

How Can Proteomics Help to Elucidate the Pathophysiological Crosstalk in Muscular Dystrophy and Associated Multi-System Dysfunction?

 , and
, and
Abstract
1. Introduction
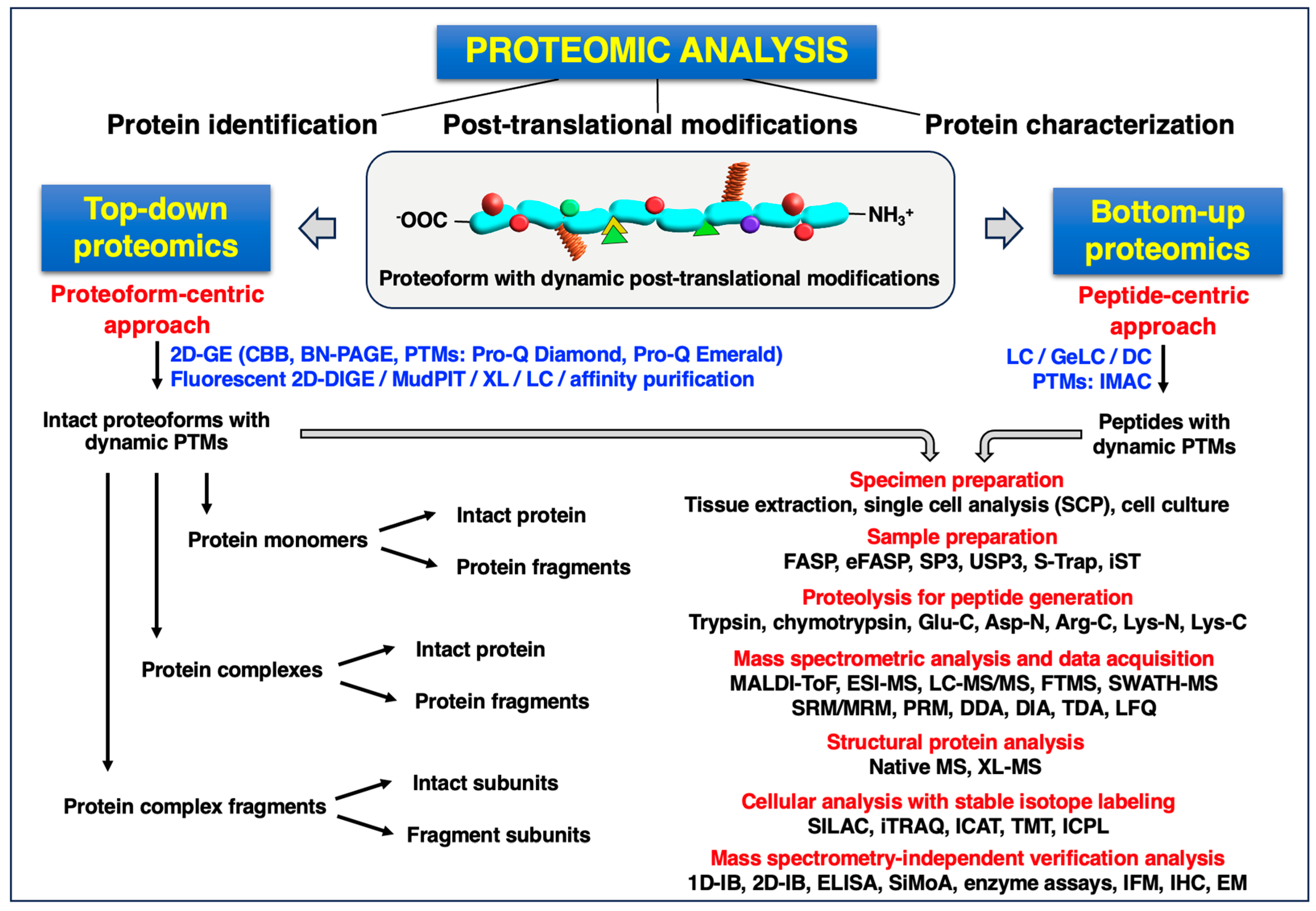
2. Mass Spectrometry-Based Proteomics: Top-Down versus Middle-Up/Down versus Bottom-Up Approaches
2.1. Top-Down Proteomic Approaches
2.2. Bottom-Up Proteomic Approaches
2.3. Mass Spectrometric Analysis and Data Acquisition Techniques

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approach | Bioanalytical Advantages | Disadvantages/Limitations | References |
|---|---|---|---|
| Two-dimensional gel electrophoresis (2D-GE) | Two-dimensional gel electrophoresis has the ability to separate intact proteoforms. Preset conditions, such as pH ranges, size of the 2D gel and staining methods, can be adjusted to increase resolution. Straightforward to interface with many powerful biochemistry techniques including immunoblotting. Two-dimensional gels can be imaged using stains or fluorescent dyes, including labeling of PTMs. | Two-dimensional gel electrophoresis exhibits a narrower dynamic range as compared to certain LC-based separation methods. Difficult to resolve very acidic or very basic proteins. Problematic analysis of very low- or extremely high-molecular-weight proteins. | [55,56,57,58,59,60,61,62,63,64,65,66,71] |
| Differential imaging gel electrophoresis using fluorescence two-dimensional difference gel electrophoresis (2D-DIGE) | Two-dimensional difference gel electrophoresis allows the simultaneous investigation and comparison of three different samples on one two-dimensional gel, thus reducing gel-to-gel variability. Normalization within an experiment can be carried out via the inclusion of an internal control (such as CyDye2) in all sample sets. | A significant number of steps are involved, taking multiple days to complete. Multiple phenotype comparison is still a challenge using the 2D-DIGE technique. | [86,87,88,89,90,91,92,93] |
| Gel electrophoresis–liquid chromatography (GeLC) methods | The initial 1D-GE step using the GeLC-MS/MS technique allows for the efficient separation of extremely large proteins that do not properly separate in conventional 2D gels. | GeLC-based methods are based on crowded 1D gel bands with the limited resolution of individual protein species. | [103,104,105] |
| Protein microarrays | Microarray technology allows high throughput of samples. Different formats are available for general and targeted custom screening approaches. Systems can be arrayed as semi-quantitative or quantitative formats. | High-quality antibodies are not available for all targets. Microarrays require two specific antibodies for each target from the specific sample of interest. | [143,144,145,146] |
| Matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectrometry | MALDI-ToF MS is characterized by a simple operation mode and good mass accuracy, as well as high resolution and sensitivity for peptide mass fingerprinting (PMF). The method can be used for profiling and imaging of proteins directly using thin tissue sections (MALDI-IMS; imaging mass spectrometry) | The sequence information provided by MALDI-ToF MS is generally not as comprehensive as that generated by LC-MS/MS. The method has reduced success rates for identifying proteins that are not in databases. | [70,147,148] |
| Surface-enhanced laser desorption/ionization time-of-flight (SELDI-ToF) mass spectrometry | SELDI-ToF MS allows for high-throughput analyses. The preanalytical sample preparation is rapid and streamlined due to the ability to achieve chromatographic separation using a variety of protein-chip surfaces. | Results are generally based on peptides and smaller proteins (<30 kDa). Additional effort is required to identify peaks of interest. Relatively low resolution of MS scans and low sensitivity. | [149,150] |
| Label-free quantification (LFQ) mass spectrometry | LFQ MS analysis does not require expensive chemicals or metabolic tags, making it a cost-effective proteomic method. The time needed for sample preparation is significantly reduced due to a straightforward workflow as compared to labeling techniques. | Factors such as the peptide or spectral count have limitations. Considerably more LC-MS time is needed for sample analysis. Low-abundance peptides may be underrepresented. | [124,125,126,127] |
| Isobaric tandem mass tagging (TMT) | The TMT method makes it possible to analyze a significant number of samples that can be labeled (18-plex). Specifically linked protocols, such as synchronous precursor selection (SPS), can be helpful in identifying and quantifying low-abundance proteins. | During TMT experiments, replication in labeling procedures and batch effects cannot be completely uniform. The method is associated with a high cost of reagents. | [128,131] |
| Stable isotope labeling by amino acids in cell culture (SILAC) | For the SILAC approach, no in vitro labeling steps are necessary in the experimental procedures. Heavy and light amino acids share the same physico-chemical properties. | SILAC has limited sample multiplexing capabilities and can only be carried out using cell culture or labeling of whole organisms. | [130,134] |
| Multiplex enzyme-linked immunosorbent assay (ELISA) | The ELISA method can be conveniently used for the verification of the proteomic identification of distinct protein species. Multiplex ELISA techniques use fewer wells and/or plates for sample handling and have increased throughput capabilities and the ability to develop custom panels. | Identifying antibodies with high specificity is a challenge due to issues with cross-reactivity. Proteins that are present at different abundance levels make linearity over a wide range of concentrations difficult. | [160,161,162] |
| Microscopical analysis | Histological, histochemical and immunofluorescence microscopical studies can be employed to confirm proteomic results. Verification analyses can be carried out with both freshly dissected or frozen tissue samples for single-cell analysis. The techniques allow the subcellular localization of protein expression levels in a tissue sample with a fast turn-around time to achieve meaningful results. Of note, the recent development of imaging mass cytometry using metal-labeled antibodies has greatly improved the scope of microscopical investigations. | Although these techniques provide data on the single-cell level, the optimization and quantifying results can be difficult. Immuno-histological studies can be subject to human error. Often, a highly trained histopathologist is needed for the proper interpretation of results. Imaging mass cytometry is associated with high costs due to the production of special antibodies. | [169,170,171,172,173,174,175] |
| Flow cytometry (FC) | FC allows simultaneous cell biological analysis with multiple parameters. The method identifies small populations of cells within complex samples and allows for the quantification of fluorescence intensities. | For a successful analysis, the method requires the careful choosing of a suitable combination of fluorochrome conjugates. Complex instruments are prone to analytical problems. | [176,177,178] |
3. Skeletal Muscle Heterogeneity and Muscle Proteomics
3.1. Tissue Heterogeneity and Cellular Complexity of Skeletal Muscles
3.2. Protein Markers of Myofiber Specification
3.3. Cellular Complexity of the Muscle Environment
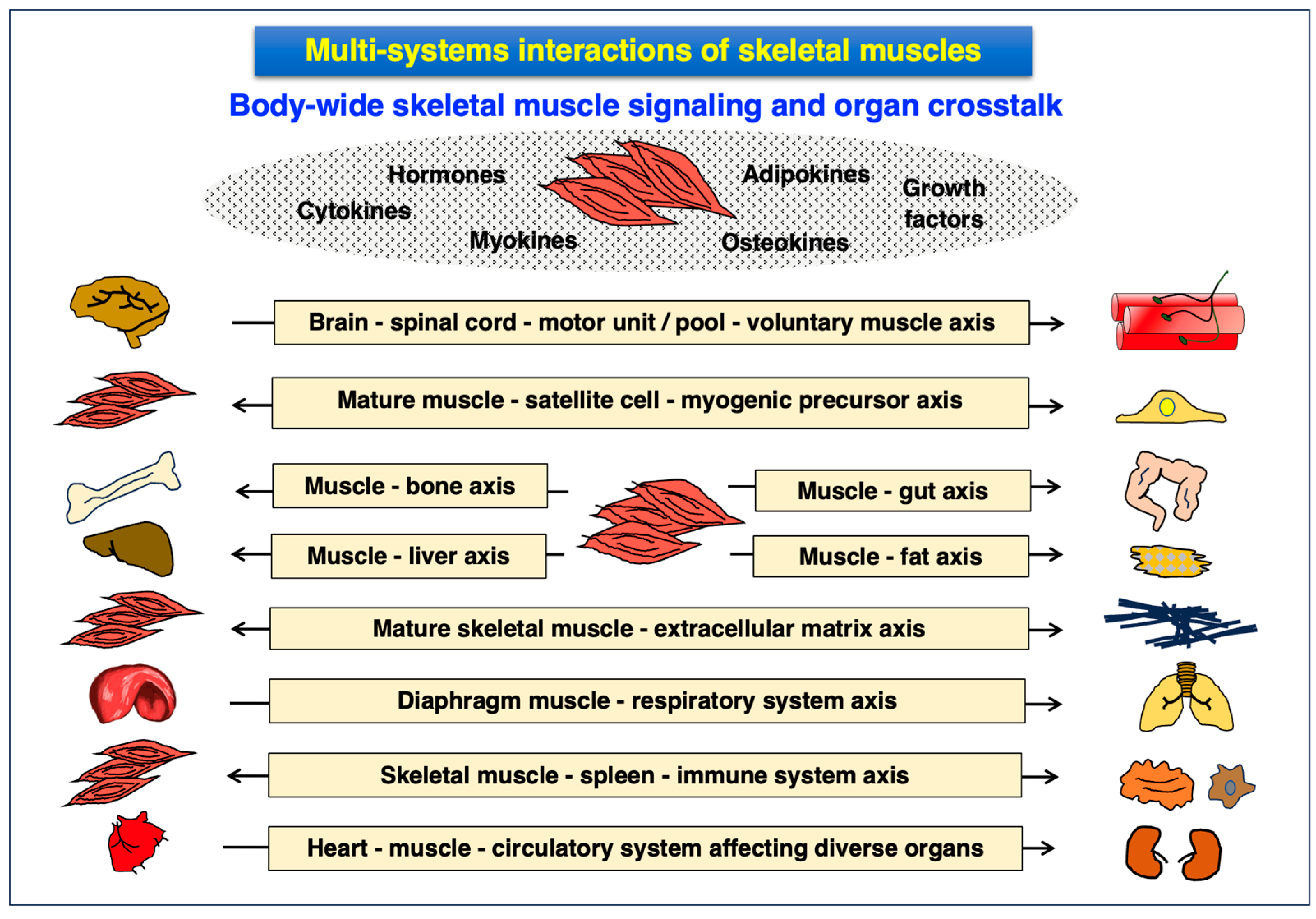
3.4. Multi-System Interactions of Skeletal Muscles and the Muscle Secretome
3.5. Skeletal Muscle Proteomics
4. The Pathoproteomic Profiling of Duchenne Muscular Dystrophy
4.1. The Genetic Basis of Dystrophinopathy
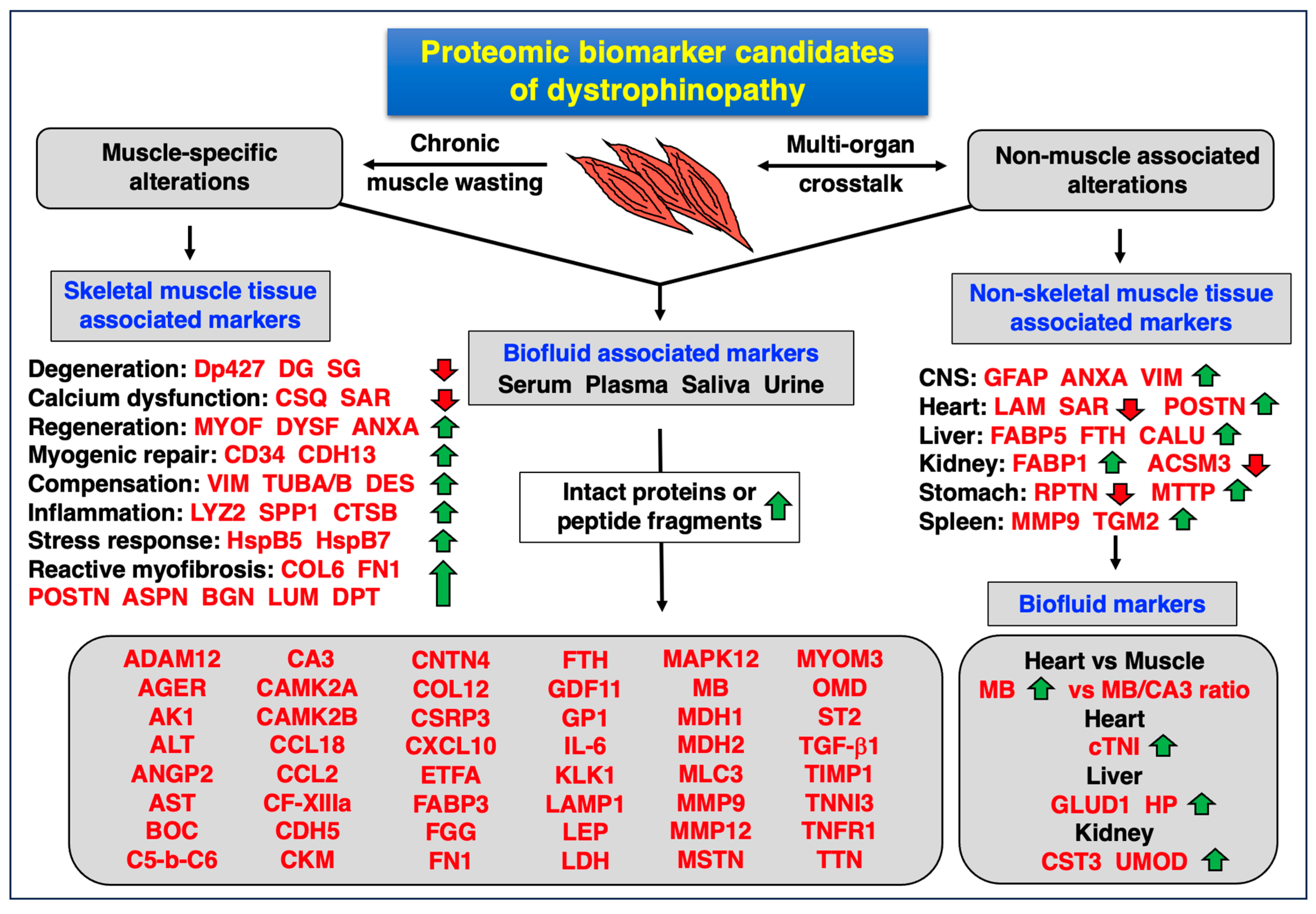
4.2. Pathoproteomics of Chronic Muscle Wasting in Dystrophinopathy
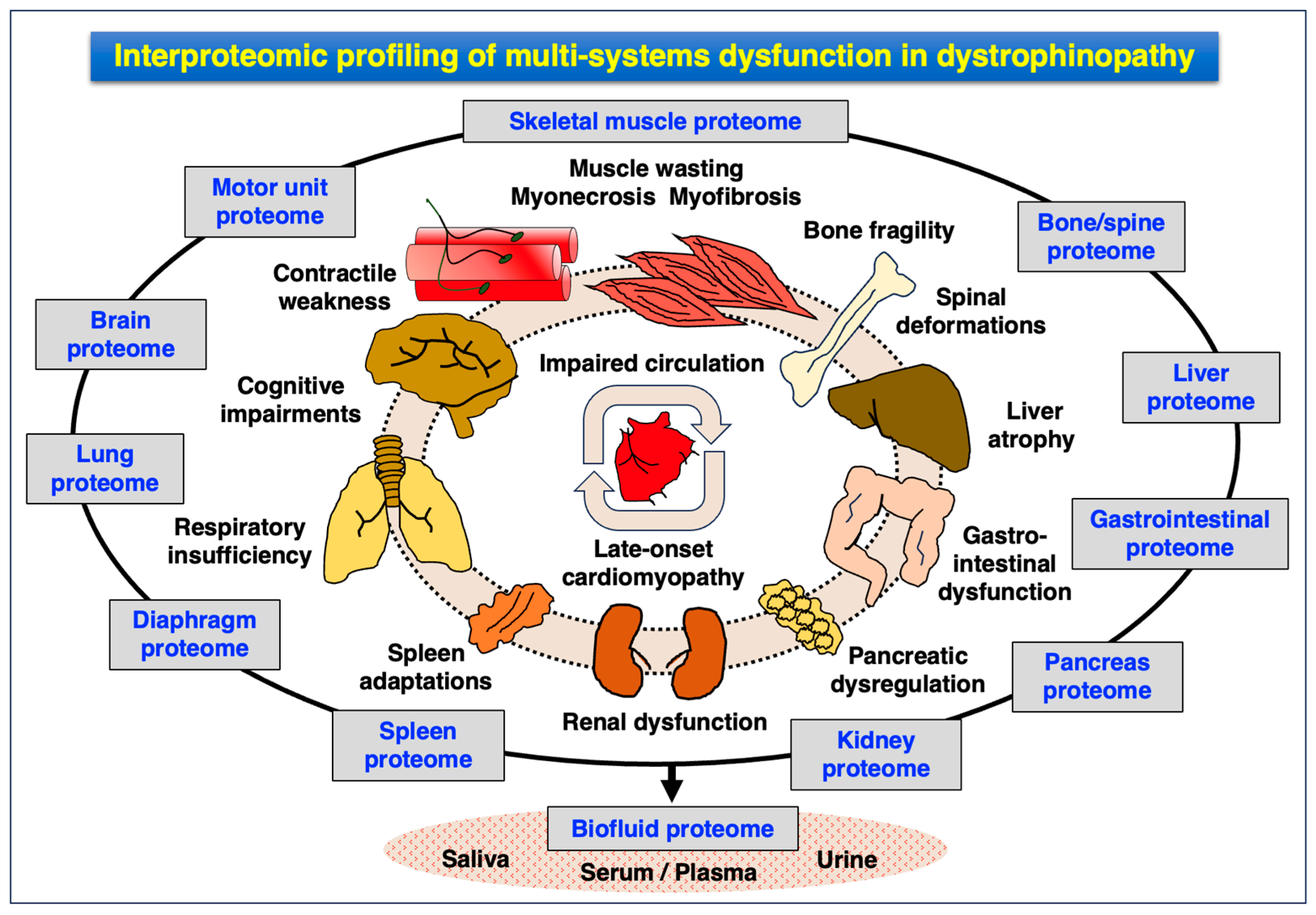
4.3. Pathoproteomics of Multi-System Dysfunction in Dystrophinopathy
- Central nervous system: cognitive impairments, attention deficit, altered emotions, impaired language, memory deficiencies and altered coordination;
- Peripheral nervous system: abnormal transmission at nerve–muscle connections;
- Cardio-respiratory system: late-onset cardiomyopathy, cardio-respiratory syndrome, respiratory insufficiency;
- Liver: enlargement, steatosis, fibrosis, atrophy and ectopic fat deposition;
- Renal system: kidney failure, cardio-renal syndrome, hyperfiltration, hypertension and ectopic fat deposition;
- Bladder: dysfunction of the urinary tract and bladder;
- Bone: increased risk of bone fragility;
- Spine: high risk of development of scoliosis;
- Gastrointestinal system: delayed gastric emptying and pancreatic dysregulation;
- Immune system: hyperactivity causing chronic inflammation, spleen adaptations.
