z.; Manevski, K.; Jørgensen, U.; Labouriau, R.; Shahbazi, S.; Lærke, P.E. Crude protein yield and theoretical extractable true protein of potential biorefinery feedstocks. Ind. Crops Prod. 2018, 115, 214–226. [Google Scholar] [CrossRef]
- Humphreys, M.; Feuerstein, U.; Vandewalle, M.; Baert, J. Ryegrasses. In Fodder Crops and Amenity Grasses; Springer: New York, NY, USA, 2010; pp. 211–260. [Google Scholar] [CrossRef]
- Larsen, S.U.; Ambye-Jensen, M.; Jørgensen, H.; Jørgensen, U. Ensiling of the pulp fraction after biorefining of grass into pulp and protein juice. Ind. Crops Prod. 2019, 139, 111576. [Google Scholar] [CrossRef]
- Santamaría-Fernández, M.; Molinuevo-Salces, B.; Lübeck, M.; Uellendahl, H. Biogas potential of green biomass after protein extraction in an organic biorefinery concept for feed, fuel and fertilizer production. Renew. Energy 2018, 129, 769–775. [Google Scholar] [CrossRef]
- Jørgensen, U.; Jensen, S.K.; Ambye-Jensen, M. Coupling the benefits of grassland crops and green biorefining to produce protein, materials and services for the green transition. Grass Forage Sci. 2022, 77, 295–306. [Google Scholar] [CrossRef]
- Sharma, H.S.S.; Lyons, G.; McRoberts, C. Biorefining of perennial grasses: A potential sustainable option for Northern Ireland grassland production. Chem. Eng. Res. Des. 2011, 89, 2309–2321. [Google Scholar] [CrossRef]
- Termansen, M.; Gylling, M.; Jørgensen, U.; Hermansen, J.; Hansen, L.B.; Knudsen, M.T.; Adamsen, A.P.S.; Ambye-Jensen, M.; Jensen, M.V.; Jensen, S.K.; et al. Green Biomass; DCA—Danish Centre for Food and Agriculture: Tjele, Danmark, 2015. [Google Scholar]
- Hermansen, J.E.; Jørgensen, U.; Lærke, P.E.; Manevski, K.; Boelt, B.; Jensen, S.K.; Weisbjerg, M.R.; Dalsgaard, T.K.; Danielsen, M.; Asp, T.; et al. Green Biomass—Protein Production Through Biorefining; DCA—Danish Centre for Food and Agriculture: Tjele, Danmark, 2017. [Google Scholar]
- Parajuli, R.; Kristensen, I.S.; Knudsen, M.T.; Mogensen, L.; Corona, A.; Birkved, M.; Peña, N.; Graversgaard, M.; Dalgaard, T. Environmental life cycle assessments of producing maize, grass-clover, ryegrass and winter wheat straw for biorefinery. J. Clean. Prod. 2017, 142, 3859–3871. [Google Scholar] [CrossRef]
- Pérez-Vila, S.; Fenelon, M.A.; O’Mahony, J.A.; Gómez-Mascaraque, L.G. Extraction of plant protein from green leaves: Biomass composition and processing considerations. Food Hydrocoll. 2022, 133, 107902. [Google Scholar] [CrossRef]
- Sharma, H.S.S.; Carmichael, E.; Muhamad, M.; McCall, D.; Andrews, F.; Lyons, G.; McRoberts, W.C.; Hornsby, P.R. Biorefining of perennial ryegrass for the production of nanofibrillated cellulose. RSC Adv. 2012, 2, 6424–6437. [Google Scholar] [CrossRef]
- Sharma, H.S.S.; McCall, D.; Andrews, F.; Carmichael, E.; Lyons, G.; McRoberts, C. Biorefining of perennial grasses: Development of visible and near infrared tools for assessing fineness of extracted fibre. Ind. Crops Prod. 2012, 36, 476–484. [Google Scholar] [CrossRef]
- Costigan, H.; Lahart, B.; Kennedy, M.; Herron, J.; Sanders, J.P.M.; Gaffey, J.; Lambrechts, B.; Shalloo, L. Biorefined perennial ryegrass press cake as an alternative feed for dairy cows in late lactation and during the dry period: A demonstration. J. Agric. Sci. 2024, 162, 528–537. [Google Scholar] [CrossRef]
