

Advancements in Oncoproteomics Technologies: Treading toward Translation into Clinical Practice


Abstract
1. Introduction
2. Advances in Proteomic Technologies Used in the Study of Cancer
2.1. Gel-Based Approaches
2.1.1. Two-Dimensional Gel Electrophoresis
2.1.2. 2D Differential in-Gel Electrophoresis
2.2. Mass Spectrometry-Based Approaches
2.2.1. Liquid Chromatography–Mass Spectrometry
2.2.2. Matrix-Assisted Laser Desorption/Ionization
2.2.3. MALDI Mass Spectrometry Imaging
2.2.4. Surface-Enhanced Laser Desorption/Ionization Time-Of-Flight Mass Spectrometry
2.2.5. Targeted/Directed Mass Spectrometry
Single Reaction Monitoring and Parallel Reaction Monitoring-Mass Spectrometry
High-Pressure and High-Resolution Separations Coupled with Intelligent Selection and Multiplexing
Parallel Reaction Monitoring
Sequential Window Acquisition of All Theoretical Fragmentation Spectra
2.2.6. Quantitative Analysis Methods
Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)
Isotope-Coded Affinity Tag
Isobaric Tags for Relative and Absolute Quantification
Tandem Mass Tag
Dimethyl Labeling
Proteolytic 18O Labeling
Label-Free
2.3. Microarrays
2.3.1. Protein Microarray
2.3.2. Antibody/Antigen Microarrays
2.3.3. Tissue Microarrays
2.3.4. Protein Domain Microarray
2.3.5. Immunosensor Arrays
3. Contemporary Technologies and Approaches
3.1. Laser Capture Microdissection
3.2. Aptamer-Based Molecular Probes for Protein Signature of Cancer Cells
3.3. Extracellular Vesicle-Based Protein Blood Test
3.4. Proximity Extension Assay
3.5. Immuno-Affinity Capillary Electrophoresis
3.6. Cancer Immunomics to Identify Autoantibody Signatures
3.7. Protein Terminomics
3.8. Single-Cell Proteomics
3.9. Nanoproteomics
3.10. PTM Enrichment Methods
4. Role of Proteomics in the Prognosis and Diagnosis of Cancer
4.1. Hepatocellular Carcinoma
4.2. Colorectal Cancer
4.3. Leukemia
4.4. Prostate Cancer
4.5. Lung Cancer
4.6. Breast Cancer
5. Proteomics Contribution to the Clinical Treatment of Cancer
6. Role of Proteomics in Drug Discovery
7. Discussion and Perspective
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, L.M.; Kelleher, N.L. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Jungblut, P.R.; Holzhütter, H.G.; Apweiler, R.; Schlüter, H. The speciation of the proteome. Chem. Cent. J. 2008, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L. Proteoforms as the next proteomics currency. Science 2018, 359, 1106–1107. [Google Scholar] [CrossRef]
- Seydel, C. Diving deeper into the proteome. Nat. Methods 2022, 19, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Agar, J.N.; Chamot-Rooke, J.; Danis, P.O.; Ge, Y.; Loo, J.A.; Paša-Tolić, L.; Tsybin, Y.O.; Kelleher, N.L. The Human Proteoform Project: Defining the Human Proteome. Sci. Adv. 2021, 7, eabk0734. [Google Scholar] [CrossRef] [PubMed]
- Forgrave, L.M.; Wang, M.; Yang, D.; DeMarco, M.L. Proteoforms and their expanding role in laboratory medicine. Pract. Lab. Med. 2021, 28, e00260. [Google Scholar] [CrossRef]
- Cancer. Available online: http://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 25 September 2022).
- Cancer Today. Available online: http://gco.iarc.fr/today/home (accessed on 25 September 2022).
- Cancer Data and Statistics CDC. Available online: https://www.cdc.gov/cancer/dcpc/data/index.htm (accessed on 25 September 2022).