4.4. Proteomic Biomarkers of Muscular and Multi-System Changes in Dystrophinopathy
- Susceptibility markers: for risk assessment of potential disease initiation;
- Diagnostic markers: for initial detection of a specific disease process;
- Prognostic markers: for envisaging disease progression and adverse clinical events;
- Predictive markers: for differential patient screening and individual sensitivities;
- Pharmacodynamic markers: for assessing the body’s response to a specific treatment;
- Therapeutic monitoring markers: for the repeated assessment of disease status following therapeutic intervention;
- Safety-related biomarkers: for determining potential adverse side effects.
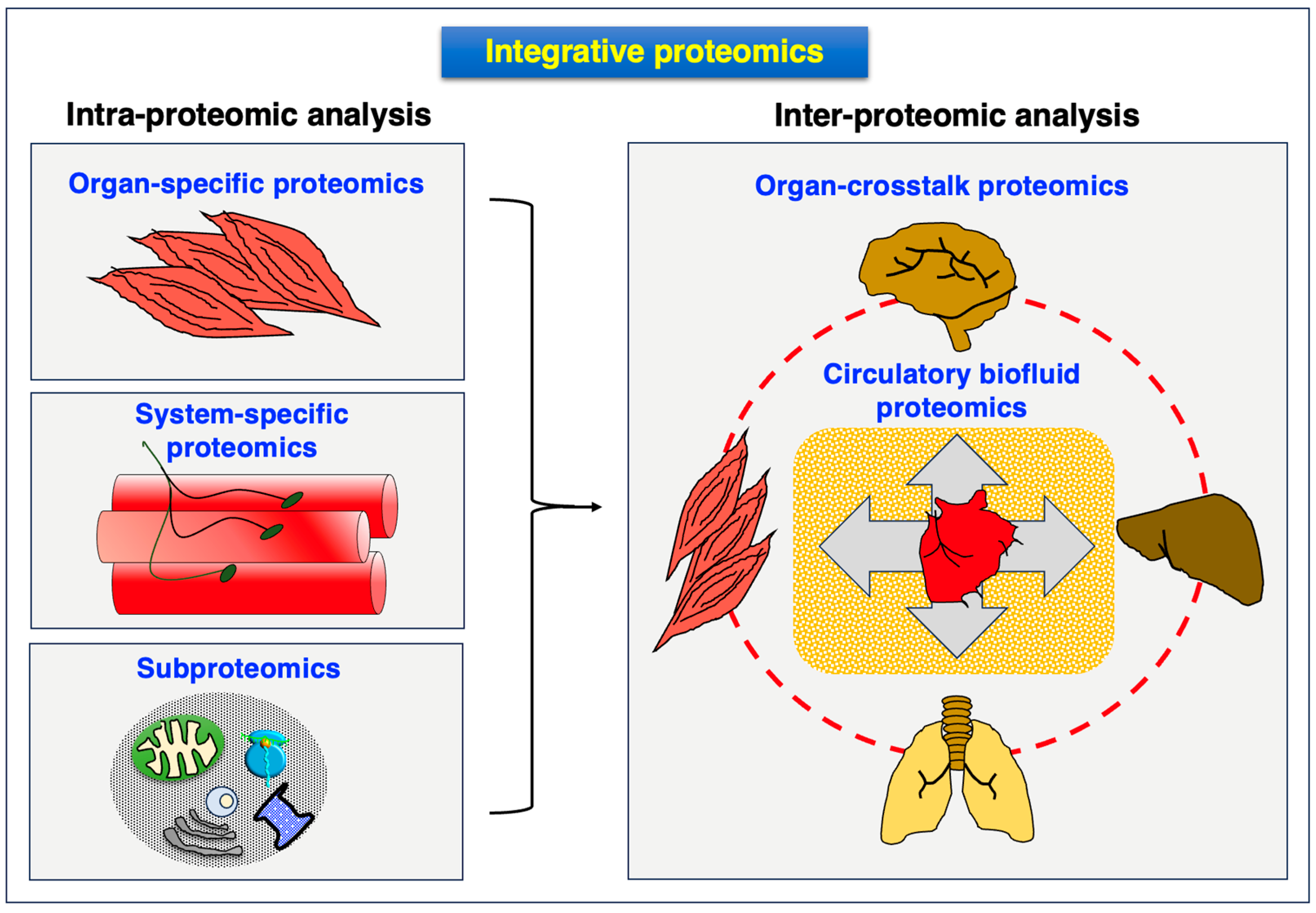
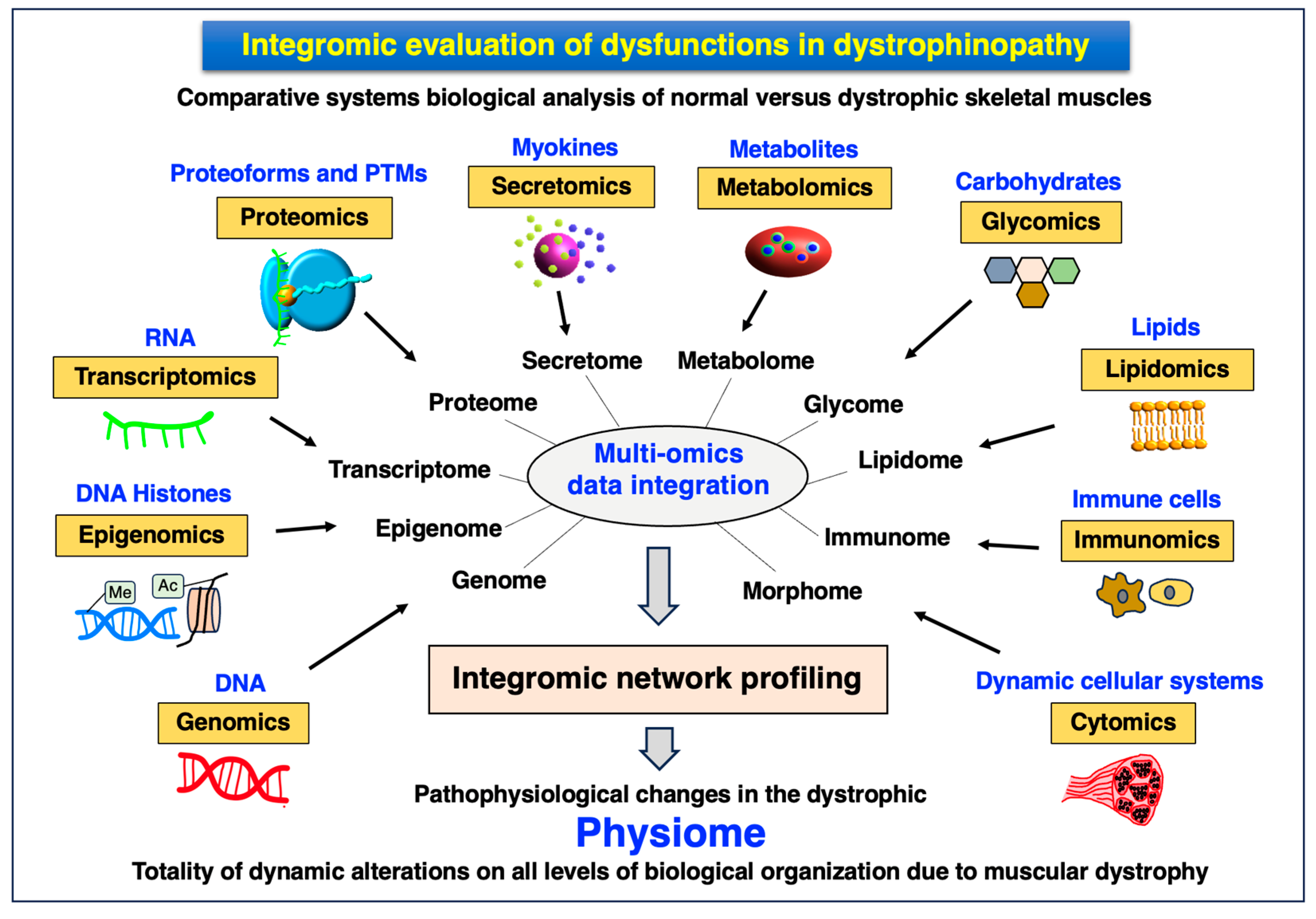
4.5. Integromics: Systems Biological Multi-Omics Analysis of Dystrophinopathy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Börner, K.; Teichmann, S.A.; Quardokus, E.M.; Gee, J.C.; Browne, K.; Osumi-Sutherland, D.; Herr, B.W., 2nd; Bueckle, A.; Paul, H.; Haniffa, M.; et al. Anatomical structures, cell types and biomarkers of the Human Reference Atlas. Nat. Cell Biol. 2021, 23, 1117–1128. [Google Scholar] [CrossRef]
- Bianconi, E.; Piovesan, A.; Facchin, F.; Beraudi, A.; Casadei, R.; Frabetti, F.; Vitale, L.; Pelleri, M.C.; Tassani, S.; Piva, F.; et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013, 40, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Hatton, I.A.; Galbraith, E.D.; Merleau, N.S.C.; Miettinen, T.P.; Smith, B.M.; Shander, J.A. The human cell count and size distribution. Proc. Natl. Acad. Sci. USA 2023, 120, e2303077120. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.; Baryshnikova, A.; Brown, G.W. Unification of Protein Abundance Datasets Yields a Quantitative Saccharomyces cerevisiae Proteome. Cell Syst. 2018, 6, 192–205.e3. [Google Scholar] [CrossRef]
- Adhikari, S.; Nice, E.C.; Deutsch, E.W.; Lane, L.; Omenn, G.S.; Pennington, S.R.; Paik, Y.K.; Overall, C.M.; Corrales, F.J.; Cristea, I.M.; et al. A high-stringency blueprint of the human proteome. Nat. Commun. 2020, 11, 5301. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, M.; Lin, S.; Jian, R.; Li, X.; Chan, J.; Dong, G.; Fang, H.; Robinson, A.E.; GTEx Consortium; et al. A Quantitative Proteome Map of the Human Body. Cell 2020, 183, 269–283.e19. [Google Scholar] [CrossRef]
- Omenn, G.S.; Lane, L.; Overall, C.M.; Cristea, I.M.; Corrales, F.J.; Lindskog, C.; Paik, Y.K.; Van Eyk, J.E.; Liu, S.; Pennington, S.R.; et al. Research on the Human Proteome Reaches a Major Milestone: >90% of Predicted Human Proteins Now Credibly Detected, According to the HUPO Human Proteome Project. J. Proteome Res. 2020, 19, 4735–4746. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Pertea, M.; Salzberg, S.L. Between a chicken and a grape: Estimating the number of human genes. Genome Biol. 2010, 11, 206. [Google Scholar] [CrossRef]
- Amaral, P.; Carbonell-Sala, S.; De La Vega, F.M.; Faial, T.; Frankish, A.; Gingeras, T.; Guigo, R.; Harrow, J.L.; Hatzigeorgiou, A.G.; Johnson, R.; et al. The status of the human gene catalogue. Nature 2023, 622, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L.; Consortium for Top Down Proteomics. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, L.V.; Millikin, R.J.; Miller, R.M.; Anderson, L.C.; Fellers, R.T.; Ge, Y.; Kelleher, N.L.; LeDuc, R.D.; Liu, X.; Payne, S.H.; et al. Identification and Quantification of Proteoforms by Mass Spectrometry. Proteomics 2019, 19, e1800361. [Google Scholar] [CrossRef] [PubMed]
- Carbonara, K.; Andonovski, M.; Coorssen, J.R. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes 2021, 9, 38. [Google Scholar] [CrossRef]
- Forgrave, L.M.; Wang, M.; Yang, D.; DeMarco, M.L. Proteoforms and their expanding role in laboratory medicine. Pract. Lab. Med. 2021, 28, e00260. [Google Scholar] [CrossRef]
- Smith, L.M.; Agar, J.N.; Chamot-Rooke, J.; Danis, P.O.; Ge, Y.; Loo, J.A.; Paša-Tolić, L.; Tsybin, Y.O.; Kelleher, N.L.; Consortium for Top-Down Proteomics. The Human Proteoform Project: Defining the human proteome. Sci. Adv. 2021, 7, eabk0734. [Google Scholar] [CrossRef]
- Capitanio, D.; Moriggi, M.; Gelfi, C. Mapping the human skeletal muscle proteome: Progress and potential. Expert Rev. Proteom. 2017, 14, 825–839. [Google Scholar] [CrossRef]
- Gonzalez-Freire, M.; Semba, R.D.; Ubaida-Mohien, C.; Fabbri, E.; Scalzo, P.; Højlund, K.; Dufresne, C.; Lyashkov, A.; Ferrucci, L. The Human Skeletal Muscle Proteome Project: A reappraisal of the current literature. J. Cachexia Sarcopenia Muscle 2017, 8, 5–18. [Google Scholar] [CrossRef]
- Hesketh, S.J.; Stansfield, B.N.; Stead, C.A.; Burniston, J.G. The application of proteomics in muscle exercise physiology. Expert Rev. Proteom. 2020, 17, 813–825. [Google Scholar] [CrossRef]
- Dowling, J.J.; Weihl, C.C.; Spencer, M.J. Molecular and cellular basis of genetically inherited skeletal muscle disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Spendiff, S.; Roos, A.; Bourque, P.R.; Warman Chardon, J.; Kirschner, J.; Horvath, R.; Lochmüller, H. Advances in the diagnosis of inherited neuromuscular diseases and implications for therapy development. Lancet Neurol. 2020, 19, 522–532. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- Bez Batti Angulski, A.; Hosny, N.; Cohen, H.; Martin, A.A.; Hahn, D.; Bauer, J.; Metzger, J.M. Duchenne muscular dystrophy: Disease mechanism and therapeutic strategies. Front. Physiol. 2023, 14, 1183101. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P. The discovery of dystrophin, the protein product of the Duchenne muscular dystrophy gene. FEBS J. 2020, 287, 3879–3887. [Google Scholar] [CrossRef]
- Roberts, R.G. Dystrophin, its gene, and the dystrophinopathies. Adv. Genet. 1995, 33, 177–231. [Google Scholar] [CrossRef]
- Fortunato, F.; Farnè, M.; Ferlini, A. The DMD gene and therapeutic approaches to restore dystrophin. Neuromuscul. Disord. 2021, 31, 1013–1020. [Google Scholar] [CrossRef]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Strehle, E.M.; Straub, V. Recent advances in the management of Duchenne muscular dystrophy. Arch. Dis. Child. 2015, 100, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Vita, G.L. Clinical management of Duchenne muscular dystrophy: The state of the art. Neurol. Sci. 2018, 39, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K.; Swandulla, D. Complexity of skeletal muscle degeneration: Multi-systems pathophysiology and organ crosstalk in dystrophinopathy. Pflugers Arch. 2021, 473, 1813–1839. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.A.; Vogel, C. New horizons in the stormy sea of multimodal single-cell data integration. Mol. Cell 2022, 82, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef]
- Mirza, S.P.; Olivier, M. Methods and approaches for the comprehensive characterization and quantification of cellular proteomes using mass spectrometry. Physiol. Genom. 2008, 33, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Val, A.; Guzmán, U.H.; Olsen, J.V. Obtaining Complete Human Proteomes. Annu. Rev. Genom. Hum. Genet. 2022, 23, 99–121. [Google Scholar] [CrossRef]
- Wheeler, C.H.; Berry, S.L.; Wilkins, M.R.; Corbett, J.M.; Ou, K.; Gooley, A.A.; Humphery-Smith, I.; Williams, K.L.; Dunn, M.J. Characterisation of proteins from two-dimensional electrophoresis gels by matrix-assisted laser desorption mass spectrometry and amino acid compositional analysis. Electrophoresis 1996, 17, 580–587. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Sanchez, J.C.; Gooley, A.A.; Appel, R.D.; Humphery-Smith, I.; Hochstrasser, D.F.; Williams, K.L. Progress with proteome projects: Why all proteins expressed by a genome should be identified and how to do it. Biotechnol. Genet. Eng. Rev. 1996, 13, 19–50. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Pasquali, C.; Appel, R.D.; Ou, K.; Golaz, O.; Sanchez, J.C.; Yan, J.X.; Gooley, A.A.; Hughes, G.; Humphery-Smith, I.; et al. From proteins to proteomes: Large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology 1996, 14, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Vistain, L.F.; Tay, S. Single-Cell Proteomics. Trends Biochem. Sci. 2021, 46, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Mund, A.; Brunner, A.D.; Mann, M. Unbiased spatial proteomics with single-cell resolution in tissues. Mol. Cell 2022, 82, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, M.; Mayer, R.L.; Mechtler, K. Label-free single cell proteomics utilizing ultrafast LC and MS instrumentation: A valuable complementary technique to multiplexing. Proteomics 2023, 23, e2200162. [Google Scholar] [CrossRef] [PubMed]
- Habeck, T.; Lermyte, F. Seeing the complete picture: Proteins in top-down mass spectrometry. Essays Biochem. 2023, 67, 283–300. [Google Scholar] [CrossRef]
- Miller, R.M.; Smith, L.M. Overview and considerations in bottom-up proteomics. Analyst 2023, 148, 475–486. [Google Scholar] [CrossRef]
- Lermyte, F.; Tsybin, Y.O.; O’Connor, P.B.; Loo, J.A. Top or Middle? Up or Down? Toward a Standard Lexicon for Protein Top-Down and Allied Mass Spectrometry Approaches. J. Am. Soc. Mass Spectrom. 2019, 30, 1149–1157. [Google Scholar] [CrossRef]
- Dowling, P.; Swandulla, S.; Ohlendieck, K. Mass spectrometry-based proteomic technology and its application to study skeletal muscle cell biology. Cells 2023, 12, 2560. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Swandulla, D.; Ohlendieck, K. Fiber-Type Shifting in Sarcopenia of Old Age: Proteomic Profiling of the Contractile Apparatus of Skeletal Muscles. Int. J. Mol. Sci. 2023, 24, 2415. [Google Scholar] [CrossRef]
- Dowling, P.; Swandulla, D.; Ohlendieck, K. Biochemical and proteomic insights into sarcoplasmic reticulum Ca2+-ATPase complexes in skeletal muscles. Expert Rev. Proteom. 2023, 20, 125–142. [Google Scholar] [CrossRef]
- Yates, J.R., 3rd. Recent technical advances in proteomics. F1000Research 2019, 8, 351. [Google Scholar] [CrossRef] [PubMed]
- Matthiesen, R.; Carvalho, A.S. Methods and Algorithms for Quantitative Proteomics by Mass Spectrometry. Methods Mol. Biol. 2020, 2051, 161–197. [Google Scholar] [CrossRef]
- Rozanova, S.; Barkovits, K.; Nikolov, M.; Schmidt, C.; Urlaub, H.; Marcus, K. Quantitative Mass Spectrometry-Based Proteomics: An Overview. Methods Mol. Biol. 2021, 2228, 85–116. [Google Scholar] [CrossRef]
- Neagu, A.N.; Jayathirtha, M.; Baxter, E.; Donnelly, M.; Petre, B.A.; Darie, C.C. Applications of Tandem Mass Spectrometry (MS/MS) in Protein Analysis for Biomedical Research. Molecules 2022, 27, 2411. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.M.; Coorssen, J.R.; Martins-de-Souza, D. 2DE: The phoenix of proteomics. J. Proteom. 2014, 104, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Westermeier, R. 2D gel-based Proteomics: There’s life in the old dog yet. Arch. Physiol. Biochem. 2016, 122, 236–237. [Google Scholar] [CrossRef]
- Zhan, X.; Li, B.; Zhan, X.; Schlüter, H.; Jungblut, P.R.; Coorssen, J.R. Innovating the Concept and Practice of Two-Dimensional Gel Electrophoresis in the Analysis of Proteomes at the Proteoform Level. Proteomes 2019, 7, 36. [Google Scholar] [CrossRef]
- Marcus, K.; Lelong, C.; Rabilloud, T. What Room for Two-Dimensional Gel-Based Proteomics in a Shotgun Proteomics World? Proteomes 2020, 8, 17. [Google Scholar] [CrossRef]
- Murphy, S.; Dowling, P.; Ohlendieck, K. Comparative Skeletal Muscle Proteomics Using Two-Dimensional Gel Electrophoresis. Proteomes 2016, 4, 27. [Google Scholar] [CrossRef]
- Dowling, P.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Characterization of Contractile Proteins from Skeletal Muscle Using Gel-Based Top-Down Proteomics. Proteomes 2019, 7, 25. [Google Scholar] [CrossRef]
- Zabel, C.; Klose, J. High-resolution large-gel 2DE. Methods Mol. Biol. 2009, 519, 311–338. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.B.; Hoving, S.; Westermeier, R. Isoelectric focusing and two-dimensional gel electrophoresis. Methods Enzymol. 2009, 463, 515–540. [Google Scholar] [CrossRef]
- Rabilloud, T.; Chevallet, M.; Luche, S.; Lelong, C. Two-dimensional gel electrophoresis in proteomics: Past, present and future. J. Proteom. 2010, 73, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Lelong, C. Two-dimensional gel electrophoresis in proteomics: A tutorial. J. Proteom. 2011, 74, 1829–1841. [Google Scholar] [CrossRef] [PubMed]
- Westermeier, R. Looking at proteins from two dimensions: A review on five decades of 2D electrophoresis. Arch. Physiol. Biochem. 2014, 120, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.Y.; Saraygord-Afshari, N.; Low, T.Y. The evolution of two-dimensional gel electrophoresis—From proteomics to emerging alternative applications. J. Chromatogr. A 2020, 1615, 460763. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, G.K.; Thelen, J.J. A high-resolution two dimensional Gel- and Pro-Q DPS-based proteomics workflow for phosphoprotein identification and quantitative profiling. Methods Mol. Biol. 2009, 527, 3–19. [Google Scholar] [CrossRef]
- Lourenço Dos Santos, S.; Baraibar, M.A.; Lundberg, S.; Eeg-Olofsson, O.; Larsson, L.; Friguet, B. Oxidative proteome alterations during skeletal muscle ageing. Redox Biol. 2015, 5, 267–274. [Google Scholar] [CrossRef]
- Rodríguez-Vázquez, R.; Mouzo, D.; Zapata, C. Phosphoproteome Analysis Using Two-Dimensional Electrophoresis Coupled with Chemical Dephosphorylation. Foods 2022, 11, 3119. [Google Scholar] [CrossRef]
- Zimny-Arndt, U.; Schmid, M.; Ackermann, R.; Jungblut, P.R. Classical proteomics: Two-dimensional electrophoresis/MALDI mass spectrometry. Methods Mol. Biol. 2009, 492, 65–91. [Google Scholar] [CrossRef]
- Reed, P.W.; Densmore, A.; Bloch, R.J. Optimization of large gel 2D electrophoresis for proteomic studies of skeletal muscle. Electrophoresis 2012, 33, 1263–1270. [Google Scholar] [CrossRef]
- Noaman, N.; Coorssen, J.R. Coomassie does it (better): A Robin Hood approach to total protein quantification. Anal. Biochem. 2018, 556, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Noaman, N.; Abbineni, P.S.; Withers, M.; Coorssen, J.R. Coomassie staining provides routine (sub)femtomole in-gel detection of intact proteoforms: Expanding opportunities for genuine Top-down Proteomics. Electrophoresis 2017, 38, 3086–3099. [Google Scholar] [CrossRef]
- Chevalier, F. Standard Dyes for Total Protein Staining in Gel-Based Proteomic Analysis. Materials 2010, 3, 4784–4792. [Google Scholar] [CrossRef] [PubMed]
- Panfoli, I.; Calzia, D.; Santucci, L.; Ravera, S.; Bruschi, M.; Candiano, G. A blue dive: From ‘blue fingers’ to ‘blue silver’. A comparative overview of staining methods for in-gel proteomics. Expert Rev. Proteom. 2012, 9, 627–634. [Google Scholar] [CrossRef]
- Sundaram, P. Protein Stains and Applications. In Protein Gel Detection and Imaging; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1853, pp. 1–14. [Google Scholar] [CrossRef]
- Krishnamoorthy, V.; Donofrio, A.J.; Martin, J.L. O-GlcNAcylation of αB-crystallin regulates its stress-induced translocation and cytoprotection. Mol. Cell. Biochem. 2013, 379, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Mehta-D’souza, P. Detection of Glycoproteins in Polyacrylamide Gels Using Pro-Q Emerald 300 Dye, a Fluorescent Periodate Schiff-Base Stain. Methods Mol. Biol. 2018, 1853, 115–119. [Google Scholar] [CrossRef]
- Gannon, J.; Staunton, L.; O’Connell, K.; Doran, P.; Ohlendieck, K. Phosphoproteomic analysis of aged skeletal muscle. Int. J. Mol. Med. 2008, 22, 33–42. [Google Scholar] [CrossRef]