- Stødkilde, L.; Ingerslev, A.K.; Ambye-Jensen, M.; Jensen, S.K. The composition and nutritional quality of biorefined lucerne protein depend on precipitation method. J. Sci. Food Agric. 2024, 104, 3405–3412. [Google Scholar] [CrossRef]
- Møller, A.H.; Hammershøj, M.; Dos Passos, N.H.M.; Tanambell, H.; Stødkilde, L.; Ambye-Jensen, M.; Danielsen, M.; Jensen, S.K.; Trine, K. Dalsgaard. Biorefinery of Green Biomass-How to Extract and Evaluate High Quality Leaf Protein for Food? J. Agric. Food Chem. 2021, 69, 14341–14357. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, R.; Koopmans, S.; Sanders, J.P.M.; McMahon, H.; Gaffey, J. Production of Green Biorefinery Protein Concentrate Derived from Perennial Ryegrass as an Alternative Feed for Pigs. Clean Technol. 2021, 3, 656–669. [Google Scholar] [CrossRef]
- Pérez-Vila, S.; Fenelon, M.A.; O’Mahony, J.A.; Gómez-Mascaraque, L.G. The emulsifying properties of protein extracts from perennial ryegrass (Lolium perenne L.) depend on the extraction method. Food Hydrocoll. 2024, 152, 109917. [Google Scholar] [CrossRef]
- Pérez-Vila, S.; Fenelon, M.; Hennessy, D.; O’Mahony, J.A.; Gómez-Mascaraque, L.G. Impact of the extraction method on the composition and solubility of leaf protein concentrates from perennial ryegrass (Lolium perenne L.). Food Hydrocoll. 2024, 147, 109372. [Google Scholar] [CrossRef]
- Ma, X.; van Polen, T.; Habibi, M.; Landman, J.; Sagis, L.M.C.; Shen, P. Rubisco at interfaces I: Conformational flexibility enhances air-water interface and foam stabilization. Food Hydrocoll. 2025, 160, 110783. [Google Scholar] [CrossRef]
- Elias, R.J.; Kellerby, S.S.; Decker, E.A. Antioxidant Activity of Proteins and Peptides. Crit. Rev. Food Sci. Nutr. 2008, 48, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, R.; Pateiro, M.; Munekata, P.E.S.; Zhang, W.; Garcia-Oliveira, P.; Carpena, M.; Prieto, M.A.; Bohrer, B.; Lorenzo, J.M. Protein Oxidation in Muscle Foods: A Comprehensive Review. Antioxidants 2021, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Gülçin, I. Antioxidant activity of food constituents: An overview. Arch. Toxicol. 2011, 86, 345–391. [Google Scholar] [CrossRef]
- Abeyrathne, E.D.N.S.; Nam, K.; Huang, X.; Ahn, D.U. Plant- and Animal-Based Antioxidants’ Structure, Efficacy, Mechanisms, and Applications: A Review. Antioxidants 2022, 11, 1025. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, W.; Santhanam, R.K.; Wang, C.; Gao, X.; Chen, Y.; Wang, C.; Xu, L.; Chen, H. Bioactive peptide with antioxidant and anticancer activities from black soybean [Glycine max (L.) Merr.] byproduct: Isolation, identification and molecular docking study. Eur. Food Res. Technol. 2019, 245, 677–689. [Google Scholar] [CrossRef]
- Wen, C.; Zhang, J.; Zhang, H.; Duan, Y.; Ma, H. Plant protein-derived antioxidant peptides: Isolation, identification, mechanism of action and application in food systems: A review. Trends Food Sci. Technol. 2020, 105, 308–322. [Google Scholar] [CrossRef]