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef]
- Haymond, A.; Davis, J.B.; Espina, V. Proteomics for cancer drug design. Expert Rev. Proteom. 2019, 16, 647–664. [Google Scholar] [CrossRef]
- Enroth, S.; Berggrund, M.; Lycke, M.; Broberg, J.; Lundberg, M.; Assarsson, E.; Olovsson, M.; Stålberg, K.; Sundfeldt, K.; Gyllensten, U. High throughput proteomics identifies a high-accuracy 11 plasma protein biomarker signature for ovarian cancer. Commun. Biol. 2019, 2, 1–12. [Google Scholar] [CrossRef]
- Chen, F.; Chandrashekar, D.S.; Varambally, S.; Creighton, C.J. Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than 500 human cancers. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Hanash, S.; Taguchi, A. Application of Proteomics to Cancer Early Detection. Cancer J. 2011, 17, 423–428. [Google Scholar] [CrossRef]
- Kwon, Y.W.; Jo, H.-S.; Bae, S.; Seo, Y.; Song, P.; Song, M.; Yoon, J.H. Application of Proteomics in Cancer: Recent Trends and Approaches for Biomarkers Discovery. Front. Med. 2021, 8, 747333. [Google Scholar] [CrossRef]
- Shenoy, A.; Nataraj, N.B.; Perry, G.; Puch, F.L.; Nagel, R.; Marin, I.; Balint, N.; Bossel, N.; Pavlovsky, A.; Barshack, I.; et al. Proteomic patterns associated with response to breast cancer neoadjuvant treatment. Mol. Syst. Biol. 2020, 16, e9443. [Google Scholar] [CrossRef] [PubMed]
- Parolo, C.; Idili, A.; Ortega, G.; Csordas, A.; Hsu, A.; Arroyo-Currás, N.; Yang, Q.; Ferguson, B.S.; Wang, J.; Plaxco, K.W. Real-Time Monitoring of a Protein Biomarker. ACS Sens. 2020, 5, 1877–1881. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Ni, J.; Beretov, J.; Wasinger, V.C.; Hao, J.; Bucci, J.; Malouf, D.; Gillatt, D.; Graham, P.H.; Li, Y. Identification of protein biomarkers and signaling pathways associated with prostate cancer radioresistance using label-free LC-MS/MS proteomic approach. Sci. Rep. 2017, 7, 41834. [Google Scholar] [CrossRef]
- Shruthi, B.S.; Vinodhkumar, P. Selvamani Proteomics: A New Perspective for Cancer. Adv. Biomed. Res. 2016, 5, 67. [Google Scholar] [CrossRef]
- Nanjundan, M.; Byers, L.A.; Carey, M.S.; Siwak, D.R.; Raso, M.G.; Diao, L.; Wang, J.; Coombes, K.R.; Roth, J.A.; Mills, G.B.; et al. Proteomic Profiling Identifies Pathways Dysregulated in Non-Small Cell Lung Cancer and an Inverse Association of AMPK and Adhesion Pathways with Recurrence. J. Thorac. Oncol. 2010, 5, 1894–1904. [Google Scholar] [CrossRef]
- Magdeldin, S.; Enany, S.; Yoshida, Y.; Xu, B.; Zhang, Y.; Zureena, Z.; Lokamani, I.; Yaoita, E.; Yamamoto, T. Basics and recent advances of two dimensional-polyacrylamide gel electrophoresis. Clin. Proteom. 2014, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Garza, S.; Moini, M. Analysis of Complex Protein Mixtures with Improved Sequence Coverage Using (CE−MS/MS)n. Anal. Chem. 2006, 78, 7309–7316. [Google Scholar] [CrossRef]
- Angel, T.E.; Aryal, U.K.; Hengel, S.M.; Baker, E.S.; Kelly, R.T.; Robinson, E.W.; Smith, R.D. Mass spectrometry-based proteomics: Existing capabilities and future directions. Chem. Soc. Rev. 2012, 41, 3912–3928. [Google Scholar] [CrossRef]