- Steinberger, B.; Mayrhofer, C. Principles and examples of gel-based approaches for phosphoprotein analysis. Methods Mol. Biol. 2015, 1295, 305–321. [Google Scholar] [CrossRef]
- Murphy, S.; Ohlendieck, K. Protein Digestion for 2D-DIGE Analysis. Methods Mol. Biol. 2023, 2596, 339–349. [Google Scholar] [CrossRef]
- Glatter, T.; Ludwig, C.; Ahrné, E.; Aebersold, R.; Heck, A.J.; Schmidt, A. Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion. J. Proteome Res. 2012, 11, 5145–5156. [Google Scholar] [CrossRef] [PubMed]
- Tsiatsiani, L.; Heck, A.J. Proteomics beyond trypsin. FEBS J. 2015, 282, 2612–2626. [Google Scholar] [CrossRef] [PubMed]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc. 2016, 11, 993–1006. [Google Scholar] [CrossRef]
- Dau, T.; Bartolomucci, G.; Rappsilber, J. Proteomics Using Protease Alternatives to Trypsin Benefits from Sequential Digestion with Trypsin. Anal. Chem. 2020, 92, 9523–9527. [Google Scholar] [CrossRef] [PubMed]
- Minden, J.S.; Dowd, S.R.; Meyer, H.E.; Stühler, K. Difference gel electrophoresis. Electrophoresis 2009, 30, S156–S161. [Google Scholar] [CrossRef] [PubMed]
- Arentz, G.; Weiland, F.; Oehler, M.K.; Hoffmann, P. State of the art of 2D DIGE. Proteom. Clin. Appl. 2015, 9, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Blundon, M.; Ganesan, V.; Redler, B.; Van, P.T.; Minden, J.S. Two-Dimensional Difference Gel Electrophoresis. Methods Mol. Biol. 2019, 1855, 229–247. [Google Scholar] [CrossRef]
- Gargan, S.; Ohlendieck, K. Sample Preparation and Protein Determination for 2D-DIGE Proteomics. Methods Mol. Biol. 2023, 2596, 325–337. [Google Scholar] [CrossRef]
- Ohlendieck, K. Top-Down Proteomics and Comparative 2D-DIGE Analysis. Methods Mol. Biol. 2023, 2596, 19–38. [Google Scholar] [CrossRef]
- Tonge, R.; Shaw, J.; Middleton, B.; Rowlinson, R.; Rayner, S.; Young, J.; Pognan, F.; Hawkins, E.; Currie, I.; Davison, M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics 2001, 1, 377–396. [Google Scholar] [CrossRef]
- Marouga, R.; David, S.; Hawkins, E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal. Bioanal. Chem. 2005, 382, 669–678. [Google Scholar] [CrossRef]
- Ohlendieck, K. Comparative 3-Sample 2D-DIGE Analysis of Skeletal Muscles. Methods Mol. Biol. 2023, 2596, 127–146. [Google Scholar] [CrossRef]
- Dowling, P. DIGE Analysis Software and Protein Identification Approaches. Methods Mol. Biol. 2023, 2596, 39–50. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Bioinformatic Analysis of the Subproteomic Profile of Cardiomyopathic Tissue. Methods Mol. Biol. 2023, 2596, 377–395. [Google Scholar] [CrossRef]
- Greengauz-Roberts, O.; Stöppler, H.; Nomura, S.; Yamaguchi, H.; Goldenring, J.R.; Podolsky, R.H.; Lee, J.R.; Dynan, W.S. Saturation labeling with cysteine-reactive cyanine fluorescent dyes provides increased sensitivity for protein expression profiling of laser-microdissected clinical specimens. Proteomics 2005, 5, 1746–1757. [Google Scholar] [CrossRef]
- Banks, C.A.; Kong, S.E.; Washburn, M.P. Affinity purification of protein complexes for analysis by multidimensional protein identification technology. Protein Expr. Purif. 2012, 86, 105–119. [Google Scholar] [CrossRef]
- Elschenbroich, S.; Ignatchenko, V.; Sharma, P.; Schmitt-Ulms, G.; Gramolini, A.O.; Kislinger, T. Peptide separations by on-line MudPIT compared to isoelectric focusing in an off-gel format: Application to a membrane-enriched fraction from C2C12 mouse skeletal muscle cells. J. Proteome Res. 2009, 8, 4860–4869. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Murphy, S.; Dowling, P.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of giant skeletal muscle proteins. Expert Rev. Proteom. 2019, 16, 241–256. [Google Scholar] [CrossRef]
- Hirabayashi, T. Agarose isoelectric focusing for the detection of many isoforms and high molecules in muscle protein analysis. Electrophoresis 2000, 21, 446–451. [Google Scholar] [CrossRef]
- Oh-Ishi, M.; Maeda, T. Disease proteomics of high-molecular-mass proteins by two-dimensional gel electrophoresis with agarose gels in the first dimension (Agarose 2-DE). J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 849, 211–222. [Google Scholar] [CrossRef]
- Nakagawa, M.; Tomioka, Y.; Sakuma, C.; Kurosawa, Y.; Shibata, T.; Arakawa, T.; Akuta, T. Development of a novel two-dimensional gel electrophoresis protocol with agarose native gel electrophoresis. Electrophoresis 2023, 44, 1446–1460. [Google Scholar] [CrossRef]
- Murphy, S.; Henry, M.; Meleady, P.; Ohlendieck, K. Utilization of dried and long-term stored polyacrylamide gels for the advanced proteomic profiling of mitochondrial contact sites from rat liver. Biol. Methods Protoc. 2018, 3, bpy008. [Google Scholar] [CrossRef]
- Goldman, A.R.; Beer, L.A.; Tang, H.Y.; Hembach, P.; Zayas-Bazan, D.; Speicher, D.W. Proteome Analysis Using Gel-LC-MS/MS. Curr. Protoc. Protein Sci. 2019, 96, e93. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Ohlendieck, K. Proteomic profiling of large myofibrillar proteins from dried and long-term stored polyacrylamide gels. Anal. Biochem. 2018, 543, 8–11. [Google Scholar] [CrossRef]
- Melby, J.A.; Brown, K.A.; Gregorich, Z.R.; Roberts, D.S.; Chapman, E.A.; Ehlers, L.E.; Gao, Z.; Larson, E.J.; Jin, Y.; Lopez, J.R.; et al. High sensitivity top-down proteomics captures single muscle cell heterogeneity in large proteoforms. Proc. Natl. Acad. Sci. USA 2023, 120, e2222081120. [Google Scholar] [CrossRef] [PubMed]
- Manes, N.P.; Nita-Lazar, A. Application of targeted mass spectrometry in bottom-up proteomics for systems biology research. J. Proteom. 2018, 189, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Drissi, R.; Dubois, M.L.; Boisvert, F.M. Proteomics methods for subcellular proteome analysis. FEBS J. 2013, 280, 5626–5634. [Google Scholar] [CrossRef]
- Lee, Y.H.; Tan, H.T.; Chung, M.C. Subcellular fractionation methods and strategies for proteomics. Proteomics 2010, 10, 3935–3956. [Google Scholar] [CrossRef]
- Plöscher, M.; Granvogl, B.; Reisinger, V.; Masanek, A.; Eichacker, L.A. Organelle proteomics. Methods Mol. Biol. 2009, 519, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Bennett, H.M.; Stephenson, W.; Rose, C.M.; Darmanis, S. Single-cell proteomics enabled by next-generation sequencing or mass spectrometry. Nat. Methods 2023, 20, 363–374. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C.; Murgia, M. Fiber type diversity in skeletal muscle explored by mass spectrometry-based single fiber proteomics. Histol. Histopathol. 2020, 35, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Sielaff, M.; Kuharev, J.; Bohn, T.; Hahlbrock, J.; Bopp, T.; Tenzer, S.; Distler, U. Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J. Proteome Res. 2017, 16, 4060–4072. [Google Scholar] [CrossRef]
- Duong, V.A.; Lee, H. Bottom-Up Proteomics: Advancements in Sample Preparation. Int. J. Mol. Sci. 2023, 24, 5350. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Zougman, A.; Selby, P.J.; Banks, R.E. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 2014, 14, 1006. [Google Scholar] [CrossRef] [PubMed]
- Brandi, J.; Noberini, R.; Bonaldi, T.; Cecconi, D. Advances in enrichment methods for mass spectrometry-based proteomics analysis of post-translational modifications. J. Chromatogr. A 2022, 1678, 463352. [Google Scholar] [CrossRef]
- Xie, Z.; Feng, Q.; Zhang, S.; Yan, Y.; Deng, C.; Ding, C.F. Advances in proteomics sample preparation and enrichment for phosphorylation and glycosylation analysis. Proteomics 2022, 22, e2200070. [Google Scholar] [CrossRef]
- Morandell, S.; Stasyk, T.; Grosstessner-Hain, K.; Roitinger, E.; Mechtler, K.; Bonn, G.K.; Huber, L.A. Phosphoproteomics strategies for the functional analysis of signal transduction. Proteomics 2006, 6, 4047–4056. [Google Scholar] [CrossRef]
- Stasyk, T.; Huber, L.A. DIGE-Based Phosphoproteomic Analysis. Methods Mol. Biol. 2023, 2596, 97–104. [Google Scholar] [CrossRef]
- Ino, Y.; Kinoshita, E.; Kinoshita-Kikuta, E.; Akiyama, T.; Nakai, Y.; Nishino, K.; Osada, M.; Ryo, A.; Hirano, H.; Koike, T.; et al. Evaluation of four phosphopeptide enrichment strategies for mass spectrometry-based proteomic analysis. Proteomics 2022, 22, e2100216. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, X. Current methodologies in protein ubiquitination characterization: From ubiquitinated protein to ubiquitin chain architecture. Cell Biosci. 2022, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Duong, V.A.; Park, J.M.; Lee, H. Review of Three-Dimensional Liquid Chromatography Platforms for Bottom-Up Proteomics. Int. J. Mol. Sci. 2020, 21, 1524. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Protocol for the Bottom-Up Proteomic Analysis of Mouse Spleen. STAR Protoc. 2020, 1, 100196. [Google Scholar] [CrossRef]
- Shah, A.D.; Goode, R.J.A.; Huang, C.; Powell, D.R.; Schittenhelm, R.B. LFQ-Analyst: An Easy-To-Use Interactive Web Platform To Analyze and Visualize Label-Free Proteomics Data Preprocessed with MaxQuant. J. Proteome Res. 2020, 19, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Distler, U.; Sielaff, M.; Tenzer, S. Label-Free Proteomics of Quantity-Limited Samples Using Ion Mobility-Assisted Data-Independent Acquisition Mass Spectrometry. Methods Mol. Biol. 2021, 2228, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Budnik, B. A review of the current state of single-cell proteomics and future perspective. Anal. Bioanal. Chem. 2023, 415, 6889–6899. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, Y.; Zhang, T.; Shu, L.; Roepstorff, P.; Yang, F. Quantitative Proteomics Using Isobaric Labeling: A Practical Guide. Genom. Proteom. Bioinform. 2021, 19, 689–706. [Google Scholar] [CrossRef]
- Xing, T.; Wang, C.; Zhao, X.; Dai, C.; Zhou, G.; Xu, X. Proteome Analysis Using Isobaric Tags for Relative and Absolute Analysis Quantitation (iTRAQ) Reveals Alterations in Stress-Induced Dysfunctional Chicken Muscle. J. Agric. Food Chem. 2017, 65, 2913–2922. [Google Scholar] [CrossRef]
- Deng, J.; Erdjument-Bromage, H.; Neubert, T.A. Quantitative Comparison of Proteomes Using SILAC. Curr. Protoc. Protein Sci. 2019, 95, e74. [Google Scholar] [CrossRef]
- Sivanich, M.K.; Gu, T.J.; Tabang, D.N.; Li, L. Recent advances in isobaric labeling and applications in quantitative proteomics. Proteomics 2022, 22, e2100256. [Google Scholar] [CrossRef]
- Lardenois, A.; Jagot, S.; Lagarrigue, M.; Guével, B.; Ledevin, M.; Larcher, T.; Dubreil, L.; Pineau, C.; Rouger, K.; Guével, L. Quantitative proteome profiling of dystrophic dog skeletal muscle reveals a stabilized muscular architecture and protection against oxidative stress after systemic delivery of MuStem cells. Proteomics 2016, 16, 2028–2042. [Google Scholar] [CrossRef] [PubMed]
- Wdowiak, A.P.; Duong, M.N.; Joyce, R.D.; Boyatzis, A.E.; Walkey, M.C.; Nealon, G.L.; Arthur, P.G.; Piggott, M.J. Isotope-Coded Maleimide Affinity Tags for Proteomics Applications. Bioconjug. Chem. 2021, 32, 1652–1666. [Google Scholar] [CrossRef] [PubMed]
- Rayavarapu, S.; Coley, W.; Cakir, E.; Jahnke, V.; Takeda, S.; Aoki, Y.; Grodish-Dressman, H.; Jaiswal, J.K.; Hoffman, E.P.; Brown, K.J.; et al. Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol. Cell. Proteom. 2013, 12, 1061–1073. [Google Scholar] [CrossRef]
- Alayi, T.D.; Tawalbeh, S.M.; Ogundele, M.; Smith, H.R.; Samsel, A.M.; Barbieri, M.L.; Hathout, Y. Tandem Mass Tag-Based Serum Proteome Profiling for Biomarker Discovery in Young Duchenne Muscular Dystrophy Boys. ACS Omega 2020, 5, 26504–26517. [Google Scholar] [CrossRef]
- Jünger, M.A.; Aebersold, R. Mass spectrometry-driven phosphoproteomics: Patterning the systems biology mosaic. Wiley Interdiscip. Rev. Dev. Biol. 2014, 3, 83–112. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Zhang, J.; Zong, C.; Wang, Y.; Lu, H.; Yang, P.; Wang, W.; Young, G.W.; Wang, Y.; Korge, P.; et al. Phosphoproteome analysis reveals regulatory sites in major pathways of cardiac mitochondria. Mol. Cell. Proteom. 2011, 10, S1–S14. [Google Scholar] [CrossRef]
- Kitata, R.B.; Yang, J.C.; Chen, Y.J. Advances in data-independent acquisition mass spectrometry towards comprehensive digital proteome landscape. Mass Spectrom. Rev. 2023, 42, 2324–2348. [Google Scholar] [CrossRef]
- Fenaille, F.; Barbier Saint-Hilaire, P.; Rousseau, K.; Junot, C. Data acquisition workflows in liquid chromatography coupled to high resolution mass spectrometry-based metabolomics: Where do we stand? J. Chromatogr. A 2017, 1526, 1–12. [Google Scholar] [CrossRef]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Sun, B.; Smialowski, P.; Aftab, W.; Schmidt, A.; Forne, I.; Straub, T.; Imhof, A. Improving SWATH-MS analysis by deep-learning. Proteomics 2023, 23, e2200179. [Google Scholar] [CrossRef]
- Öhman, T.; Teppo, J.; Datta, N.; Mäkinen, S.; Varjosalo, M.; Koistinen, H.A. Skeletal muscle proteomes reveal downregulation of mitochondrial proteins in transition from prediabetes into type 2 diabetes. iScience 2021, 24, 102712. [Google Scholar] [CrossRef] [PubMed]
- Cretich, M.; Damin, F.; Chiari, M. Protein microarray technology: How far off is routine diagnostics? Analyst 2014, 139, 528–542. [Google Scholar] [CrossRef]
- Ayoglu, B.; Chaouch, A.; Lochmüller, H.; Politano, L.; Bertini, E.; Spitali, P.; Hiller, M.; Niks, E.H.; Gualandi, F.; Pontén, F.; et al. Affinity proteomics within rare diseases: A BIO-NMD study for blood biomarkers of muscular dystrophies. EMBO Mol. Med. 2014, 6, 918–936. [Google Scholar] [CrossRef] [PubMed]
- Cosemans, G.; Merckx, C.; De Bleecker, J.L.; De Paepe, B. Inducible Heat Shock Protein 70 Levels in Patients and the mdx Mouse Affirm Regulation during Skeletal Muscle Regeneration in Muscular Dystrophy. Front. Biosci. (Schol. Ed.) 2022, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Aparna, G.M.; Tetala, K.K.R. Recent Progress in Development and Application of DNA, Protein, Peptide, Glycan, Antibody, and Aptamer Microarrays. Biomolecules 2023, 13, 602. [Google Scholar] [CrossRef]
- Domon, B.; Aebersold, R. Mass spectrometry and protein analysis. Science 2006, 312, 212–217. [Google Scholar] [CrossRef]
- Doran, P.; Wilton, S.D.; Fletcher, S.; Ohlendieck, K. Proteomic profiling of antisense-induced exon skipping reveals reversal of pathobiochemical abnormalities in dystrophic mdx diaphragm. Proteomics 2009, 9, 671–685. [Google Scholar] [CrossRef]
- Clarke, M.S. Proteomic analysis of skeletal muscle tissue using SELDI-TOF MS: Application to disuse atrophy. Methods Mol. Biol. 2012, 818, 131–141. [Google Scholar] [CrossRef]
- Dare, T.O.; Davies, H.A.; Turton, J.A.; Lomas, L.; Williams, T.C.; York, M.J. Application of surface-enhanced laser desorption/ionization technology to the detection and identification of urinary parvalbumin-alpha: A biomarker of compound-induced skeletal muscle toxicity in the rat. Electrophoresis 2002, 23, 3241–3251. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Vitek, O.; Aebersold, R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods 2007, 4, 787–797. [Google Scholar] [CrossRef]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.J.; Thibert, S.; Zhou, M. Dissecting the structural heterogeneity of proteins by native mass spectrometry. Protein Sci. 2023, 32, e4612. [Google Scholar] [CrossRef]