- Bjørlie, M.; Hartmann, J.C.; Rasmussen, L.H.; Yesiltas, B.; Sørensen, A.-D.M.; Echers, S.G.; Jacobsen, C. Screening for Metal-Chelating Activity in Potato Protein Hydrolysates Using Surface Plasmon Resonance and Peptidomics. Antioxidants 2024, 13, 346. [Google Scholar] [CrossRef] [PubMed]
- Varona, E.; García-Moreno, P.J.; Echers, S.G.; Olsen, T.H.; Marcatili, P.; Guardiola, F.; Overgaard, M.T.; Hansen, E.B.; Jacobsen, C.; Yesiltas, B. Antioxidant peptides from alternative sources reduce lipid oxidation in 5% fish oil-in-water emulsions (pH 4) and fish oil-enriched mayonnaise. Food Chem. 2023, 426, 136498. [Google Scholar] [CrossRef] [PubMed]
- Yesiltas, B.; García-Moreno, P.J.; Gregersen, S.; Olsen, T.H.; Jones, N.C.; Hoffmann, S.V.; Marcatili, P.; Overgaard, M.T.; Hansen, E.B.; Jacobsen, C. Antioxidant peptides derived from potato, seaweed, microbial and spinach proteins: Oxidative stability of 5% fish oil-in-water emulsions. Food Chem. 2022, 385, 132699. [Google Scholar] [CrossRef] [PubMed]
- César, A.P.C.; Lopes, F.E.S.; Azevedo, F.F.N.; Pinto, Y.O.; Andrade, C.R.; Mesquita, F.P.; Silva, G.O.; Freitas, C.D.T.; Souza, P.F.N. Antioxidant peptides from plants: A review. Phytochem. Rev. 2024, 23, 95–104. [Google Scholar] [CrossRef]
- Vanani, F.R.; Shabani, L.; Sabzalian, M.R.; Dehghanian, F.; Winner, L. Comparative physiological and proteomic analysis indicates lower shock response to drought stress conditions in a self-pollinating perennial ryegrass. PLoS ONE 2020, 15, e0234317. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Woo, J.H.; Song, Y.; Lee, S.-H.; Hasan, M.; Azad, A.K.; Lee, K.-W. Heat Shock Proteins and Antioxidant Genes Involved in Heat Combined with Drought Stress Responses in Perennial Rye Grass. Life 2022, 12, 1426. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Li, Z.; Yang, J.; Zu, Y. Responses of antioxidant enzymes and key resistant substances in perennial ryegrass (Lolium perenne L.) to cadmium and arsenic stresses. BMC Plant Biol. 2022, 22, 145. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Q.; Xue, B.; Wu, H.; Song, G.; Zhang, X. Arsenic and nutrient absorption characteristics and antioxidant response in different leaves of two ryegrass (Lolium perenne) species under arsenic stress. PLoS ONE 2019, 14, e0225373. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.J.; Hoernstein, S.N.W.; Reski, R. Approaches to characterize organelle, compartment, or structure purity. Methods Mol. Biol. 2017, 1511, 13–28. [Google Scholar] [CrossRef]
- Nicklisch, S.C.T.; Waite, J.H. Optimized DPPH assay in a detergent-based buffer system for measuring antioxidant activity of proteins. MethodsX 2014, 1, e233–e238. [Google Scholar] [CrossRef] [PubMed]
- Sabeena Farvin, K.H.; Baron, C.P.; Nielsen, N.S.; Jacobsen, C. Antioxidant activity of yoghurt peptides: Part 1-in vitro assays and evaluation in ω-3 enriched milk. Food Chem. 2010, 123, 1081–1089. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Quaglieri, A.; Bloom, P.; Triantafyllidis, P.; Green, B.; Condina, P.R.; Ngov, P.B.; Infusini, P.; Webb, P.I. Mass Dynamics 2.0: An improved modular web-based platform for accelerated proteomics insight generation and decision making. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tanambell, H.; Møller, A.H.; Roman, L.; Corredig, M.; Dalsgaard, T.K. Supramolecular structure modification of RuBisCO from alfalfa during removal of chloroplastic materials. Innov. Food Sci. Emerg. Technol. 2023, 87, 103408. [Google Scholar] [CrossRef]