- Padula, M.; Berry, I.; O′rourke, M.; Raymond, B.; Santos, J.; Djordjevic, S.P. A Comprehensive Guide for Performing Sample Preparation and Top-Down Protein Analysis. Proteomes 2017, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- DuPree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad and the Future of This Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Burian, A.; Lujber, L.; Gerlinger, I.; Jarai, T.; Orosz, E.; Turiak, L.; Acs, A.; Hegedus, Z.; Peter, A.K.; Tornoczki, T.; et al. Label-Free Semiquantitative Liquid Chromatography-Tandem Mass Spectrometry Proteomics Analysis of Laryngeal/Hypopharyngeal Squamous Cell Carcinoma on Formalin-Fixed, Paraffin-Embedded Tissue Samples—A Pilot Study. Pathol. Oncol. Res. 2020, 26, 2801–2807. [Google Scholar] [CrossRef] [PubMed]
- Negishi, A.; Ono, M.; Handa, Y.; Kato, H.; Yamashita, K.; Honda, K.; Shitashige, M.; Satow, R.; Sakuma, T.; Kuwabara, H.; et al. Large-scale quantitative clinical proteomics by label-free liquid chromatography and mass spectrometry. Cancer Sci. 2009, 100, 514–519. [Google Scholar] [CrossRef]
- Smit, N.P.M.; Ruhaak, L.R.; Romijn, F.P.H.T.M.; Pieterse, M.M.; van der Burgt, Y.E.M.; Cobbaert, C.M. The Time Has Come for Quantitative Protein Mass Spectrometry Tests That Target Unmet Clinical Needs. J. Am. Soc. Mass Spectrom. 2021, 32, 636–647. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.; Zhang, T.; Shu, L.; Roepstorff, P.; Yang, F. Quantitative Proteomics Using Isobaric Labeling: A Practical Guide. Genom. Proteom. Bioinform. 2021, 19, 689–706. [Google Scholar] [CrossRef]
- Zhu, Y.; Piehowski, P.; Kelly, R.T.; Qian, W.J. Nanoproteomics comes of age. Expert Rev. Proteom. 2018, 15, 865–871. [Google Scholar] [CrossRef]
- Mesri, M. Advances in Proteomic Technologies and Its Contribution to the Field of Cancer. Adv. Med. 2014, 2014, 1–25. [Google Scholar] [CrossRef]
- Macklin, A.; Khan, S.; Kislinger, T. Recent advances in mass spectrometry based clinical proteomics: Applications to cancer research. Clin. Proteom. 2020, 17, 17. [Google Scholar] [CrossRef]
- Xie, F.; Liu, T.; Qian, W.J.; Petyuk, V.; Smith, R.D. Liquid Chromatography-Mass Spectrometry-Based Quantitative Proteomics. J. Biol. Chem. 2011, 286, 25443–25449. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, L.; Iavarone, F.; Olianas, A.; Greco, V.; Desiderio, C.; Martelli, C.; Manconi, B.; Sanna, M.T.; Messana, I.; Castagnola, M.; et al. Enrichments of post-translational modifications in proteomic studies. J. Sep. Sci. 2019, 43, 313–336. [Google Scholar] [CrossRef]
- He, Y.; Mohamedali, A.; Huang, C.; Baker, M.S.; Nice, E.C. Oncoproteomics: Current status and future opportunities. Clin. Chim. Acta 2019, 495, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Jain, K. Oncoproteomics for Personalized Management of Cancer. Cancer Proteom. 2007, 81–99. [Google Scholar] [CrossRef]
- O’Farrell, P. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [CrossRef]
- Zhan, X.; Li, B.; Zhan, X.; Schlüter, H.; Jungblut, P.R.; Coorssen, J.R. Innovating the Concept and Practice of Two-Dimensional Gel Electrophoresis in the Analysis of Proteomes at the Proteoform Level. Proteomes 2019, 7, 36. [Google Scholar] [CrossRef]
- Revival of 2DE-LC/MS in Proteomics and Its Potential for Large-Scale Study of Human Proteoforms. Med One 2018, 3, e180008. [CrossRef]