- van Schaick, G.; Haselberg, R.; Somsen, G.W.; Wuhrer, M.; Domínguez-Vega, E. Studying protein structure and function by native separation-mass spectrometry. Nat. Rev. Chem. 2022, 6, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.F.C.; Greatorex, R.E.; Gidman, C.E.; Rahman, S.; Griffiths, R.L. Surface-sampling mass spectrometry to study proteins and protein complexes. Essays Biochem. 2023, 67, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Can, T.; Faas, L.; Ashford, D.A.; Dowle, A.; Thomas, J.; O’Toole, P.; Blanco, G. Proteomic analysis of laser capture microscopy purified myotendinous junction regions from muscle sections. Proteome Sci. 2014, 12, 25. [Google Scholar] [CrossRef]
- Stuart, C.A.; Stone, W.L.; Howell, M.E.; Brannon, M.F.; Hall, H.K.; Gibson, A.L.; Stone, M.H. Myosin content of individual human muscle fibers isolated by laser capture microdissection. Am. J. Physiol. Cell Physiol. 2016, 310, C381–C389. [Google Scholar] [CrossRef]
- Guo, W.; Hu, Y.; Qian, J.; Zhu, L.; Cheng, J.; Liao, J.; Fan, X. Laser capture microdissection for biomedical research: Towards high-throughput, multi-omics, and single-cell resolution. J. Genet. Genom. 2023, 50, 641–651. [Google Scholar] [CrossRef]
- Murgia, M.; Ciciliot, S.; Nagaraj, N.; Reggiani, C.; Schiaffino, S.; Franchi, M.V.; Pišot, R.; Šimunič, B.; Toniolo, L.; Blaauw, B.; et al. Signatures of muscle disuse in spaceflight and bed rest revealed by single muscle fiber proteomics. PNAS Nexus 2022, 1, pgac086. [Google Scholar] [CrossRef]
- Tighe, P.J.; Ryder, R.R.; Todd, I.; Fairclough, L.C. ELISA in the multiplex era: Potentials and pitfalls. Proteom. Clin. Appl. 2015, 9, 406–422. [Google Scholar] [CrossRef]
- Tabatabaei, M.S.; Ahmed, M. Enzyme-Linked Immunosorbent Assay (ELISA). Methods Mol. Biol. 2022, 2508, 115–134. [Google Scholar] [CrossRef]
- Hakim, C.H.; Kumar, S.R.P.; Pérez-López, D.; Teixeira, J.; Herzog, R.W.; Duan, D. Assessment of the Gene Therapy Immune Response in the Canine Muscular Dystrophy Model. Methods Mol. Biol. 2023, 2587, 353–375. [Google Scholar] [CrossRef] [PubMed]
- Landsberger, M.; Brinkmeier, H. Immunoblot Analysis of DIGE-Based Proteomics. Methods Mol. Biol. 2023, 2596, 429–443. [Google Scholar] [CrossRef]
- Mishra, M.; Tiwari, S.; Gomes, A.V. Protein purification and analysis: Next generation Western blotting techniques. Expert Rev. Proteom. 2017, 14, 1037–1053. [Google Scholar] [CrossRef]
- Carberry, S.; Ohlendieck, K. Gel electrophoresis-based proteomics of senescent tissues. Methods Mol. Biol. 2013, 1048, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.; Given Chunyk, A.; Dysinger, M.; Purushothama, S.; Ricks, C.; Osterlund, K.; Theobald, V. Next generation ligand binding assays-review of emerging technologies’ capabilities to enhance throughput and multiplexing. AAPS J. 2014, 16, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, R.; Baker, D.; Brumm, J.; Davancaze, T.; Harp, C.; Herman, A.; Büdingen, H.V.; Townsend, M.; Fischer, S.K. Establishment of neurofilament light chain Simoa assay in cerebrospinal fluid and blood. Bioanalysis 2019, 11, 1405–1418. [Google Scholar] [CrossRef]
- Cui, M.; Cheng, C.; Zhang, L. High-throughput proteomics: A methodological mini-review. Lab. Invest. 2022, 102, 1170–1181. [Google Scholar] [CrossRef]
- Zweyer, M.; Ohlendieck, K.; Swandulla, D. Histological and Histochemical Microscopy Used to Verify 2D-DIGE Pathoproteomics. Methods Mol. Biol. 2023, 2596, 465–480. [Google Scholar] [CrossRef]
- Zweyer, M.; Ohlendieck, K.; Swandulla, D. Verification of Protein Changes Determined by 2D-DIGE Based Proteomics Using Immunofluorescence Microscopy. Methods Mol. Biol. 2023, 2596, 445–464. [Google Scholar] [CrossRef]
- Sawano, S.; Mizunoya, W. History and development of staining methods for skeletal muscle fiber types. Histol. Histopathol. 2022, 37, 493–503. [Google Scholar] [CrossRef]
- Zweyer, M.; Sabir, H.; Dowling, P.; Gargan, S.; Murphy, S.; Swandulla, D.; Ohlendieck, K. Histopathology of Duchenne muscular dystrophy in correlation with changes in proteomic biomarkers. Histol. Histopathol. 2022, 37, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.; McDonald, D.; Capaldi, R.; Deehan, D.; Taylor, R.W.; Filby, A.; Turnbull, D.M.; Lawless, C.; Vincent, A.E. Decoding mitochondrial heterogeneity in single muscle fibres by imaging mass cytometry. Sci. Rep. 2020, 10, 15336. [Google Scholar] [CrossRef] [PubMed]
- Thirumal, S.; Jamzad, A.; Cotechini, T.; Hindmarch, C.T.; Graham, C.H.; Siemens, D.R.; Mousavi, P. TITAN: An end-to-end data analysis environment for the Hyperion™ imaging system. Cytometry A 2022, 101, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, J.; Lewy, T.; Rice, C.M.; Elemento, O.; Rendeiro, A.F.; Mason, C.E. Spatial omics technologies at multimodal and single cell/subcellular level. Genome Biol. 2022, 23, 256. [Google Scholar] [CrossRef] [PubMed]
- Manohar, S.M.; Shah, P.; Nair, A. Flow cytometry: Principles, applications and recent advances. Bioanalysis 2021, 13, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Dell’Orso, S.; Juan, A.H.; Ko, K.D.; Naz, F.; Perovanovic, J.; Gutierrez-Cruz, G.; Feng, X.; Sartorelli, V. Single cell analysis of adult mouse skeletal muscle stem cells in homeostatic and regenerative conditions. Development 2019, 146, dev174177. [Google Scholar] [CrossRef]
- Li, N.; Parkes, J.E.; Spathis, R.; Morales, M.; Mcdonald, J.; Kendra, R.M.; Ott, E.M.; Brown, K.J.; Lawlor, M.W.; Nagaraju, K. The Effect of Immunomodulatory Treatments on Anti-Dystrophin Immune Response After AAV Gene Therapy in Dystrophin Deficient mdx Mice. J. Neuromuscul. Dis. 2021, 8, S325–S340. [Google Scholar] [CrossRef]
- Arora, B.; Tandon, R.; Attri, P.; Bhatia, R. Chemical Crosslinking: Role in Protein and Peptide Science. Curr. Protein Pept. Sci. 2017, 18, 946–955. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Chemical crosslinking analysis of β-dystroglycan in dystrophin-deficient skeletal muscle. HRB Open Res. 2018, 1, 17. [Google Scholar] [CrossRef]
- Jayachandran, B.; Parvin, T.N.; Alam, M.M.; Chanda, K.; Mm, B. Insights on Chemical Crosslinking Strategies for Proteins. Molecules 2022, 27, 8124. [Google Scholar] [CrossRef]
- Dowd, A. Elucidating Cellular Metabolism and Protein Difference Data from DIGE Proteomics Experiments Using Enzyme Assays. Methods Mol. Biol. 2023, 2596, 399–419. [Google Scholar] [CrossRef]
- O’Connell, K.; Ohlendieck, K. Proteomic DIGE analysis of the mitochondria-enriched fraction from aged rat skeletal muscle. Proteomics 2009, 9, 5509–5524. [Google Scholar] [CrossRef]
- Dowd, A. Enzyme Assay Methods to Validate DIGE Proteomics Data. Methods Mol. Biol. 2023, 2596, 421–428. [Google Scholar] [CrossRef]
- Ohlendieck, K. Skeletal muscle proteomics: Current approaches, technical challenges and emerging techniques. Skelet. Muscle 2011, 1, 6. [Google Scholar] [CrossRef]
- Nakka, K.; Ghigna, C.; Gabellini, D.; Dilworth, F.J. Diversification of the muscle proteome through alternative splicing. Skelet. Muscle 2018, 8, 8. [Google Scholar] [CrossRef]
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef]
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1462. [Google Scholar] [CrossRef]
- Brooks, S.V.; Guzman, S.D.; Ruiz, L.P. Skeletal muscle structure, physiology, and function. Handb. Clin. Neurol. 2023, 195, 3–16. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Schiaffino, S. Fibre types in skeletal muscle: A personal account. Acta Physiol. 2010, 199, 451–463. [Google Scholar] [CrossRef]
- Bottinelli, R.; Reggiani, C. Human skeletal muscle fibres: Molecular and functional diversity. Prog. Biophys. Mol. Biol. 2000, 73, 195–262. [Google Scholar] [CrossRef]
- Swiderski, K.; Lynch, G.S. Murine models of Duchenne muscular dystrophy: Is there a best model? Am. J. Physiol. Cell Physiol. 2021, 321, C409–C412. [Google Scholar] [CrossRef]
- Schiaffino, S.; Gorza, L.; Pitton, G.; Saggin, L.; Ausoni, S.; Sartore, S.; Lømo, T. Embryonic and neonatal myosin heavy chain in denervated and paralyzed rat skeletal muscle. Dev. Biol. 1988, 127, 1–11. [Google Scholar] [CrossRef]
- Agarwal, M.; Sharma, A.; Kumar, P.; Kumar, A.; Bharadwaj, A.; Saini, M.; Kardon, G.; Mathew, S.J. Myosin heavy chain-embryonic regulates skeletal muscle differentiation during mammalian development. Development 2020, 147, dev184507. [Google Scholar] [CrossRef]
- Gargan, S.; Dowling, P.; Zweyer, M.; Reimann, J.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Mass Spectrometric Profiling of Extraocular Muscle and Proteomic Adaptations in the mdx-4cv Model of Duchenne Muscular Dystrophy. Life 2021, 11, 595. [Google Scholar] [CrossRef]
- Hoh, J.F.Y. Developmental, physiologic and phylogenetic perspectives on the expression and regulation of myosin heavy chains in mammalian skeletal muscles. J. Comp. Physiol. B 2023, 193, 355–382. [Google Scholar] [CrossRef]
- Pette, D.; Staron, R.S. Transitions of muscle fiber phenotypic profiles. Histochem. Cell Biol. 2001, 115, 359–372. [Google Scholar] [CrossRef]
- Ohlendieck, K. Proteomic profiling of skeletal muscle plasticity. Muscles Ligaments Tendons J. 2012, 1, 119–216. [Google Scholar]
- McGlory, C.; van Vliet, S.; Stokes, T.; Mittendorfer, B.; Phillips, S.M. The impact of exercise and nutrition on the regulation of skeletal muscle mass. J. Physiol. 2019, 597, 1251–1258. [Google Scholar] [CrossRef]
- Murach, K.A.; Dungan, C.M.; Kosmac, K.; Voigt, T.B.; Tourville, T.W.; Miller, M.S.; Bamman, M.M.; Peterson, C.A.; Toth, M.J. Fiber typing human skeletal muscle with fluorescent immunohistochemistry. J. Appl. Physiol. 2019, 127, 1632–1639. [Google Scholar] [CrossRef]
- Kallabis, S.; Abraham, L.; Müller, S.; Dzialas, V.; Türk, C.; Wiederstein, J.L.; Bock, T.; Nolte, H.; Nogara, L.; Blaauw, B.; et al. High-throughput proteomics fiber typing (ProFiT) for comprehensive characterization of single skeletal muscle fibers. Skelet. Muscle 2020, 10, 7. [Google Scholar] [CrossRef]
- Momenzadeh, A.; Jiang, Y.; Kreimer, S.; Teigen, L.E.; Zepeda, C.S.; Haghani, A.; Mastali, M.; Song, Y.; Hutton, A.; Parker, S.J.; et al. A Complete Workflow for High Throughput Human Single Skeletal Muscle Fiber Proteomics. J. Am. Soc. Mass Spectrom. 2023, 34, 1858–1867. [Google Scholar] [CrossRef]
- Yablonka-Reuveni, Z. The skeletal muscle satellite cell: Still young and fascinating at 50. J. Histochem. Cytochem. 2011, 59, 1041–1059. [Google Scholar] [CrossRef]
- Negroni, E.; Kondili, M.; Muraine, L.; Bensalah, M.; Butler-Browne, G.S.; Mouly, V.; Bigot, A.; Trollet, C. Muscle fibro-adipogenic progenitors from a single-cell perspective: Focus on their “virtual” secretome. Front. Cell Dev. Biol. 2022, 10, 952041. [Google Scholar] [CrossRef]
- Bernard, C.; Zavoriti, A.; Pucelle, Q.; Chazaud, B.; Gondin, J. Role of macrophages during skeletal muscle regeneration and hypertrophy-Implications for immunomodulatory strategies. Physiol. Rep. 2022, 10, e15480. [Google Scholar] [CrossRef]
- Engquist, E.N.; Zammit, P.S. The Satellite Cell at 60: The Foundation Years. J. Neuromuscul. Dis. 2021, 8, S183–S203. [Google Scholar] [CrossRef]
- Morgan, J.E.; Partridge, T.A. Muscle satellite cells. Int. J. Biochem. Cell Biol. 2003, 35, 1151–1156. [Google Scholar] [CrossRef]
- Buckingham, M.; Relaix, F. PAX3 and PAX7 as upstream regulators of myogenesis. Semin. Cell Dev. Biol. 2015, 44, 115–125. [Google Scholar] [CrossRef]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef]
- Beauchamp, J.R.; Heslop, L.; Yu, D.S.; Tajbakhsh, S.; Kelly, R.G.; Wernig, A.; Buckingham, M.E.; Partridge, T.A.; Zammit, P.S. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J. Cell Biol. 2000, 151, 1221–1234. [Google Scholar] [CrossRef]
- Khurram, O.U.; Pearcey, G.E.P.; Chardon, M.K.; Kim, E.H.; García, M.; Heckman, C.J. The Cellular Basis for the Generation of Firing Patterns in Human Motor Units. Adv. Neurobiol. 2022, 28, 233–258. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Swandulla, D.; Ohlendieck, K. Identification of Subproteomic Markers for Skeletal Muscle Profiling. Methods Mol. Biol. 2023, 2596, 291–302. [Google Scholar] [CrossRef]
- Severinsen, M.C.K.; Pedersen, B.K. Muscle-Organ Crosstalk: The Emerging Roles of Myokines. Endocr. Rev. 2020, 41, 594–609. [Google Scholar] [CrossRef]
- Kirk, B.; Feehan, J.; Lombardi, G.; Duque, G. Muscle, Bone, and Fat Crosstalk: The Biological Role of Myokines, Osteokines, and Adipokines. Curr. Osteoporos. Rep. 2020, 18, 388–400. [Google Scholar] [CrossRef]
- Lara-Castillo, N.; Johnson, M.L. Bone-Muscle Mutual Interactions. Curr. Osteoporos. Rep. 2020, 18, 408–421. [Google Scholar] [CrossRef]
- Gomarasca, M.; Banfi, G.; Lombardi, G. Myokines: The endocrine coupling of skeletal muscle and bone. Adv. Clin. Chem. 2020, 94, 155–218. [Google Scholar] [CrossRef]
- Florin, A.; Lambert, C.; Sanchez, C.; Zappia, J.; Durieux, N.; Tieppo, A.M.; Mobasheri, A.; Henrotin, Y. The secretome of skeletal muscle cells: A systematic review. Osteoarthr. Cartil. Open. 2020, 2, 100019. [Google Scholar] [CrossRef]
- Le Bihan, M.C.; Bigot, A.; Jensen, S.S.; Dennis, J.L.; Rogowska-Wrzesinska, A.; Lainé, J.; Gache, V.; Furling, D.; Jensen, O.N.; Voit, T.; et al. In-depth analysis of the secretome identifies three major independent secretory pathways in differentiating human myoblasts. J. Proteom. 2012, 77, 344–356. [Google Scholar] [CrossRef]
- Hartwig, S.; Raschke, S.; Knebel, B.; Scheler, M.; Irmler, M.; Passlack, W.; Muller, S.; Hanisch, F.G.; Franz, T.; Li, X.; et al. Secretome profiling of primary human skeletal muscle cells. Biochim. Biophys. Acta 2014, 1844, 1011–1017. [Google Scholar] [CrossRef]
- Leuchtmann, A.B.; Adak, V.; Dilbaz, S.; Handschin, C. The Role of the Skeletal Muscle Secretome in Mediating Endurance and Resistance Training Adaptations. Front. Physiol. 2021, 12, 709807. [Google Scholar] [CrossRef]
- Baeza-Trinidad, R. Rhabdomyolysis: A syndrome to be considered. Rabdomiólisis: Un síndrome a tener en cuenta. Med. Clin. 2022, 158, 277–283. [Google Scholar] [CrossRef]
- Cabrera-Serrano, M.; Ravenscroft, G. Recent advances in our understanding of genetic rhabdomyolysis. Curr. Opin. Neurol. 2022, 35, 651–657. [Google Scholar] [CrossRef]
- Cabral, B.M.I.; Edding, S.N.; Portocarrero, J.P.; Lerma, E.V. Rhabdomyolysis. Dis. Mon. 2020, 66, 101015. [Google Scholar] [CrossRef]
- Carneiro, A.; Macedo-da-Silva, J.; Santiago, V.F.; de Oliveira, G.S.; Guimarães, T.; Mendonça, C.F.; de Oliveira Branquinho, J.L.; Lucena, C.V.; Osório, J.; Pernambuco, E. Urine proteomics as a non-invasive approach to monitor exertional rhabdomyolysis during military training. J. Proteom. 2022, 258, 104498. [Google Scholar] [CrossRef]
- Deshmukh, A.S.; Murgia, M.; Nagaraj, N.; Treebak, J.T.; Cox, J.; Mann, M. Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors. Mol. Cell. Proteom. 2015, 14, 841–853. [Google Scholar] [CrossRef]
- Højlund, K.; Yi, Z.; Hwang, H.; Bowen, B.; Lefort, N.; Flynn, C.R.; Langlais, P.; Weintraub, S.T.; Mandarino, L.J. Characterization of the human skeletal muscle proteome by one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. Mol. Cell. Proteom. 2008, 7, 257–267. [Google Scholar] [CrossRef]
- Parker, K.C.; Walsh, R.J.; Salajegheh, M.; Amato, A.A.; Krastins, B.; Sarracino, D.A.; Greenberg, S.A. Characterization of human skeletal muscle biopsy samples using shotgun proteomics. J. Proteome Res. 2009, 8, 3265–3277. [Google Scholar] [CrossRef]
- Raddatz, K.; Albrecht, D.; Hochgräfe, F.; Hecker, M.; Gotthardt, M. A proteome map of murine heart and skeletal muscle. Proteomics 2008, 8, 1885–1897. [Google Scholar] [CrossRef]
- Burniston, J.G.; Connolly, J.; Kainulainen, H.; Britton, S.L.; Koch, L.G. Label-free profiling of skeletal muscle using high-definition mass spectrometry. Proteomics 2014, 14, 2339–2344. [Google Scholar] [CrossRef]