- Chen, Z.; Bertin, R.; Froldi, G. EC50 estimation of antioxidant activity in DPPH assay using several statistical programs. Food Chem. 2013, 138, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Krey, J.F.; Scheffer, D.I.; Choi, D.; Reddy, A.; David, L.L.; Corey, D.P.; Barr-Gillespie, P.G. Mass spectrometry quantitation of proteins from small pools of developing auditory and vestibular cells. Sci. Data 2018, 5, 180128. [Google Scholar] [CrossRef] [PubMed]
- Echers, S.G.; Abdul-Khalek, N.; Mikkelsen, R.K.; Holdt, S.L.; Jacobsen, C.; Hansen, E.B.; Olsen, T.H.; Sejberg, J.J.; Overgaard, M.T. Is Gigartina a potential source of food protein and functional peptide-based ingredients? Evaluating an industrial, pilot-scale extract by proteomics and bioinformatics. Future Foods 2022, 6, 100189. [Google Scholar] [CrossRef]
- Stoykov, Y.M.; Pavlov, A.I.; Krastanov, A.I. Chitinase biotechnology: Production, purification, and application. Eng. Life Sci. 2015, 15, 30–38. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2011, 35, 211. [Google Scholar] [CrossRef] [PubMed]
- Karapetyan, H.; Marutyan, S.; Muradyan, A.; Badalyan, H.; Marutyan, S.V.; Trchounian, K. Changes in ATPase activity, antioxidant enzymes and proline biosynthesis in yeast Candida guilliermondii NP-4 under X-irradiation. J. Bioenerg. Biomembr. 2024, 56, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Schröder, E.; Harris, J.R.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, M.E.; Mata-Cabana, A.; Sánchez-Riego, A.M.; Lindahl, M.; Florencio, F.J. A comprehensive analysis of the peroxiredoxin reduction system in the cyanobacterium Synechocystis sp. strain PCC 6803 reveals that all five peroxiredoxins are thioredoxin dependent. J. Bacteriol. 2009, 191, 7477–7489. [Google Scholar] [CrossRef]
- Zheng, M.; Liu, Y.; Zhang, G.; Yang, Z.; Xu, W.; Chen, Q. The Applications and Mechanisms of Superoxide Dismutase in Medicine, Food, and Cosmetics. Antioxidants 2023, 12, 1675. [Google Scholar] [CrossRef] [PubMed]
- Kozuleva, M.; Goss, T.; Twachtmann, M.; Rudi, K.; Trapka, J.; Selinski, J.; Ivanov, B.; Garapati, P.; Steinhoff, H.-J.; Hase, T.; et al. Ferredoxin:NADP(H) Oxidoreductase Abundance and Location Influences Redox Poise and Stress Tolerance. Plant Physiol. 2016, 172, 1480–1493. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Du, J.; Yue, L.; Zhan, X. Proteomic analysis of plasma membrane proteins in wheat roots exposed to phenanthrene. Environ. Sci. Pollut. Res. 2016, 23, 10863–10871. [Google Scholar] [CrossRef]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 121942. [Google Scholar] [CrossRef]
- Rouhier, N.; Gelhaye, E.; Jacquot, J.P. Glutaredoxin-dependent Peroxiredoxin from Poplar: Protein-Protein Interaction and Catalytic Mechanism. J. Biol. Chem. 2002, 277, 13609–13614. [Google Scholar] [CrossRef]