- Zhan, X.; Yang, H.; Peng, F.; Li, J.; Mu, Y.; Long, Y.; Cheng, T.; Huang, Y.; Li, Z.; Lu, M.; et al. How many proteins can be identified in a 2DE gel spot within an analysis of a complex human cancer tissue proteome? Electrophoresis 2018, 39, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Yang, Y.; Li, N.; Cheng, T.; Wang, X.; Liu, J.; Li, X.; Desiderio, D.M.; Zhan, X. Prolactin Variants in Human Pituitaries and Pituitary Adenomas Identified with Two-Dimensional Gel Electrophoresis and Mass Spectrometry. Front. Endocrinol. 2018, 9, 468. [Google Scholar] [CrossRef] [PubMed]
- Issaq, H.J.; Veenstra, T.D. Two-dimensional polyacrylamide gel electrophoresis (2D-PAGE): Advances and perspectives. BioTechniques 2008, 44, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Hirohashi, S. Application of 2D-DIGE in Cancer Proteomics toward Personalized Medicine. Methods Mol. Biol. 2009, 577, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; Kim, K.-I.; Min, B.-H.; Lee, G.M. Differential Protein Expression in Human Articular Chondrocytes Expanded in Serum-Free Media of Different Medium Osmolalities by DIGE. J. Proteome Res. 2010, 9, 2480–2487. [Google Scholar] [CrossRef]
- Ma, Z.-Q.; Dasari, S.; Chambers, M.C.; Litton, M.D.; Sobecki, S.M.; Zimmerman, L.J.; Halvey, P.J.; Schilling, B.; Drake, P.M.; Gibson, B.W.; et al. IDPicker 2.0: Improved Protein Assembly with High Discrimination Peptide Identification Filtering. J. Proteome Res. 2009, 8, 3872–3881. [Google Scholar] [CrossRef]
- Ummanni, R.; Mundt, F.; Pospisil, H.; Venz, S.; Scharf, C.; Barett, C.; Fälth, M.; Köllermann, J.; Walther, R.; Schlomm, T.; et al. Identification of Clinically Relevant Protein Targets in Prostate Cancer with 2D-DIGE Coupled Mass Spectrometry and Systems Biology Network Platform. PLoS ONE 2011, 6, e16833. [Google Scholar] [CrossRef] [PubMed]
- Thiede, B.; Koehler, C.J.; Strozynski, M.; Treumann, A.; Stein, R.; Zimny-Arndt, U.; Schmid, M.; Jungblut, P.R. High resolution quantitative proteomics of HeLa cells protein species using stable isotope labeling with amino acids in cell culture (SILAC), two-dimensional gel electrophoresis (2DE) and nano-liquid chromatography coupled to an LTQ-Orbitrap mass spectrometer. Mol. Cell. Proteom. 2014, 13, 1900. [Google Scholar] [CrossRef][Green Version]
- Ciereszko, A.; Dietrich, M.A.; Słowińska, M.; Nynca, J.; Ciborowski, M.; Kaczmarek, M.M.; Myszczyński, K.; Kiśluk, J.; Majewska, A.; Michalska-Falkowska, A.; et al. Application of two-dimensional difference gel electrophoresis to identify protein changes between center, margin, and adjacent non-tumor tissues obtained from non-small-cell lung cancer with adenocarcinoma or squamous cell carcinoma subtype. PLoS ONE 2022, 17, e0268073. [Google Scholar] [CrossRef] [PubMed]
- Kiseleva, O.; Zgoda, V.; Naryzhny, S.; Poverennaya, E.; Ponomarenko, E. Empowering Shotgun Mass Spectrometry with 2DE: A HepG2 Study. Int. J. Mol. Sci. 2020, 21, 3813. [Google Scholar] [CrossRef]
- Hariharan, D.; Weeks, M.E.; Crnogorac-Jurcevic, T. Application of Proteomics in Cancer Gene Profiling: Two-Dimensional Difference in Gel Electrophoresis (2D-DIGE). Methods Mol. Biol. 2009, 576, 197–211. [Google Scholar] [CrossRef]