- Malik, Z.A.; Cobley, J.N.; Morton, J.P.; Close, G.L.; Edwards, B.J.; Koch, L.G.; Britton, S.L.; Burniston, J.G. Label-Free LC-MS Profiling of Skeletal Muscle Reveals Heart-Type Fatty Acid Binding Protein as a Candidate Biomarker of Aerobic Capacity. Proteomes 2013, 1, 290–308. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of the mouse diaphragm and refined mass spectrometric analysis of the dystrophic phenotype. J. Muscle Res. Cell Motil. 2019, 40, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, D.; Viganò, A.; Ricci, E.; Cerretelli, P.; Wait, R.; Gelfi, C. Comparison of protein expression in human deltoideus and vastus lateralis muscles using two-dimensional gel electrophoresis. Proteomics 2005, 5, 2577–2586. [Google Scholar] [CrossRef]
- Drexler, H.C.; Ruhs, A.; Konzer, A.; Mendler, L.; Bruckskotten, M.; Looso, M.; Günther, S.; Boettger, T.; Krüger, M.; Braun, T. On marathons and Sprints: An integrated quantitative proteomics and transcriptomics analysis of differences between slow and fast muscle fibers. Mol. Cell. Proteom. 2012, 11, M111.010801. [Google Scholar] [CrossRef]
- Gelfi, C.; Viganò, A.; De Palma, S.; Ripamonti, M.; Begum, S.; Cerretelli, P.; Wait, R. 2-D protein maps of rat gastrocnemius and soleus muscles: A tool for muscle plasticity assessment. Proteomics 2006, 6, 321–340. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Hashida-Okumura, A.; Kita, K.; Matsubae, M.; Matsubara, T.; Takao, T.; Nagai, K. Proteomic analysis of slow- and fast-twitch skeletal muscles. Proteomics 2005, 5, 2896–2906. [Google Scholar] [CrossRef] [PubMed]
- Eggers, B.; Schork, K.; Turewicz, M.; Barkovits, K.; Eisenacher, M.; Schröder, R.; Clemen, C.S.; Marcus, K. Advanced fiber type- specific protein profiles derived from adult murine skeletal muscle. Proteomes 2021, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Toniolo, L.; Nagaraj, N.; Ciciliot, S.; Vindigni, V.; Schiaffino, S.; Reggiani, C.; Mann, M. Single Muscle Fiber Proteomics Reveals Fiber-Type-Specific Features of Human Muscle Aging. Cell Rep. 2017, 19, 2396–2409. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Nogara, L.; Baraldo, M.; Reggiani, C.; Mann, M.; Schiaffino, S. Protein profile of fiber types in human skeletal muscle: A single-fiber proteomics study. Skelet. Muscle 2021, 11, 24. [Google Scholar] [CrossRef]
- Deshmukh, A.S.; Steenberg, D.E.; Hostrup, M.; Birk, J.B.; Larsen, J.K.; Santos, A.; Kjøbsted, R.; Hingst, J.R.; Schéele, C.C.; Murgia, M.; et al. Deep muscle-proteomic analysis of freeze-dried human muscle biopsies reveals fiber type-specific adaptations to exercise training. Nat. Commun. 2021, 12, 304. [Google Scholar] [CrossRef]
- Hadrévi, J.; Hellström, F.; Kieselbach, T.; Malm, C.; Pedrosa-Domellöf, F. Protein differences between human trapezius and vastus lateralis muscles determined with a proteomic approach. BMC Musculoskelet. Disord. 2011, 12, 181. [Google Scholar] [CrossRef]
- Tan, X.; He, Y.; He, Y.; Yan, Z.; Chen, J.; Zhao, R.; Sui, X.; Zhang, L.; Du, X.; Irwin, D.M.; et al. Comparative Proteomic Analysis of Glycolytic and Oxidative Muscle in Pigs. Genes 2023, 14, 361. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Guglieri, M. An update on Becker muscular dystrophy. Curr. Opin. Neurol. 2023, 36, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Gappmaier, E.; Howard, M.T.; Sampson, J.B.; Mendell, J.R.; Wall, C.; King, W.M.; et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort. Hum. Mutat. 2009, 30, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Juan-Mateu, J.; Gonzalez-Quereda, L.; Rodriguez, M.J.; Baena, M.; Verdura, E.; Nascimento, A.; Ortez, C.; Baiget, M.; Gallano, P. DMD Mutations in 576 Dystrophinopathy Families: A Step Forward in Genotype-Phenotype Correlations. PLoS ONE 2015, 10, e0135189. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Ohlendieck, K. Towards an understanding of the dystrophin-glycoprotein complex: Linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. Eur. J. Cell Biol. 1996, 69, 1–10. [Google Scholar]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Campbell, K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J. Cell Biol. 1991, 115, 1685–1694. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Matsumura, K.; Ionasescu, V.V.; Towbin, J.A.; Bosch, E.P.; Weinstein, S.L.; Sernett, S.W.; Campbell, K.P. Duchenne muscular dystrophy: Deficiency of dystrophin-associated proteins in the sarcolemma. Neurology 1993, 43, 795–800. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef]
- Murphy, S.; Ohlendieck, K. The biochemical and mass spectrometric profiling of the dystrophin complexome from skeletal muscle. Comput. Struct. Biotechnol. J. 2015, 14, 20–27. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Murphy, S.; Zweyer, M.; Sabir, H.; Swandulla, D.; Ohlendieck, K. The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle. Proteomes 2021, 9, 9. [Google Scholar] [CrossRef]
- Zabłocka, B.; Górecki, D.C.; Zabłocki, K. Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences. Int. J. Mol. Sci. 2021, 22, 11040. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of impaired excitation-contraction coupling and abnormal calcium handling in muscular dystrophy. Proteomics 2022, 22, e2200003. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Belosludtsev, K.N. Ion Channels of the Sarcolemma and Intracellular Organelles in Duchenne Muscular Dystrophy: A Role in the Dysregulation of Ion Homeostasis and a Possible Target for Therapy. Int. J. Mol. Sci. 2023, 24, 2229. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Swandulla, D.; Ohlendieck, K. Cellular pathogenesis of Duchenne muscular dystrophy: Progressive myofibre degeneration, chronic inflammation, reactive myofibrosis and satellite cell dysfunction. Eur. J. Transl. Myol. 2023, 33, 11856. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Gargan, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Extracellular Matrix Proteomics: The mdx-4cv Mouse Diaphragm as a Surrogate for Studying Myofibrosis in Dystrophinopathy. Biomolecules 2023, 13, 1108. [Google Scholar] [CrossRef]
- Holland, A.; Carberry, S.; Ohlendieck, K. Proteomics of the dystrophin-glycoprotein complex and dystrophinopathy. Curr. Protein Pept. Sci. 2013, 14, 680–697. [Google Scholar] [CrossRef]
- Fuller, H.R.; Graham, L.C.; Llavero Hurtado, M.; Wishart, T.M. Understanding the molecular consequences of inherited muscular dystrophies: Advancements through proteomic experimentation. Expert Rev. Proteom. 2016, 13, 659–671. [Google Scholar] [CrossRef]
- Holland, A.; Murphy, S.; Dowling, P.; Ohlendieck, K. Pathoproteomic profiling of the skeletal muscle matrisome in dystrophinopathy associated myofibrosis. Proteomics 2016, 16, 345–366. [Google Scholar] [CrossRef] [PubMed]
- Carr, S.J.; Zahedi, R.P.; Lochmüller, H.; Roos, A. Mass spectrometry-based protein analysis to unravel the tissue pathophysiology in Duchenne muscular dystrophy. Proteom. Clin. Appl. 2018, 12, 1700071. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Murphy, S.; Zweyer, M.; Raucamp, M.; Swandulla, D.; Ohlendieck, K. Emerging proteomic biomarkers of X-linked muscular dystrophy. Expert Rev. Mol. Diagn. 2019, 19, 739–755. [Google Scholar] [CrossRef]
- Al-Khalili Szigyarto, C. Duchenne muscular dystrophy: Recent advances in protein biomarkers and the clinical application. Expert Rev. Proteom. 2020, 17, 365–375. [Google Scholar] [CrossRef]
- Ge, Y.; Molloy, M.P.; Chamberlain, J.S.; Andrews, P.C. Proteomic analysis of mdx skeletal muscle: Great reduction of adenylate kinase 1 expression and enzymatic activity. Proteomics 2003, 3, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Martin, G.; Dowling, P.; Jockusch, H.; Ohlendieck, K. Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics 2006, 6, 4610–4621. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Dowling, P.; Donoghue, P.; Buffini, M.; Ohlendieck, K. Reduced expression of regucalcin in young and aged mdx diaphragm indicates abnormal cytosolic calcium handling in dystrophin-deficient muscle. Biochim. Biophys. Acta 2006, 1764, 773–785. [Google Scholar] [CrossRef]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Proteomics reveals drastic increase of extracellular matrix proteins collagen and dermatopontin in the aged mdx diaphragm model of Duchenne muscular dystrophy. Int. J. Mol. Med. 2012, 30, 229–234. [Google Scholar] [CrossRef]
- Gardan-Salmon, D.; Dixon, J.M.; Lonergan, S.M.; Selsby, J.T. Proteomic assessment of the acute phase of dystrophin deficiency in mdx mice. Eur. J. Appl. Physiol. 2011, 111, 2763–2773. [Google Scholar] [CrossRef]
- Guevel, L.; Lavoie, J.R.; Perez-Iratxeta, C.; Rouger, K.; Dubreil, L.; Feron, M.; Talon, S.; Brand, M.; Megeney, L.A. Quantitative proteomic analysis of dystrophic dog muscle. J. Proteome Res. 2011, 10, 2465–2478. [Google Scholar] [CrossRef]
- Yoon, J.H.; Johnson, E.; Xu, R.; Martin, L.T.; Martin, P.T.; Montanaro, F. Comparative proteomic profiling of dystroglycan-associated proteins in wild type, mdx, and Galgt2 transgenic mouse skeletal muscle. J. Proteome Res. 2012, 11, 4413–4424. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Molloy, M.P.; Chamberlain, J.S.; Andrews, P.C. Differential expression of the skeletal muscle proteome in mdx mice at different ages. Electrophoresis 2004, 25, 2576–2585. [Google Scholar] [CrossRef]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Application of fluorescence two-dimensional difference in-gel electrophoresis as a proteomic biomarker discovery tool in muscular dystrophy research. Biology 2013, 2, 1438–1464. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, C.Y.; Menezes de Oliveira, B.; Durbeej, M.; Marques, M.J. Isobaric Tagging-Based Quantification for Proteomic Analysis: A Comparative Study of Spared and Affected Muscles from mdx Mice at the Early Phase of Dystrophy. PLoS ONE 2013, 8, e65831. [Google Scholar] [CrossRef]
- Holland, A.; Dowling, P.; Meleady, P.; Henry, M.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis of the mdx-4cv diaphragm identifies the matricellular protein periostin as a potential factor involved in dystrophinopathy-related fibrosis. Proteomics 2015, 15, 2318–2331. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Johansson, H.J.; McClorey, G.; Godfrey, C.; Blomberg, K.E.; Coursindel, T.; Gait, M.J.; Smith, C.I.; Lehtiö, J.; El Andaloussi, S.; et al. Multi-level omics analysis in a murine model of dystrophin loss and therapeutic restoration. Hum. Mol. Genet. 2015, 24, 6756–6768. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Henry, M.; Meleady, P.; Winkler, C.K.; Krautwald, M.; Brinkmeier, H.; Ohlendieck, K. Comparative Label-Free Mass Spectrometric Analysis of Mildly versus Severely Affected mdx Mouse Skeletal Muscles Identifies Annexin, Lamin, and Vimentin as Universal Dystrophic Markers. Molecules 2015, 20, 11317–11344. [Google Scholar] [CrossRef]
- Murphy, S.; Henry, M.; Meleady, P.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Simultaneous Pathoproteomic Evaluation of the Dystrophin-Glycoprotein Complex and Secondary Changes in the mdx-4cv Mouse Model of Duchenne Muscular Dystrophy. Biology 2015, 4, 397–423. [Google Scholar] [CrossRef]
- Fröhlich, T.; Kemter, E.; Flenkenthaler, F.; Klymiuk, N.; Otte, K.A.; Blutke, A.; Krause, S.; Walter, M.C.; Wanke, R.; Wolf, E.; et al. Progressive muscle proteome changes in a clinically relevant pig model of Duchenne muscular dystrophy. Sci. Rep. 2016, 6, 33362. [Google Scholar] [CrossRef]
- Turk, R.; Hsiao, J.J.; Smits, M.M.; Ng, B.H.; Pospisil, T.C.; Jones, K.S.; Campbell, K.P.; Wright, M.E. Molecular Signatures of Membrane Protein Complexes Underlying Muscular Dystrophy. Mol. Cell. Proteom. 2016, 15, 2169–2185. [Google Scholar] [CrossRef]
- Murphy, S.; Brinkmeier, H.; Krautwald, M.; Henry, M.; Meleady, P.; Ohlendieck, K. Proteomic profiling of the dystrophin complex and membrane fraction from dystrophic mdx muscle reveals decreases in the cytolinker desmoglein and increases in the extracellular matrix stabilizers biglycan and fibronectin. J. Muscle Res. Cell Motil. 2017, 38, 251–268. [Google Scholar] [CrossRef]
- Gamberi, T.; Fiaschi, T.; Valocchia, E.; Modesti, A.; Mantuano, P.; Rolland, J.F.; Sanarica, F.; De Luca, A.; Magherini, F. Proteome analysis in dystrophic mdx mouse muscle reveals a drastic alteration of key metabolic and contractile proteins after chronic exercise and the potential modulation by anti-oxidant compounds. J. Proteom. 2018, 170, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Comparative gel-based proteomic analysis of chemically crosslinked complexes in dystrophic skeletal muscle. Electrophoresis 2018, 39, 1735–1744. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of the sarcolemma-enriched fraction from dystrophic mdx-4cv skeletal muscle. J. Proteom. 2019, 191, 212–227. [Google Scholar] [CrossRef]
- Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; De Palma, S.; Viganò, A.; Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; et al. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: Changes contributing to preserve muscle function in Becker muscular dystrophy patients. J. Cachexia Sarcopenia Muscle 2020, 11, 547–563. [Google Scholar] [CrossRef]
- Capitanio, D.; Moriggi, M.; Barbacini, P.; Torretta, E.; Moroni, I.; Blasevich, F.; Morandi, L.; Mora, M.; Gelfi, C. Molecular fingerprint of BMD patients lacking a portion in the rod domain of dystrophin. Int. J. Mol. Sci. 2020, 23, 2624. [Google Scholar] [CrossRef] [PubMed]
- van Westering, T.L.E.; Johansson, H.J.; Hanson, B.; Coenen-Stass, A.M.L.; Lomonosova, Y.; Tanihata, J.; Motohashi, N.; Yokota, T.; Takeda, S.; Lehtiö, J.; et al. Mutation-independent proteomic signatures of pathological progression in murine models of Duchenne muscular dystrophy. Mol. Cell. Proteom. 2020, 19, 2047–2068. [Google Scholar] [CrossRef] [PubMed]
- Stirm, M.; Fonteyne, L.M.; Shashikadze, B.; Lindner, M.; Chirivi, M.; Lange, A.; Kaufhold, C.; Mayer, C.; Medugorac, I.; Kessler, B.; et al. A scalable, clinically severe pig model for Duchenne muscular dystrophy. Dis. Models Mech. 2021, 14, dmm049285. [Google Scholar] [CrossRef]
- Gargan, S.; Dowling, P.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic Identification of Markers of Membrane Repair, Regeneration and Fibrosis in the Aged and Dystrophic Diaphragm. Life 2022, 12, 1679. [Google Scholar] [CrossRef]
- Day, N.J.; Zhang, T.; Gaffrey, M.J.; Zhao, R.; Fillmore, T.L.; Moore, R.J.; Rodney, G.G.; Qian, W.J. A deep redox proteome profiling workflow and its application to skeletal muscle of a Duchenne Muscular Dystrophy model. Free Radic. Biol. Med. 2022, 193, 373–384. [Google Scholar] [CrossRef]
- Mucha, O.; Myszka, M.; Podkalicka, P.; Świderska, B.; Malinowska, A.; Dulak, J.; Łoboda, A. Proteome Profiling of the Dystrophic mdx Mice Diaphragm. Biomolecules 2023, 13, 1648. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Ljubicic, V. Recent insights into neuromuscular junction biology in Duchenne muscular dystrophy: Impacts, challenges, and opportunities. EBioMedicine 2020, 61, 103032. [Google Scholar] [CrossRef]
- Meyers, T.A.; Townsend, D. Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef] [PubMed]
- Florczyk-Soluch, U.; Polak, K.; Dulak, J. The multifaceted view of heart problem in Duchenne muscular dystrophy. Cell. Mol. Life Sci. 2021, 78, 5447–5468. [Google Scholar] [CrossRef] [PubMed]
- Lechner, A.; Herzig, J.J.; Kientsch, J.G.; Kohler, M.; Bloch, K.E.; Ulrich, S.; Schwarz, E.I. Cardiomyopathy as cause of death in Duchenne muscular dystrophy: A longitudinal observational study. ERJ Open Res. 2023, 9, 00176–02023. [Google Scholar] [CrossRef]