- Singhal, S.S.; Singh, S.P.; Singhal, P.; Horne, D.; Singhal, J.; Awasthi, S. Antioxidant Role of Glutathione S-Transferases: 4-Hydroxynonenal, a Key Molecule in Stress-Mediated Signaling. Toxicol. Appl. Pharmacol. 2015, 289, 361. [Google Scholar] [CrossRef]
- de Oliveira, F.K.; Santos, L.O.; Buffon, J.G. Mechanism of action, sources, and application of peroxidases. Food Res. Int. 2021, 143, 110266. [Google Scholar] [CrossRef] [PubMed]
- Vadassery, J.; Tripathi, S.; Prasad, R.; Varma, A.; Oelmüller, R. Monodehydroascorbate reductase 2 and dehydroascorbate reductase 5 are crucial for a mutualistic interaction between Piriformospora indica and Arabidopsis. J. Plant Physiol. 2009, 166, 1263–1274. [Google Scholar] [CrossRef]
- Rouhier, N.; Gelhaye, E.; Gualberto, J.M.; Jordy, M.N.; De Fay, E.; Hirasawa, M.; Duplessis, S.; Lemaire, S.D.; Frey, P.; Martin, F.; et al. Poplar peroxiredoxin Q. A Thioredoxin-Linked Chloroplast Antioxidant Functional in Pathogen Defense. Plant Physiol. 2004, 134, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Peng, L.; Wang, X.; Zhang, L. Reduction in chloroplastic ribulose-5-phosphate-3-epimerase decreases photosynthetic capacity in Arabidopsis. Front. Plant Sci. 2022, 13, 813241. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Panda, D.; Mohanty, S.; Biswal, M.; Dey, P.; Dash, M.; Sah, R.P.; Kumar, S.; Baig, M.J.; Behera, L. Role of sedoheptulose-1,7 bisphosphatase in low light tolerance of rice (Oryza sativa L.). Physiol. Mol. Biol. Plants 2020, 26, 2465–2485. [Google Scholar] [CrossRef]
- Bathellier, C.; Yu, L.J.; Farquhar, G.D.; Coote, M.L.; Lorimer, G.H.; Tcherkez, G. Ribulose 1,5-bisphosphate carboxylase/oxygenase activates O2 by electron transfer. Proc. Natl. Acad. Sci. USA 2020, 117, 24234–24242. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, M.; Tripathy, J.N.; Sommer, F.; Somasundaram, R.; Chung, J.-S.; Nestander, M.; Kruthiventi, M.; Zabet-Moghaddam, M.; Johnson, M.K.; Merchant, S.S.; et al. Enzymatic properties of the ferredoxin-dependent nitrite reductase from Chlamydomonas reinhardtii. Evidence for hydroxylamine as a late intermediate in ammonia production. Photosynth. Res. 2010, 103, 67–77. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crude Fraction | Mass (g) | DM (% w/w) | DM Distribution | Crude Protein 1 (%, DM Basis) | Crude Protein Distribution |
|---|---|---|---|---|---|
| Grass | 75.2 | 19.2 ± 1.1 a | 100% | 11.2 ± 0.4 a | 100% |
| Pulp | 32.9 | 27.7 ± 3.1 b | 63.0% | 10.4 ± 0.4 a | 58.7% |
| Green Juice | 34.4 | 15.5 ± 2.1 a | 36.9% | 12.5 ± 0.5 b | 41.0% |
| DPPH | Iron Chelation | ||||
|---|---|---|---|---|---|
| Protein Fraction | MWavg (kDa) | EC50 (μg/mL) | EC50 (μM) | EC50 (μg/mL) | EC50 (μM) |
| 3 | n.d. 1 | 130 | n.d. 1 | n.d. 2 | n.d. 1,2 |
| 5 | 407 | 120 | 0.29 | n.d. 2 | n.d. 2 |
| 7 | 278 | 150 | 0.53 | 6.5 | 0.023 |
| 16 | 50.0 | n.d. 2 | n.d. 2 | 9.0 | 0.18 |
| 19 | 28.2 | n.d. 2 | n.d. 2 | 7.4 | 0.26 |
| 22 | 15.9 | n.d. 2 | n.d. 2 | 5.9 | 0.37 |
| 25 | 9.00 | n.d. 2 | n.d. 2 | 7.0 | 0.78 |
| 28 | 5.08 | n.d. 2 | n.d. 2 | 7.2 | 1.4 |
| 34 | 1.62 | 191 | 120 | 4.8 | 3.0 |
| 37 | 0.914 | 63 | 69 | 0.57 | 0.62 |
| 39 | 0.624 | 38 | 61 | 0.44 | 0.71 |