- Ura, B.; Biffi, S.; Monasta, L.; Arrigoni, G.; Battisti, I.; Di Lorenzo, G.; Romano, F.; Aloisio, M.; Celsi, F.; Addobbati, R.; et al. Two Dimensional-Difference in Gel Electrophoresis (2D-DIGE) Proteomic Approach for the Identification of Biomarkers in Endometrial Cancer Serum. Cancers 2021, 13, 3639. [Google Scholar] [CrossRef]
- Kondo, T. Cancer biomarker development and two-dimensional difference gel electrophoresis (2D-DIGE). Biochim. Biophys. Acta Proteins Proteom. 2018, 1867, 2–8. [Google Scholar] [CrossRef]
- Kaufmann, A. High-resolution mass spectrometry for bioanalytical applications: Is this the new gold standard? J. Mass Spectrom. 2020, 55, e4533. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Wang, Y. Advances in high-resolution mass spectrometry techniques for analysis of high mass-to-charge ions. Mass Spectrom. Rev. 2022, e21790. [Google Scholar] [CrossRef]
- Rubakhin, S.S.; Sweedler, J.V. A Mass Spectrometry Primer for Mass Spectrometry Imaging. Methods Mol. Biol. 2010, 656, 21–49. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef]
- Borrebaeck, C.A.K. Precision diagnostics: Moving towards protein biomarker signatures of clinical utility in cancer. Nat. Rev. Cancer 2017, 17, 199–204. [Google Scholar] [CrossRef]
- Ding, Z.; Wang, N.; Ji, N.; Chen, Z.-S. Proteomics technologies for cancer liquid biopsies. Mol. Cancer 2022, 21, 53. [Google Scholar] [CrossRef]
- Allen, D.R.; McWhinney, B.C. Quadrupole Time-of-Flight Mass Spectrometry: A Paradigm Shift in Toxicology Screening Applications. Clin. Biochem. Rev. 2019, 40, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, J.R.; Yoon, O.K.; Zuleta, I.A.; Trapp, O.; Zare, R.N. Peak Height Precision in Hadamard Transform Time-of-Flight Mass Spectra. J. Am. Soc. Mass Spectrom. 2005, 16, 1117–1130. [Google Scholar] [CrossRef][Green Version]
- Pittenauer, E.; Allmaier, G. High-Energy Collision Induced Dissociation of Biomolecules: MALDITOF/RTOF Mass Spectrometry in Comparison to Tandem Sector Mass Spectrometry. Comb. Chem. High Throughput Screen. 2009, 12, 137–155. [Google Scholar] [CrossRef]
- Medzihradszky, K.F.; Campbell, J.M.; Baldwin, M.A.; Falick, A.M.; Juhasz, P.; Vestal, M.L.; Burlingame, A.L. The Characteristics of Peptide Collision-Induced Dissociation Using a High-Performance MALDI-TOF/TOF Tandem Mass Spectrometer. Anal. Chem. 1999, 72, 552–558. [Google Scholar] [CrossRef]
- Mbasu, R.J.; Heaney, L.M.; Molloy, B.J.; Hughes, C.J.; Ng, L.L.; Vissers, J.P.C.; Langridge, J.I.; Jones, D.J.L. Advances in quadrupole and time-of-flight mass spectrometry for peptide MRM based translational research analysis. Proteomics 2016, 16, 2206–2220. [Google Scholar] [CrossRef]
- Ranasinghe, A.; Ramanathan, R.; Jemal, M.; D’Arienzo, C.J.; Humphreys, W.G.; Olah, T.V. Integrated quantitative and qualitative workflow for in vivo bioanalytical support in drug discovery using hybrid Q-TOF-MS. Bioanalysis 2012, 4, 511–528. [Google Scholar] [CrossRef]
- Zambonin, C.; Aresta, A. MALDI-TOF/MS Analysis of Non-Invasive Human Urine and Saliva Samples for the Identification of New Cancer Biomarkers. Molecules 2022, 27, 1925. [Google Scholar] [CrossRef]