- Ricotti, V.; Selby, V.; Ridout, D.; Domingos, J.; Decostre, V.; Mayhew, A.; Eagle, M.; Butler, J.; Guglieri, M.; Van der Holst, M.; et al. Respiratory and upper limb function as outcome measures in ambulant and non- ambulant subjects with Duchenne muscular dystrophy: A prospective multicentre study. Neuromuscul. Disord. 2019, 29, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.M.; Lott, D.J.; Batra, A.; Triplett, W.T.; Forbes, S.C.; Riehl, S.L.; Willcocks, R.J.; Smith, B.K.; Vandenborne, K.; Walter, G.A. Imaging respiratory muscle quality and function in Duchenne muscular dystrophy. J. Neurol. 2019, 266, 2752–2763. [Google Scholar] [CrossRef] [PubMed]
- Pennati, F.; LoMauro, A.; D’Angelo, M.G.; Aliverti, A. Non-Invasive Respiratory Assessment in Duchenne Muscular Dystrophy: From Clinical Research to Outcome Measures. Life 2019, 11, 947. [Google Scholar] [CrossRef]
- Fayssoil, A.; Chaffaut, C.; Ogna, A.; Stojkovic, T.; Lamothe, L.; Mompoint, D.; Meng, P.; Prigent, H.; Clair, B.; Behin, A.; et al. Echographic assessment of diaphragmatic function in Duchenne muscular dystrophy from childhood to adulthood. J. Neuromuscul. Dis. 2019, 6, 55–64. [Google Scholar] [CrossRef]
- Moriuchi, T.; Fujii, Y.; Kagawa, N.; Hizawa, K. Autopsy study on the weight of the heart, liver, kidney and brain in Duchenne muscular dystrophy. Tokushima J. Exp. Med. 1991, 38, 5–13. [Google Scholar]
- Naume, M.M.; Jørgensen, M.H.; Høi-Hansen, C.E.; Born, A.P.; Vissing, J.; Borgwardt, L.; Staerk, D.M.R.; Ørngreen, M.C. Metabolic assessment in children with neuromuscular disorders shows risk of liver enlargement, steatosis and fibrosis. Acta Paediatr. 2023, 112, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Motoki, T.; Shimizu-Motohashi, Y.; Komaki, H.; Mori-Yoshimura, M.; Oya, Y.; Takeshita, E.; Ishiyama, A.; Saito, T.; Nakagawa, E.; Sugai, K.; et al. Treatable renal failure found in non-ambulatory Duchenne muscular dystrophy patients. Neuromuscul. Disord. 2015, 25, 754–757. [Google Scholar] [CrossRef] [PubMed]
- Braat, E.; Hoste, L.; De Waele, L.; Gheysens, O.; Vermeersch, P.; Goffin, K.; Pottel, H.; Goemans, N.; Levtchenko, E. Renal function in children and adolescents with Duchenne muscular dystrophy. Neuromuscul. Disord. 2015, 25, 381–387. [Google Scholar] [CrossRef]
- Kutluk, M.G.; Doğan, Ç.S. Kidney involvement and associated risk factors in children with Duchenne muscular dystrophy. Pediatr. Nephrol. 2020, 35, 1953–1958. [Google Scholar] [CrossRef]
- Villa, C.R.; Kaddourah, A.; Mathew, J.; Ryan, T.D.; Wong, B.L.; Goldstein, S.L.; Jefferies, J.L. Identifying evidence of cardio-renal syndrome in patients with Duchenne muscular dystrophy using cystatin C. Neuromuscul. Disord. 2016, 26, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Caress, J.B.; Kothari, M.J.; Bauer, S.B.; Shefner, J.M. Urinary dysfunction in Duchenne muscular dystrophy. Muscle Nerve 1996, 19, 819–822. [Google Scholar] [CrossRef]
- MacLeod, M.; Kelly, R.; Robb, S.A.; Borzyskowski, M. Bladder dysfunction in Duchenne muscular dystrophy. Arch. Dis. Child. 2003, 88, 347–349. [Google Scholar] [CrossRef]
- Lionarons, J.M.; de Groot, I.J.M.; Fock, J.M.; Klinkenberg, S.; Vrijens, D.M.J.; Vreugdenhil, A.C.E.; Medici-van den Herik, E.G.; Cuppen, I.; Jaeger, B.; Niks, E.H.; et al. Prevalence of Bladder and Bowel Dysfunction in Duchenne Muscular Dystrophy Using the Childhood Bladder and Bowel Dysfunction Questionnaire. Life 2021, 11, 772. [Google Scholar] [CrossRef]
- Manokaran, R.K.; Aggarwala, S.; Kumar, R.; Gupta, A.K.; Chakrabarty, B.; Jauhari, P.; Pandey, R.M.; Gulati, S. Prevalence of smooth muscle dysfunction among children with Duchenne muscular dystrophy. Muscle Nerve 2020, 62, 699–704. [Google Scholar] [CrossRef]
- Jaffe, K.M.; McDonald, C.M.; Ingman, E.; Haas, J. Symptoms of upper gastrointestinal dysfunction in Duchenne muscular dystrophy: Case-control study. Arch. Phys. Med. Rehabil. 1990, 71, 742–744. [Google Scholar]
- Borrelli, O.; Salvia, G.; Mancini, V.; Santoro, L.; Tagliente, F.; Romeo, E.F.; Cucchiara, S. Evolution of gastric electrical features and gastric emptying in children with Duchenne and Becker muscular dystrophy. Am. J. Gastroenterol. 2005, 100, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Lo Cascio, C.M.; Goetze, O.; Latshang, T.D.; Bluemel, S.; Frauenfelder, T.; Bloch, K.E. Gastrointestinal Dysfunction in Patients with Duchenne Muscular Dystrophy. PLoS ONE 2016, 11, e0163779. [Google Scholar] [CrossRef] [PubMed]
- Rufo, A.; Del Fattore, A.; Capulli, M.; Carvello, F.; De Pasquale, L.; Ferrari, S.; Pierroz, D.; Morandi, L.; De Simone, M.; Rucci, N.; et al. Mechanisms inducing low bone density in Duchenne muscular dystrophy in mice and humans. J. Bone Miner. Res. 2011, 26, 1891–1903. [Google Scholar] [CrossRef] [PubMed]
- Garg, S. Management of scoliosis in patients with Duchenne muscular dystrophy and spinal muscular atrophy: A literature review. J. Pediatr. Rehabil. Med. 2016, 9, 23–29. [Google Scholar] [CrossRef]
- Roberts, S.; Arshad, A.; Tsirikos, A.I. Surgical and long-term functional outcomes of patients with Duchenne muscular dystrophy following spinal deformity correction. World J. Orthop. 2023, 14, 411–426. [Google Scholar] [CrossRef]
- Arshad, A.; Tsirikos, A.I. Survival of patients with Duchenne muscular dystrophy who underwent spinal deformity correction. Dev. Med. Child Neurol. 2024, 66, 187–194. [Google Scholar] [CrossRef]
- Mehler, M.F. Brain dystrophin, neurogenetics and mental retardation. Brain Res. Rev. 2000, 32, 277–307. [Google Scholar] [CrossRef]
- Pane, M.; Lombardo, M.E.; Alfieri, P.; D’Amico, A.; Bianco, F.; Vasco, G.; Piccini, G.; Mallardi, M.; Romeo, D.M.; Ricotti, V.; et al. Attention deficit hyperactivity disorder and cognitive function in Duchenne muscular dystrophy: Phenotype-genotype correlation. J. Pediatr. 2012, 161, 705–709.e1. [Google Scholar] [CrossRef]
- Ricotti, V.; Mandy, W.P.; Scoto, M.; Pane, M.; Deconinck, N.; Messina, S.; Mercuri, E.; Skuse, D.H.; Muntoni, F. Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev. Med. Child Neurol. 2016, 58, 77–84. [Google Scholar] [CrossRef]
- Parisi, L.; Di Filippo, T.; Glorioso, P.; La Grutta, S.; Epifanio, M.S.; Roccella, M. Autism spectrum disorders in children affected by Duchenne muscular dystrophy. Minerva Pediatr. 2018, 70, 233–239. [Google Scholar] [CrossRef]
- Pascual-Morena, C.; Cavero-Redondo, I.; Álvarez-Bueno, C.; Jiménez-López, E.; Saz-Lara, A.; Martínez-García, I.; Martínez-Vizcaíno, V. Global prevalence of intellectual developmental disorder in dystrophinopathies: A systematic review and meta-analysis. Dev. Med. Child Neurol. 2023, 65, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Ohlendieck, K. Proteomic profiling of the dystrophin-deficient mdx phenocopy of dystrophinopathy-associated cardiomyopathy. Biomed. Res. Int. 2014, 2014, 246195. [Google Scholar] [CrossRef] [PubMed]
- Gowran, A.; Brioschi, M.; Rovina, D.; Chiesa, M.; Piacentini, L.; Mallia, S.; Banfi, C.; Pompilio, G.; Santoro, R. Multiomic Approaches to Uncover the Complexities of Dystrophin-Associated Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 8954. [Google Scholar] [CrossRef] [PubMed]
- Lee-Gannon, T.; Jiang, X.; Tassin, T.C.; Mammen, P.P.A. Biomarkers in Duchenne Muscular Dystrophy. Curr. Heart Fail. Rep. 2022, 19, 52–62. [Google Scholar] [CrossRef]
- Matkovich, S.J. Multiomic approaches to delineate the pathogenesis of cardiac disease. Curr. Opin. Cardiol. 2019, 34, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Sohag, M.M.H.; Raqib, S.M.; Akhmad, S.A. OMICS approaches in cardiovascular diseases: A mini review. Genom. Inform. 2021, 19, e13. [Google Scholar] [CrossRef]
- Sarhene, M.; Wang, Y.; Wei, J.; Huang, Y.; Li, M.; Li, L.; Acheampong, E.; Zhengcan, Z.; Xiaoyan, Q.; Yunsheng, X.; et al. Biomarkers in heart failure: The past, current and future. Heart Fail. Rev. 2019, 24, 867–903. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Rienks, M.; Theofilatos, K.; Mayr, M. Systems biology in cardiovascular disease: A multiomics approach. Nat. Rev. Cardiol. 2021, 18, 313–330. [Google Scholar] [CrossRef]
- Lohan, J.; Ohlendieck, K. Drastic reduction in the luminal Ca2+-binding proteins calsequestrin and sarcalumenin in dystrophin-deficient cardiac muscle. Biochim. Biophys. Acta 2004, 1689, 252–258. [Google Scholar] [CrossRef]
- Gulston, M.K.; Rubtsov, D.V.; Atherton, H.J.; Clarke, K.; Davies, K.E.; Lilley, K.S.; Griffin, J.L. A combined metabolomic and proteomic investigation of the effects of a failure to express dystrophin in the mouse heart. J. Proteome Res. 2008, 7, 2069–2077. [Google Scholar] [CrossRef]
- Lewis, C.; Jockusch, H.; Ohlendieck, K. Proteomic Profiling of the Dystrophin-Deficient MDX Heart Reveals Drastically Altered Levels of Key Metabolic and Contractile Proteins. J. Biomed. Biotechnol. 2010, 2010, 648501. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.K.; Zhang, L.; Adams, M.E.; Phillips, A.; Freitas, M.A.; Froehner, S.C.; Green-Church, K.B.; Montanaro, F. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS ONE 2012, 7, e43515. [Google Scholar] [CrossRef]
- Holland, A.; Dowling, P.; Zweyer, M.; Swandulla, D.; Henry, M.; Clynes, M.; Ohlendieck, K. Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics 2013, 13, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Dowling, P.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of dystrophin deficiency and associated changes in the aged mdx-4cv heart model of dystrophinopathy-related cardiomyopathy. J. Proteom. 2016, 145, 24–36. [Google Scholar] [CrossRef]
- Chung, H.S.; Kim, G.E.; Holewinski, R.J.; Venkatraman, V.; Zhu, G.; Bedja, D.; Kass, D.A.; Van Eyk, J.E. Transient receptor potential channel 6 regulates abnormal cardiac S-nitrosylation in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, E10763–E10771. [Google Scholar] [CrossRef] [PubMed]
- Tamiyakul, H.; Kemter, E.; Kösters, M.; Ebner, S.; Blutke, A.; Klymiuk, N.; Flenkenthaler, F.; Wolf, E.; Arnold, G.J.; Fröhlich, T. Progressive Proteome Changes in the Myocardium of a Pig Model for Duchenne Muscular Dystrophy. iScience 2020, 23, 101516. [Google Scholar] [CrossRef]
- Jung, C.; Martins, A.S.; Niggli, E.; Shirokova, N. Dystrophic cardiomyopathy: Amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc. Res. 2008, 77, 766–773. [Google Scholar] [CrossRef]
- Klietsch, R.; Ervasti, J.M.; Arnold, W.; Campbell, K.P.; Jorgensen, A.O. Dystrophin-glycoprotein complex and laminin colocalize to the sarcolemma and transverse tubules of cardiac muscle. Circ. Res. 1993, 72, 349–360. [Google Scholar] [CrossRef]
- Wang, H.; Marrosu, E.; Brayson, D.; Wasala, N.B.; Johnson, E.K.; Scott, C.S.; Yue, Y.; Hau, K.L.; Trask, A.J.; Froehner, S.C.; et al. Proteomic analysis identifies key differences in the cardiac interactomes of dystrophin and micro-dystrophin. Hum. Mol. Genet. 2021, 30, 1321–1336. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of liver tissue from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin. Proteom. 2018, 15, 34. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of fatty acid binding proteins in muscular dystrophy. Expert Rev. Proteom. 2020, 17, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Dowling, P.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of mdx-4cv serum reveals highly elevated levels of the inflammation-induced plasma marker haptoglobin in muscular dystrophy. Int. J. Mol. Med. 2017, 39, 1357–1370. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Dataset on the comparative proteomic profiling of mouse saliva and serum from wild type versus the dystrophic mdx-4cv mouse model of dystrophinopathy. Data Brief. 2018, 21, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic serum biomarkers for neuromuscular diseases. Expert Rev. Proteom. 2018, 15, 277–291. [Google Scholar] [CrossRef]
- Dowling, P.; Zweyer, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic and cell biological profiling of the renal phenotype of the mdx-4cv mouse model of Duchenne muscular dystrophy. Eur. J. Cell Biol. 2020, 99, 151059. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Zweyer, M.; Raucamp, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Dataset on the mass spectrometry-based proteomic profiling of the kidney from wild type and the dystrophic mdx-4cv mouse model of X-linked muscular dystrophy. Data Brief 2020, 28, 105067. [Google Scholar] [CrossRef] [PubMed]
- Hathout, Y.; Marathi, R.L.; Rayavarapu, S.; Zhang, A.; Brown, K.J.; Seol, H.; Gordish-Dressman, H.; Cirak, S.; Bello, L.; Nagaraju, K.; et al. Discovery of serum protein biomarkers in the mdx mouse model and cross-species comparison to Duchenne muscular dystrophy patients. Hum. Mol. Genet. 2014, 23, 6458–6469. [Google Scholar] [CrossRef]
- Coenen-Stass, A.M.; McClorey, G.; Manzano, R.; Betts, C.A.; Blain, A.; Saleh, A.F.; Gait, M.J.; Lochmüller, H.; Wood, M.J.; Roberts, T.C. Identification of novel, therapy-responsive protein biomarkers in a mouse model of Duchenne muscular dystrophy by aptamer-based serum proteomics. Sci. Rep. 2015, 5, 17014. [Google Scholar] [CrossRef]
- Spitali, P.; Hettne, K.; Tsonaka, R.; Charrout, M.; van den Bergen, J.; Koeks, Z.; Kan, H.E.; Hooijmans, M.T.; Roos, A.; Straub, V.; et al. Tracking disease progression non-invasively in Duchenne and Becker muscular dystrophies. J. Cachexia Sarcopenia Muscle 2018, 9, 715–726. [Google Scholar] [CrossRef]
- Parolo, S.; Marchetti, L.; Lauria, M.; Misselbeck, K.; Scott-Boyer, M.P.; Caberlotto, L.; Priami, C. Combined use of protein biomarkers and network analysis unveils deregulated regulatory circuits in Duchenne muscular dystrophy. PLoS ONE 2018, 13, e0194225. [Google Scholar] [CrossRef]
- Villalta, S.A.; Rosenberg, A.S.; Bluestone, J.A. The immune system in Duchenne muscular dystrophy: Friend or foe. Rare Dis. 2015, 3, e1010966. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299rv4. [Google Scholar] [CrossRef]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of Inherited Muscular Dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Sitzia, C.; Cassani, B.; Cassinelli, L.; Rigoni, R.; Colleoni, F.; Fusco, N.; Gatti, S.; Bella, P.; Villa, C.; et al. Therapeutic Potential of Immunoproteasome Inhibition in Duchenne Muscular Dystrophy. Mol. Ther. 2014, 24, 1898–1912. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Mojumdar, K.; Liang, F.; Lemaire, C.; Li, T.; Richardson, J.; Divangahi, M.; Qureshi, S.; Petrof, B.J. Toll-like receptor 4 ablation in mdx mice reveals innate immunity as a therapeutic target in Duchenne muscular dystrophy. Hum. Mol. Genet. 2015, 24, 2147–6212. [Google Scholar] [CrossRef] [PubMed]
- Ouisse, L.H.; Remy, S.; Lafoux, A.; Larcher, T.; Tesson, L.; Chenouard, V.; Guillonneau, C.; Brusselle, L.; Vimond, N.; Rouger, K.; et al. Immunophenotype of a Rat Model of Duchenne’s Disease and Demonstration of Improved Muscle Strength After Anti-CD45RC Antibody Treatment. Front. Immunol. 2019, 10, 2131. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, L.; Molinaro, D.; Fortunato, F.; Mella, C.; Cassani, B.; Torrente, Y.; Farini, A. Immunoproteasome Inhibition Ameliorates Aged Dystrophic Mouse Muscle Environment. Int. J. Mol. Sci. 2022, 23, 14657. [Google Scholar] [CrossRef]
- Tripodi, L.; Villa, C.; Molinaro, D.; Torrente, Y.; Farini, A. The Immune System in Duchenne Muscular Dystrophy Pathogenesis. Biomedicines 2021, 9, 1447. [Google Scholar] [CrossRef]