| 43 | 0.291 | 51 | 170 | 2.1 | 7.1 |
| 48 | 0.112 | 28 | 250 | 1.6 | 14 |
| 51 | 0.0634 | 13 | 210 | 0.60 | 9.5 |
| 55 | 0.030 | 26 | 880 | 0.31 | 11 |
| Trolox | 0.251 | 37 ± 18 | 150 ± 74 | ||
| EDTA | 0.292 | 2.5 ± 1.5 | 8.5 ± 5.1 | ||
| Protein | Lead AC#s | Cat. | Mechanism | Ref. |
|---|---|---|---|---|
| ATP synthase (α, β, and γ subunits) | A8Y9G7 A8Y9H7 I1GU08 | N | Formation of ATP | [54] |
| ATPase (V-type α subunit) | I1GVU2 | N | Formation of ADP by ATP hydrolysis | [55] |
| Thioredoxin-dependent peroxiredoxin (TPx) | I1IXL1 I1IAA5 | A | Reduction of hydrogen peroxide and hydroperoxides. | [56,57] |
| Superoxide dismutase (SOD) | I1I9J4 | A | Oxygen radical scavenging | [58] |
| Ferredoxin-NADP reductase (FNR) | I1H1Z5 I1HW30 | I | NADPH regeneration | [59] |
| Lactoylglutathione lyase (Glyoxalase I, Glo1) | A0A0Q3HLX8 | I | Glutathione formation | [60] |
| L-ascorbate peroxidase (APX) | H6BDN2 | A | Reduction of hydrogen peroxide | [61] |
| Glutaredoxin-dependent peroxiredoxin (GPx) | I1HY81 | A | Reduction of hydrogen peroxide | [62] |
| Glutathione S-transferase (GST) | A0A165FYU2 | I | Reduction of lipid hydroperoxides by facilitating GSH binding | [63] |
| Peroxidase (Px) | I1HZ46 | A | Reduction of hydrogen peroxide | [64] |
| Putative dehydroascorbate reductase (DHAR) | H6BDN5 | I | Regeneration of ascorbate | [65] |
| Peroxiredoxin Q-like (ycf33) 1 | A0A0Q3H912 | A | Reduction of alkyl hydroperoxides | [66] |
| Ribulose-phosphate 3-epimerase (RPE) | I1H9A1 | N | Calvin cycle enzyme | [67] |
| Sedoheptulose-1,7-biphosphatase (SBP) | I1HTG2 | N | Calvin cycle enzyme | [68] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedersen, K.D.A.; Andersen, L.T.; Heiselberg, M.; Brigsted, C.A.; Støvring, F.L.; Mikkelsen, L.M.; Hansen, S.A.; Rusbjerg-Weberskov, C.E.; Lübeck, M.; Gregersen Echers, S. Identifying Endogenous Proteins of Perennial Ryegrass (Lolium perenne) with Ex Vivo Antioxidant Activity. Proteomes 2025, 13, 8. https://doi.org/10.3390/proteomes13010008
Pedersen KDA, Andersen LT, Heiselberg M, Brigsted CA, Støvring FL, Mikkelsen LM, Hansen SA, Rusbjerg-Weberskov CE, Lübeck M, Gregersen Echers S. Identifying Endogenous Proteins of Perennial Ryegrass (Lolium perenne) with Ex Vivo Antioxidant Activity. Proteomes. 2025; 13(1):8. https://doi.org/10.3390/proteomes13010008
Chicago/Turabian StylePedersen, Kathrine Danner Aakjær, Line Thopholm Andersen, Mads Heiselberg, Camilla Agerskov Brigsted, Freja Lyngs Støvring, Louise Mailund Mikkelsen, Sofie Albrekt Hansen, Christian Enrico Rusbjerg-Weberskov, Mette Lübeck, and Simon Gregersen Echers. 2025. "Identifying Endogenous Proteins of Perennial Ryegrass (Lolium perenne) with Ex Vivo Antioxidant Activity" Proteomes 13, no. 1: 8. https://doi.org/10.3390/proteomes13010008
APA StylePedersen, K. D. A., Andersen, L. T., Heiselberg, M., Brigsted, C. A., Støvring, F. L., Mikkelsen, L. M., Hansen, S. A., Rusbjerg-Weberskov, C. E., Lübeck, M., & Gregersen Echers, S. (2025). Identifying Endogenous Proteins of Perennial Ryegrass (Lolium perenne) with Ex Vivo Antioxidant Activity. Proteomes, 13(1), 8. https://doi.org/10.3390/proteomes13010008