- Mojumdar, K.; Liang, F.; Giordano, C.; Lemaire, C.; Danialou, G.; Okazaki, T.; Bourdon, J.; Rafei, M.; Galipeau, J.; Divangahi, M.; et al. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol. Med. 2014, 6, 1476–1492. [Google Scholar] [CrossRef]
- Mojumdar, K.; Giordano, C.; Lemaire, C.; Liang, F.; Divangahi, M.; Qureshi, S.T.; Petrof, B.J. Divergent impact of Toll-like receptor 2 deficiency on repair mechanisms in healthy muscle versus Duchenne muscular dystrophy. J. Pathol. 2016, 239, 10–22. [Google Scholar] [CrossRef]
- Lozanoska-Ochser, B.; Benedetti, A.; Rizzo, G.; Marrocco, V.; Di Maggio, R.; Fiore, P.; Bouche, M. Targeting early PKCθ-dependent T-cell infiltration of dystrophic muscle reduces disease severity in a mouse model of muscular dystrophy. J. Pathol. 2018, 244, 323–333. [Google Scholar] [CrossRef]
- Lemos, J.P.; Tenório, L.P.G.; Mouly, V.; Butler-Browne, G.; Mendes-da-Cruz, D.A.; Savino, W.; Smeriglio, P. T cell biology in neuromuscular disorders: A focus on Duchenne Muscular Dystrophy and Amyotrophic Lateral Sclerosis. Front. Immunol. 2023, 14, 1202834. [Google Scholar] [CrossRef]
- Petrof, B.J.; Podolsky, T.; Bhattarai, S.; Tan, J.; Ding, J. Trained immunity as a potential target for therapeutic immunomodulation in Duchenne muscular dystrophy. Front. Immunol. 2023, 14, 1183066. [Google Scholar] [CrossRef]
- Rizzo, G.; Di Maggio, R.; Benedetti, A.; Morroni, J.; Bouche, M.; Lozanoska-Ochser, B. Splenic Ly6Chi monocytes are critical players in dystrophic muscle injury and repair. JCI Insight 2020, 5, 130807. [Google Scholar] [CrossRef]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef]
- Lewis, S.M.; Williams, A.; Eisenbarth, S.C. Structure and function of the immune system in the spleen. Sci. Immunol. 2019, 4, eaau6085. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Zweyer, M.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteome-wide Changes in the mdx-4cv Spleen due to Pathophysiological Cross Talk with Dystrophin-Deficient Skeletal Muscle. iScience 2020, 23, 101500. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Zweyer, M.; Sabir, H.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of the interface between the stomach wall and the pancreas in dystrophinopathy. Eur. J. Transl. Myol. 2021, 31, 9627. [Google Scholar] [CrossRef]
- Dowling, P.; Zweyer, M.; Sabir, H.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Mass spectrometry-based proteomic characterization of the middle-aged mouse brain for animal model research of neuromuscular diseases. Eur. J. Transl. Myol. 2023, 33, 11553. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis reveals complex changes in the brain proteome from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin. Proteom. 2015, 12, 27. [Google Scholar] [CrossRef]
- Murphy, S.; Ohlendieck, K. Proteomic Profiling of the Dystrophin-Deficient Brain. Methods Mol. Biol. 2018, 1687, 91–105. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kadluczka, J.; Kuter, K.Z. Beyond the GFAP-Astrocyte Protein Markers in the Brain. Biomolecules 2021, 11, 1361. [Google Scholar] [CrossRef]
- Murphy, S.; Schmitt-John, T.; Dowling, P.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of the brain from the wobbler mouse model of amyotrophic lateral sclerosis reveals elevated levels of the astrogliosis marker glial fibrillary acidic protein. Eur. J. Transl. Myol. 2023, 33, 11555. [Google Scholar] [CrossRef]
- Chesshyre, M.; Ridout, D.; Hashimoto, Y.; Ookubo, Y.; Torelli, S.; Maresh, K.; Ricotti, V.; Abbott, L.; Gupta, V.A.; Main, M.; et al. Investigating the role of dystrophin isoform deficiency in motor function in Duchenne muscular dystrophy. J. Cachexia Sarcopenia Muscle 2022, 13, 1360–1372. [Google Scholar] [CrossRef]
- De Stefano, M.E.; Ferretti, V.; Mozzetta, C. Synaptic alterations as a neurodevelopmental trait of Duchenne muscular dystrophy. Neurobiol. Dis. 2022, 168, 105718. [Google Scholar] [CrossRef]
- Wijekoon, N.; Gonawala, L.; Ratnayake, P.; Amaratunga, D.; Hathout, Y.; Mohan, C.; Steinbusch, H.W.M.; Dalal, A.; Hoffman, E.P.; de Silva, K.R.D. Duchenne Muscular Dystrophy from Brain to Muscle: The Role of Brain Dystrophin Isoforms in Motor Functions. J. Clin. Med. 2023, 12, 5637. [Google Scholar] [CrossRef]
- Andrews, J.G.; Galindo, M.K.; Thomas, S.; Mathews, K.D.; Whitehead, N. DMD Gene and Dystrophinopathy Phenotypes Associated with Mutations: A Systematic Review for Clinicians. J. Clin. Neuromuscul. Dis. 2023, 24, 171–187. [Google Scholar] [CrossRef]
- Hoffman, E.P. Pharmacotherapy of Duchenne Muscular Dystrophy. Handb. Exp. Pharmacol. 2020, 261, 25–37. [Google Scholar] [CrossRef]
- Grages, S.M.; Bell, M.; Berlau, D.J. New and emerging pharmacotherapy for duchenne muscular dystrophy: A focus on synthetic therapeutics. Expert Opin. Pharmacother. 2020, 21, 841–851. [Google Scholar] [CrossRef]
- Deng, J.; Zhang, J.; Shi, K.; Liu, Z. Drug development progress in duchenne muscular dystrophy. Front. Pharmacol. 2022, 13, 950651. [Google Scholar] [CrossRef]
- Rawls, A.; Diviak, B.K.; Smith, C.I.; Severson, G.W.; Acosta, S.A.; Wilson-Rawls, J. Pharmacotherapeutic Approaches to Treatment of Muscular Dystrophies. Biomolecules 2023, 13, 1536. [Google Scholar] [CrossRef]
- Fortunato, F.; Rossi, R.; Falzarano, M.S.; Ferlini, A. Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy. J. Clin. Med. 2021, 10, 820. [Google Scholar] [CrossRef]
- Markati, T.; Oskoui, M.; Farrar, M.A.; Duong, T.; Goemans, N.; Servais, L. Emerging therapies for Duchenne muscular dystrophy. Lancet Neurol. 2022, 21, 814–829. [Google Scholar] [CrossRef]
- Chung Liang, L.; Sulaiman, N.; Yazid, M.D. A Decade of Progress in Gene Targeted Therapeutic Strategies in Duchenne Muscular Dystrophy: A Systematic Review. Front. Bioeng. Biotechnol. 2022, 10, 833833. [Google Scholar] [CrossRef]
- Chang, M.; Cai, Y.; Gao, Z.; Chen, X.; Liu, B.; Zhang, C.; Yu, W.; Cao, Q.; Shen, Y.; Yao, X.; et al. Duchenne muscular dystrophy: Pathogenesis and promising therapies. J. Neurol. 2023, 270, 3733–3749. [Google Scholar] [CrossRef]
- Duan, D. Duchenne Muscular Dystrophy Gene Therapy in 2023: Status, Perspective, and Beyond. Hum. Gene Ther. 2023, 34, 345–349. [Google Scholar] [CrossRef]
- Clemens, P.R.; Rao, V.K.; Connolly, A.M.; Harper, A.D.; Mah, J.K.; Smith, E.C.; McDonald, C.M.; Zaidman, C.M.; Morgenroth, L.P.; Osaki, H.; et al. Safety, Tolerability, and Efficacy of Viltolarsen in Boys With Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020, 77, 982–991. [Google Scholar] [CrossRef]
- Novak, J.S.; Spathis, R.; Dang, U.J.; Fiorillo, A.A.; Hindupur, R.; Tully, C.B.; Mázala, D.A.G.; Canessa, E.; Brown, K.J.; Partridge, T.A.; et al. Interrogation of Dystrophin and Dystroglycan Complex Protein Turnover After Exon Skipping Therapy. J. Neuromuscul. Dis. 2021, 8, S383–S402. [Google Scholar] [CrossRef]
- Eser, G.; Topaloğlu, H. Current Outline of Exon Skipping Trials in Duchenne Muscular Dystrophy. Genes 2022, 13, 1241. [Google Scholar] [CrossRef]
- Mercuri, E.; Seferian, A.M.; Servais, L.; Deconinck, N.; Stevenson, H.; Ni, X.; Zhang, W.; East, L.; Yonren, S.; Muntoni, F.; et al. Safety, tolerability and pharmacokinetics of eteplirsen in young boys aged 6-48 months with Duchenne muscular dystrophy amenable to exon 51 skipping. Neuromuscul. Disord. 2023, 33, 476–483. [Google Scholar] [CrossRef]
- Min, Y.L.; Bassel-Duby, R.; Olson, E.N. CRISPR Correction of Duchenne Muscular Dystrophy. Annu. Rev. Med. 2019, 70, 239–255. [Google Scholar] [CrossRef]
- Kupatt, C.; Windisch, A.; Moretti, A.; Wolf, E.; Wurst, W.; Walter, M.C. Genome editing for Duchenne muscular dystrophy: A glimpse of the future? Gene Ther. 2021, 28, 542–548. [Google Scholar] [CrossRef]
- Chemello, F.; Olson, E.N.; Bassel-Duby, R. CRISPR- Editing Therapy for Duchenne Muscular Dystrophy. Hum. Gene Ther. 2023, 34, 379–387. [Google Scholar] [CrossRef]
- Politano, L. Read-through approach for stop mutations in Duchenne muscular dystrophy. An update. Acta Myol. 2021, 40, 43–50. [Google Scholar] [CrossRef]
- Seto, J.T.; Ramos, J.N.; Muir, L.; Chamberlain, J.S.; Odom, G.L. Gene replacement therapies for duchenne muscular dystrophy using adeno-associated viral vectors. Curr. Gene Ther. 2012, 12, 139–151. [Google Scholar] [CrossRef]
- Nance, M.E.; Duan, D. Perspective on Adeno-Associated Virus Capsid Modification for Duchenne Muscular Dystrophy Gene Therapy. Hum. Gene Ther. 2015, 26, 786–800. [Google Scholar] [CrossRef]
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children with Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020, 77, 1122–1131. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Hirai, Y.; Hashido, K.; Okada, T. Therapeutic Application of Extracellular Vesicles-Capsulated Adeno-Associated Virus Vector via nSMase2/Smpd3, Satellite, and Immune Cells in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2022, 23, 1551. [Google Scholar] [CrossRef] [PubMed]
- Loro, E.; Sengupta, K.; Bogdanovich, S.; Whig, K.; Schultz, D.C.; Huryn, D.M.; Khurana, T.S. High-throughput identification of post-transcriptional utrophin up-regulators for Duchenne muscle dystrophy (DMD) therapy. Sci. Rep. 2020, 10, 2132. [Google Scholar] [CrossRef]
- Soblechero-Martín, P.; López-Martínez, A.; de la Puente-Ovejero, L.; Vallejo-Illarramendi, A.; Arechavala-Gomeza, V. Utrophin modulator drugs as potential therapies for Duchenne and Becker muscular dystrophies. Neuropathol. Appl. Neurobiol. 2021, 47, 711–723. [Google Scholar] [CrossRef]
- Vuorinen, A.; Wilkinson, I.V.L.; Chatzopoulou, M.; Edwards, B.; Squire, S.E.; Fairclough, R.J.; Bazan, N.A.; Milner, J.A.; Conole, D.; Donald, J.R.; et al. Discovery and mechanism of action studies of 4,6-diphenylpyrimidine-2-carbohydrazides as utrophin modulators for the treatment of Duchenne muscular dystrophy. Eur. J. Med. Chem. 2021, 220, 113431. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Mouly, V.; Butler-Browne, G.; Cossu, G. Challenges in cell transplantation for muscular dystrophy. Exp. Cell Res. 2021, 409, 112908. [Google Scholar] [CrossRef]
- Boyer, O.; Butler-Browne, G.; Chinoy, H.; Cossu, G.; Galli, F.; Lilleker, J.B.; Magli, A.; Mouly, V.; Perlingeiro, R.C.R.; Previtali, S.C.; et al. Myogenic Cell Transplantation in Genetic and Acquired Diseases of Skeletal Muscle. Front. Genet. 2021, 12, 702547. [Google Scholar] [CrossRef]
- Ausems, C.R.M.; van Engelen, B.G.M.; van Bokhoven, H.; Wansink, D.G. Systemic cell therapy for muscular dystrophies: The ultimate transplantable muscle progenitor cell and current challenges for clinical efficacy. Stem Cell Rev. Rep. 2021, 17, 878–899. [Google Scholar] [CrossRef]
- Saleh, K.K.; Switzler, C.; Hicks, M.R.; Gane, L.; Gibbs, D.E.; Pyle, A.D. Duchenne muscular dystrophy disease severity impacts skeletal muscle progenitor cells systemic delivery. Front. Physiol. 2023, 14, 1190524. [Google Scholar] [CrossRef]
- Anaya-Segura, M.A.; García-Martínez, F.A.; Montes-Almanza, L.A.; Díaz, B.G.; Avila-Ramírez, G.; Alvarez-Maya, I.; Coral-Vazquez, R.M.; Mondragón-Terán, P.; Escobar-Cedillo, R.E.; García-Calderón, N.; et al. Non-Invasive Biomarkers for Duchenne Muscular Dystrophy and Carrier Detection. Molecules 2015, 20, 11154–11172. [Google Scholar] [CrossRef]
- Fortunato, F.; Ferlini, A. Biomarkers in Duchenne Muscular Dystrophy: Current Status and Future Directions. J. Neuromuscul. Dis. 2023, 10, 987–1002. [Google Scholar] [CrossRef]
- O’Sullivan, E.M.; Dowling, P.; Swandulla, D.; Ohlendieck, K. Proteomic Identification of Saliva Proteins as Noninvasive Diagnostic Biomarkers. Methods Mol. Biol. 2023, 2596, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomic identification of biomarkers of skeletal muscle disorders. Biomark. Med. 2013, 7, 169–186. [Google Scholar] [CrossRef]
- Joyce, N.C.; Oskarsson, B.; Jin, L.W. Muscle biopsy evaluation in neuromuscular disorders. Med. Rehabil. Clin. N. Am. 2012, 23, 609–631. [Google Scholar] [CrossRef] [PubMed]
- Meola, G.; Bugiardini, E.; Cardani, R. Muscle biopsy. J. Neurol. 2012, 259, 601–610. [Google Scholar] [CrossRef]
- Shanely, R.A.; Zwetsloot, K.A.; Triplett, N.T.; Meaney, M.P.; Farris, G.E.; Nieman, D.C. Human skeletal muscle biopsy procedures using the modified Bergström technique. J. Vis. Exp. 2014, 10, 51812. [Google Scholar] [CrossRef]
- Nix, J.S.; Moore, S.A. What Every Neuropathologist Needs to Know: The Muscle Biopsy. J. Neuropathol. Exp. Neurol. 2020, 79, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, P.; Lippi, G.; Maffulli, N. Biochemical markers of muscular damage. Clin. Chem. Lab. Med. 2010, 48, 757–767. [Google Scholar] [CrossRef]
- Lippi, G.; Schena, F.; Ceriotti, F. Diagnostic biomarkers of muscle injury and exertional rhabdomyolysis. Clin. Chem. Lab. Med. 2018, 57, 175–182. [Google Scholar] [CrossRef]
- Burch, P.M.; Pogoryelova, O.; Goldstein, R.; Bennett, D.; Guglieri, M.; Straub, V.; Bushby, K.; Lochmüller, H.; Morris, C. Muscle-Derived Proteins as Serum Biomarkers for Monitoring Disease Progression in Three Forms of Muscular Dystrophy. J. Neuromuscul. Dis. 2015, 2, 241–255. [Google Scholar] [CrossRef]
- Szigyarto, C.A.; Spitali, P. Biomarkers of Duchenne muscular dystrophy: Current findings. Degener. Neurol. Neuromuscul. Dis. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Model Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Gargan, S.; Zweyer, M.; Sabir, H.; Swandulla, D.; Ohlendieck, K. Proteomic profiling of carbonic anhydrase CA3 in skeletal muscle. Expert Rev. Proteom. 2021, 18, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Johansson, C.; Heywood, W.; Vinette, H.; Jensen, G.; Tegel, H.; Jiménez-Requena, A.; Torelli, S.; Al-Khalili Szigyarto, C.; Ferlini, A. A Proof of Principle Proteomic Study Detects Dystrophin in Human Plasma: Implications in DMD Diagnosis and Clinical Monitoring. Int. J. Mol. Sci. 2023, 24, 5215. [Google Scholar] [CrossRef] [PubMed]
- Ogundele, M.; Zhang, J.S.; Goswami, M.V.; Barbieri, M.L.; Dang, U.J.; Novak, J.S.; Hoffman, E.P.; Nagaraju, K.; Cinrg-Dnhs Investigators; Hathout, Y. Validation of Chemokine Biomarkers in Duchenne Muscular Dystrophy. Life 2021, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Hunt, H.; Signorelli, M.; Edfors, F.; Hober, A.; Svensson, A.S.; Tegel, H.; Forstström, B.; Aartsma-Rus, A.; Niks, E.; et al. Orthogonal proteomics methods warrant the development of Duchenne muscular dystrophy biomarkers. Clin. Proteom. 2023, 20, 23. [Google Scholar] [CrossRef] [PubMed]
- Rouillon, J.; Poupiot, J.; Zocevic, A.; Amor, F.; Léger, T.; Garcia, C.; Camadro, J.M.; Wong, B.; Pinilla, R.; Cosette, J.; et al. Serum proteomic profiling reveals fragments of MYOM3 as potential biomarkers for monitoring the outcome of therapeutic interventions in muscular dystrophies. Hum. Mol. Genet. 2015, 24, 4916–4932. [Google Scholar] [CrossRef]
- Rouillon, J.; Zocevic, A.; Leger, T.; Garcia, C.; Camadro, J.M.; Udd, B.; Wong, B.; Servais, L.; Voit, T.; Svinartchouk, F. Proteomics profiling of urine reveals specific titin fragments as biomarkers of Duchenne muscular dystrophy. Neuromuscul. Disord. 2014, 24, 563–573. [Google Scholar] [CrossRef]
- Robertson, A.S.; Majchrzak, M.J.; Smith, C.M.; Gagnon, R.C.; Devidze, N.; Banks, G.B.; Little, S.C.; Nabbie, F.; Bounous, D.I.; DiPiero, J.; et al. Dramatic elevation in urinary amino terminal titin fragment excretion quantified by immunoassay in Duchenne muscular dystrophy patients and in dystrophin deficient rodents. Neuromuscul. Disord. 2017, 27, 635–645. [Google Scholar] [CrossRef]
- Gargan, S.; Dowling, P.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Identification of marker proteins of muscular dystrophy in the urine proteome from the mdx-4cv model of dystrophinopathy. Mol. Omics 2020, 16, 268–278. [Google Scholar] [CrossRef]
- Matsuo, M.; Awano, H.; Maruyama, N.; Nishio, H. Titin fragment in urine: A noninvasive biomarker of muscle degradation. Adv. Clin. Chem. 2019, 90, 1–23. [Google Scholar] [CrossRef]
- Ishii, M.N.; Nakashima, M.; Kamiguchi, H.; Zach, N.; Kuboki, R.; Baba, R.; Hirakawa, T.; Suzuki, K.; Quinton, M. Urine titin as a novel biomarker for Duchenne muscular dystrophy. Neuromuscul. Disord. 2023, 33, 302–308. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic identification of elevated saliva kallikrein levels in the mdx-4cv mouse model of Duchenne muscular dystrophy. Biochem. Biophys. Rep. 2018, 18, 100541. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Wang, Z. Precision Medicine: Disease Subtyping and Tailored Treatment. Cancers 2023, 15, 3837. [Google Scholar] [CrossRef]
- Bernas, T.; Grégori, G.; Asem, E.K.; Robinson, J.P. Integrating cytomics and proteomics. Mol. Cell. Proteom. 2006, 5, 2–13. [Google Scholar] [CrossRef]
- Gomase, V.S.; Tagore, S. Cytomics. Curr. Drug Metab. 2008, 9, 263–266. [Google Scholar] [CrossRef]
- Larance, M.; Lamond, A.I. Multidimensional proteomics for cell biology. Nat. Rev. Mol. Cell Biol. 2015, 16, 269–280. [Google Scholar] [CrossRef]
- O’Connor, L.M.; O’Connor, B.A.; Lim, S.B.; Zeng, J.; Lo, C.H. Integrative multi-omics and systems bioinformatics in translational neuroscience: A data mining perspective. J. Pharm. Anal. 2023, 13, 836–850. [Google Scholar] [CrossRef]
- Alto, S.I.; Chang, C.N.; Brown, K.; Kioussi, C.; Filtz, T.M. Gene Expression Profiling of Skeletal Muscles. Genes 2021, 12, 1718. [Google Scholar] [CrossRef]
- Erbe, L.S.; Hoffjan, S.; Janßen, S.; Kneifel, M.; Krause, K.; Gerding, W.M.; Döring, K.; Güttsches, A.K.; Roos, A.; Buena Atienza, E.; et al. Exome Sequencing and Optical Genome Mapping in Molecularly Unsolved Cases of Duchenne Muscular Dystrophy: Identification of a Causative X-Chromosomal Inversion Disrupting the DMD Gene. Int. J. Mol. Sci. 2023, 24, 14716. [Google Scholar] [CrossRef]
- Pluta, N.; von Moers, A.; Pechmann, A.; Stenzel, W.; Goebel, H.H.; Atlan, D.; Wolf, B.; Nanda, I.; Zaum, A.K.; Rost, S. Whole-Genome Sequencing Identified New Structural Variations in the DMD Gene That Cause Duchenne Muscular Dystrophy in Two Girls. Int. J. Mol. Sci. 2023, 24, 13567. [Google Scholar] [CrossRef]
- Baßler, K.; Günther, P.; Schulte-Schrepping, J.; Becker, M.; Biernat, P. A Bioinformatic Toolkit for Single-Cell mRNA Analysis. Methods Mol. Biol. 2019, 1979, 433–455. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Rao, A.; Barkley, D.; França, G.S.; Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 2021, 596, 211–220. [Google Scholar] [CrossRef]
- Sandonà, M.; Saccone, V. Post-translational Modification in Muscular Dystrophies. Adv. Exp. Med. Biol. 2022, 1382, 71–84. [Google Scholar] [CrossRef]
- Iwasaki, T.; Terrill, J.R.; Kawarai, K.; Miyata, Y.; Tagami, T.; Maeda, N.; Hasegawa, Y.; Watanabe, T.; Grounds, M.D.; Arthur, P.G. The location of protein oxidation in dystrophic skeletal muscle from the mdx mouse model of Duchenne muscular dystrophy. Acta Histochem. 2022, 124, 151959. [Google Scholar] [CrossRef]
- Xu, H.; Cai, X.; Xu, K.; Wu, Q.; Xu, B. The metabolomic plasma profile of patients with Duchenne muscular dystrophy: Providing new evidence for its pathogenesis. Orphanet J. Rare Dis. 2023, 18, 273. [Google Scholar] [CrossRef]
- Dabaj, I.; Ferey, J.; Marguet, F.; Gilard, V.; Basset, C.; Bahri, Y.; Brehin, A.C.; Vanhulle, C.; Leturcq, F.; Marret, S.; et al. Muscle metabolic remodelling patterns in Duchenne muscular dystrophy revealed by ultra-high-resolution mass spectrometry imaging. Sci. Rep. 2021, 11, 1906. [Google Scholar] [CrossRef]
- Tsonaka, R.; Signorelli, M.; Sabir, E.; Seyer, A.; Hettne, K.; Aartsma-Rus, A.; Spitali, P. Longitudinal metabolomic analysis of plasma enables modeling disease progression in Duchenne muscular dystrophy mouse models. Hum. Mol. Genet. 2020, 29, 745–755. [Google Scholar] [CrossRef]
- Milad, N.; White, Z.; Tehrani, A.Y.; Sellers, S.; Rossi, F.M.V.; Bernatchez, P. Increased plasma lipid levels exacerbate muscle pathology in the mdx mouse model of Duchenne muscular dystrophy. Skelet. Muscle 2017, 7, 19. [Google Scholar] [CrossRef]
- Tsonaka, R.; Seyer, A.; Aartsma-Rus, A.; Spitali, P. Plasma lipidomic analysis shows a disease progression signature in mdx mice. Sci. Rep. 2021, 11, 12993. [Google Scholar] [CrossRef]
- Rancourt, A.; Dufresne, S.S.; St-Pierre, G.; Lévesque, J.C.; Nakamura, H.; Kikuchi, Y.; Satoh, M.S.; Frenette, J.; Sato, S. Galectin-3 and N-acetylglucosamine promote myogenesis and improve skeletal muscle function in the mdx model of Duchenne muscular dystrophy. FASEB J. 2018, 32, 6445. [Google Scholar] [CrossRef] [PubMed]
- Bonaguro, L.; Schulte-Schrepping, J.; Ulas, T.; Aschenbrenner, A.C.; Beyer, M.; Schultze, J.L. A guide to systems-level immunomics. Nat. Immunol. 2022, 23, 1412–1423. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Riley, N.M.; Yang, A.C.; Kim, J.T.; Terrell, S.M.; Li, V.L.; Garcia-Contreras, M.; Bertozzi, C.R.; Long, J.Z. Cell type-selective secretome profiling in vivo. Nat. Chem. Biol. 2021, 17, 326–334. [Google Scholar] [CrossRef]
- Coulton, G. Are histochemistry and cytochemistry ‘Omics’? J. Mol. Histol. 2004, 35, 603–613. [Google Scholar] [CrossRef]
- Robinson, J.P.; Ostafe, R.; Iyengar, S.N.; Rajwa, B.; Fischer, R. Flow Cytometry: The Next Revolution. Cells 2023, 12, 1875. [Google Scholar] [CrossRef] [PubMed]
- Ercan, H.; Resch, U.; Hsu, F.; Mitulovic, G.; Bileck, A.; Gerner, C.; Yang, J.W.; Geiger, M.; Miller, I.; Zellner, M. A Practical and Analytical Comparative Study of Gel-Based Top-Down and Gel-Free Bottom-Up Proteomics Including Unbiased Proteoform Detection. Cells 2023, 12, 747. [Google Scholar] [CrossRef]
- Schaffer, L.V.; Millikin, R.J.; Shortreed, M.R.; Scalf, M.; Smith, L.M. Improving Proteoform Identifications in Complex Systems Through Integration of Bottom-Up and Top-Down Data. J. Proteome Res. 2020, 19, 3510–3517. [Google Scholar] [CrossRef]
- Starosta, A.; Konieczny, P. Therapeutic aspects of cell signaling and communication in Duchenne muscular dystrophy. Cell. Mol. Life Sci. 2021, 78, 4867–4891. [Google Scholar] [CrossRef]
- Mund, A.; Coscia, F.; Kriston, A.; Hollandi, R.; Kovács, F.; Brunner, A.D.; Migh, E.; Schweizer, L.; Santos, A.; Bzorek, M.; et al. Deep Visual Proteomics defines single-cell identity and heterogeneity. Nat. Biotechnol. 2022, 40, 1231–1240. [Google Scholar] [CrossRef]
- Rosenberger, F.A.; Thielert, M.; Strauss, M.T.; Schweizer, L.; Ammar, C.; Mädler, S.C.; Metousis, A.; Skowronek, P.; Wahle, M.; Madden, K.; et al. Spatial single-cell mass spectrometry defines zonation of the hepatocyte proteome. Nat. Methods 2023, 20, 1530–1536. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, G.; Stegle, O.; Reik, W. Single-cell epigenomics: Recording the past and predicting the future. Science 2017, 358, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Epigenomics: Technologies and Applications. Circ. Res. 2018, 122, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, I.; Torres-Padilla, M.E.; Almouzni, G. Epigenomics in the single cell era, an important read out for genome function and cell identity. Epigenomics 2021, 13, 981–984. [Google Scholar] [CrossRef]
- Li, Y. Modern epigenetics methods in biological research. Methods 2021, 187, 104–113. [Google Scholar] [CrossRef]
- Robinson, D.C.L.; Dilworth, F.J. Epigenetic Regulation of Adult Myogenesis. Curr. Top. Dev. Biol. 2018, 126, 235–284. [Google Scholar] [CrossRef]
- Plaza-Diaz, J.; Izquierdo, D.; Torres-Martos, Á.; Baig, A.T.; Aguilera, C.M.; Ruiz-Ojeda, F.J. Impact of Physical Activity and Exercise on the Epigenome in Skeletal Muscle and Effects on Systemic Metabolism. Biomedicines 2022, 10, 126. [Google Scholar] [CrossRef]
- Massenet, J.; Gardner, E.; Chazaud, B.; Dilworth, F.J. Epigenetic regulation of satellite cell fate during skeletal muscle regeneration. Skelet. Muscle 2021, 11, 4. [Google Scholar] [CrossRef]
- Consalvi, S.; Saccone, V.; Mozzetta, C. Histone deacetylase inhibitors: A potential epigenetic treatment for Duchenne muscular dystrophy. Epigenomics 2014, 6, 547–560. [Google Scholar] [CrossRef]
- Schreyer, L.; Reilly, J.; McConkey, H.; Kerkhof, J.; Levy, M.A.; Hu, J.; Hnaini, M.; Sadikovic, B.; Campbell, C. The discovery of the DNA methylation episignature for Duchenne muscular dystrophy. Neuromuscul. Disord. 2023, 33, 5–14. [Google Scholar] [CrossRef]
- Sandonà, M.; Cavioli, G.; Renzini, A.; Cedola, A.; Gigli, G.; Coletti, D.; McKinsey, T.A.; Moresi, V.; Saccone, V. Histone Deacetylases: Molecular Mechanisms and Therapeutic Implications for Muscular Dystrophies. Int. J. Mol. Sci. 2023, 24, 4306. [Google Scholar] [CrossRef]
- Chen, C.; Wang, J.; Pan, D.; Wang, X.; Xu, Y.; Yan, J.; Wang, L.; Yang, X.; Yang, M.; Liu, G.P. Applications of multi-omics analysis in human diseases. MedComm 2023, 4, e315. [Google Scholar] [CrossRef] [PubMed]
- Tavassoly, I.; Goldfarb, J.; Iyengar, R. Systems biology primer: The basic methods and approaches. Essays Biochem. 2018, 62, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Greising, S.M.; Gransee, H.M.; Mantilla, C.B.; Sieck, G.C. Systems biology of skeletal muscle: Fiber type as an organizing principle. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.R.; Zhang, A.; Nguyen, N.Y.; Tully, C.B.; Panigrahi, A.; Gordish-Dressman, H.; Pandey, S.N.; Guglieri, M.; Ryan, M.M.; Clemens, P.R.; et al. Multi-Omics Identifies Circulating miRNA and Protein Biomarkers for Facioscapulohumeral Dystrophy. J. Pers. Med. 2020, 10, 236. [Google Scholar] [CrossRef]
- Liu, J.C.; Dong, S.S.; Shen, H.; Yang, D.Y.; Chen, B.B.; Ma, X.Y.; Peng, Y.R.; Xiao, H.M.; Deng, H.W. Multi-omics research in sarcopenia: Current progress and future prospects. Ageing Res. Rev. 2022, 76, 101576. [Google Scholar] [CrossRef] [PubMed]
- McCourt, J.L.; Stearns-Reider, K.M.; Mamsa, H.; Kannan, P.; Afsharinia, M.H.; Shu, C.; Gibbs, E.M.; Shin, K.M.; Kurmangaliyev, Y.Z.; Schmitt, L.R.; et al. Multi-omics analysis of sarcospan overexpression in mdx skeletal muscle reveals compensatory remodeling of cytoskeleton-matrix interactions that promote mechanotransduction pathways. Skelet. Muscle 2023, 13, 1. [Google Scholar] [CrossRef]
- Bornstein, B.; Heinemann-Yerushalmi, L.; Krief, S.; Adler, R.; Dassa, B.; Leshkowitz, D.; Kim, M.; Bewick, G.; Banks, R.W.; Zelzer, E. Molecular characterization of the intact mouse muscle spindle using a multi-omics approach. eLife 2023, 12, e81843. [Google Scholar] [CrossRef]
- Van Pelt, D.W.; Kharaz, Y.A.; Sarver, D.C.; Eckhardt, L.R.; Dzierzawski, J.T.; Disser, N.P.; Piacentini, A.N.; Comerford, E.; McDonagh, B.; Mendias, C.L. Multiomics analysis of the mdx/mTR mouse model of Duchenne muscular dystrophy. Connect. Tissue Res. 2021, 62, 24–39. [Google Scholar] [CrossRef]
- Elasbali, A.M.; Al-Soud, W.A.; Adnan, M.; Alhassan, H.H.; Mohammad, T.; Hassan, M.I. Discovering Promising Biomarkers and Therapeutic Targets for Duchenne Muscular Dystrophy: A Multiomics Meta-Analysis Approach. Mol. Neurobiol. 2024. [Google Scholar] [CrossRef]
- Signorelli, M.; Tsonaka, R.; Aartsma-Rus, A.; Spitali, P. Multiomic characterization of disease progression in mice lacking dystrophin. PLoS ONE 2023, 18, e0283869. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dong, N.; Yu, L.; Liu, M.; Jiang, J.; Tang, T.; Zhao, H.; Fang, Q. Identification of immune-related features involved in Duchenne muscular dystrophy: A bidirectional transcriptome and proteome-driven analysis. Front. Immunol. 2022, 13, 1017423. [Google Scholar] [CrossRef] [PubMed]
- Mournetas, V.; Massouridès, E.; Dupont, J.B.; Kornobis, E.; Polvèche, H.; Jarrige, M.; Dorval, A.R.L.; Gosselin, M.R.F.; Manousopoulou, A.; Garbis, S.D.; et al. Myogenesis modelled by human pluripotent stem cells: A multi-omic study of Duchenne myopathy early onset. J. Cachexia Sarcopenia Muscle 2021, 12, 209–232. [Google Scholar] [CrossRef]
- Wijekoon, N.; Gonawala, L.; Ratnayake, P.; Dissanayaka, P.; Gunarathne, I.; Amaratunga, D.; Liyanage, R.; Senanayaka, S.; Wijesekara, S.; Gunasekara, H.H.; et al. Integrated genomic, proteomic and cognitive assessment in Duchenne Muscular Dystrophy suggest astrocyte centric pathology. Heliyon 2023, 9, e18530. [Google Scholar] [CrossRef] [PubMed]
- Rajczewski, A.T.; Jagtap, P.D.; Griffin, T.J. An overview of technologies for MS-based proteomics-centric multi-omics. Expert Rev. Proteom. 2022, 19, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- Mann, M.; Kumar, C.; Zeng, W.F.; Strauss, M.T. Artificial intelligence for proteomics and biomarker discovery. Cell Syst. 2021, 12, 759–770. [Google Scholar] [CrossRef]
- Vera, C.D.; Zhang, A.; Pang, P.D.; Wu, J.C. Treating Duchenne Muscular Dystrophy: The Promise of Stem Cells, Artificial Intelligence, and Multi-Omics. Front Cardiovasc. Med. 2022, 9, 851491. [Google Scholar] [CrossRef]
- Torun, F.M.; Virreira Winter, S.; Doll, S.; Riese, F.M.; Vorobyev, A.; Mueller-Reif, J.B.; Geyer, P.E.; Strauss, M.T. Transparent Exploration of Machine Learning for Biomarker Discovery from Proteomics and Omics Data. J. Proteome Res. 2023, 22, 359–367. [Google Scholar] [CrossRef]
- Procopio, A.; Cesarelli, G.; Donisi, L.; Merola, A.; Amato, F.; Cosentino, C. Combined mechanistic modeling and machine-learning approaches in systems biology—A systematic literature review. Comput. Methods Programs Biomed. 2023, 240, 107681. [Google Scholar] [CrossRef]
- Fortunato, F.; Tonelli, L.; Farnè, M.; Selvatici, R.; Ferlini, A. DMD deletions underlining mild dystrophinopathies: Literature review highlights phenotype-related mutation clusters and provides insights about genetic mechanisms and prognosis. Front. Neurol. 2024, 14, 1288721. [Google Scholar] [CrossRef]
- Marques, M.J.; Ferretti, R.; Vomero, V.U.; Minatel, E.; Neto, H.S. Intrinsic laryngeal muscles are spared from myonecrosis in the mdx mouse model of Duchenne muscular dystrophy. Muscle Nerve 2007, 35, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Lorena, M.D.S.V.; Santos, E.K.D.; Ferretti, R.; Nagana Gowda, G.A.; Odom, G.L.; Chamberlain, J.S.; Matsumura, C.Y. Biomarkers for Duchenne muscular dystrophy progression: Impact of age in the mdx tongue spared muscle. Skelet. Muscle 2023, 13, 16. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dowling, P.; Trollet, C.; Negroni, E.; Swandulla, D.; Ohlendieck, K. How Can Proteomics Help to Elucidate the Pathophysiological Crosstalk in Muscular Dystrophy and Associated Multi-System Dysfunction? Proteomes 2024, 12, 4. https://doi.org/10.3390/proteomes12010004
Dowling P, Trollet C, Negroni E, Swandulla D, Ohlendieck K. How Can Proteomics Help to Elucidate the Pathophysiological Crosstalk in Muscular Dystrophy and Associated Multi-System Dysfunction? Proteomes. 2024; 12(1):4. https://doi.org/10.3390/proteomes12010004
Chicago/Turabian StyleDowling, Paul, Capucine Trollet, Elisa Negroni, Dieter Swandulla, and Kay Ohlendieck. 2024. "How Can Proteomics Help to Elucidate the Pathophysiological Crosstalk in Muscular Dystrophy and Associated Multi-System Dysfunction?" Proteomes 12, no. 1: 4. https://doi.org/10.3390/proteomes12010004
APA StyleDowling, P., Trollet, C., Negroni, E., Swandulla, D., & Ohlendieck, K. (2024). How Can Proteomics Help to Elucidate the Pathophysiological Crosstalk in Muscular Dystrophy and Associated Multi-System Dysfunction? Proteomes, 12(1), 4. https://doi.org/10.3390/proteomes12010004