- Lv, P.; Liu, Z.; Xu, B.; Tang, C.; Li, X.; Qin, H.; Yang, S.; Gao, H.; He, K.; Liu, X. Exploratory study on application of MALDI-TOF-MS to detect serum and urine peptides related to small cell lung carcinoma. Mol. Med. Rep. 2019, 21, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-M.; Kim, M.-J.; Noh, J.-Y.; Yun, T.G.; Kang, M.-J.; Lee, S.-G.; Yoo, B.C.; Pyun, J.-C. Simultaneous Analysis of Multiple Cancer Biomarkers Using MALDI-TOF Mass Spectrometry Based on a Parylene-Matrix Chip. J. Am. Soc. Mass Spectrom. 2020, 31, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Timms, J.F.; Menon, U.; Devetyarov, D.; Tiss, A.; Camuzeaux, S.; McCurrie, K.; Nouretdinov, I.; Burford, B.; Smith, C.; Gentry-Maharaj, A.; et al. Early detection of ovarian cancer in samples pre-diagnosis using CA125 and MALDI-MS peaks. Cancer Genom. Proteom. 2011, 8, 289–305. [Google Scholar]
- Pais, R.J.; Zmuidinaite, R.; Lacey, J.C.; Jardine, C.S.; Iles, R.K. A Rapid and Affordable Screening Tool for Early-Stage Ovarian Cancer Detection Based on MALDI-ToF MS of Blood Serum. Appl. Sci. 2022, 12, 3030. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoo, B.C.; Kim, Y.H.; Ahn, S.-A.; Yeo, S.-G.; Cho, J.Y.; Kim, K.-H.; Kim, S.C. Low-mass-ion discriminant equation (LOME) for ovarian cancer screening. BioData Min. 2016, 9, 1–14. [Google Scholar] [CrossRef]
- Sun, J.; Yu, G.; Yang, Y.; Qiao, L.; Xu, B.; Ding, C.; Liu, Y.; Yu, S. Evaluation of prostate cancer based on MALDI-TOF MS fingerprinting of nanoparticle-treated serum proteins/peptides. Talanta 2020, 220, 121331. [Google Scholar] [CrossRef]
- Periyasamy, A.; Gopisetty, G.; Veluswami, S.; Subramanium, M.J.; Thangarajan, R. Identification of candidate biomarker mass (m/z) ranges in serous ovarian adenocarcinoma using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry profiling. Biomarkers 2015, 20, 292–298. [Google Scholar] [CrossRef]
- Swiatly, A.; Horala, A.; Matysiak, J.; Hajduk, J.; Nowak-Markwitz, E.; Kokot, Z.J. Understanding Ovarian Cancer: iTRAQ-Based Proteomics for Biomarker Discovery. Int. J. Mol. Sci. 2018, 19, 2240. [Google Scholar] [CrossRef]
- Buszewska-Forajta, M.; Pomastowski, P.; Monedeiro, F.; Król-Górniak, A.; Adamczyk, P.; Markuszewski, M.; Buszewski, B. New approach in determination of urinary diagnostic markers for prostate cancer by MALDI-TOF/MS. Talanta 2021, 236, 122843. [Google Scholar] [CrossRef]
- Park, H.-G.; Jang, K.-S.; Park, H.-M.; Song, W.-S.; Jeong, Y.-Y.; Ahn, D.-H.; Kim, S.-M.; Yang, Y.-H.; Kim, Y.-G. MALDI-TOF MS-based total serum protein fingerprinting for liver cancer diagnosis. Analyst 2019, 144, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Long, S.; Qin, Q.; Wang, Y.; Yang, Y.; Wang, Y.; Deng, A.; Qiao, L.; Liu, B. Nanoporous silica coupled MALDI-TOF MS detection of Bence-Jones proteins in human urine for diagnosis of multiple myeloma. Talanta 2019, 200, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Yi, J.; Yang, Y.; Li, D.; Peng, C.; Long, S.; Peng, X.; Shen, Y.; Liu, B.; Qiao, L. SERS and MALDI-TOF MS based plasma exosome profiling for rapid detection of osteosarcoma. Analyst 2021, 146, 6496–6505. [Google Scholar] [CrossRef]
- Li, D.; Yi, J.; Han, G.; Qiao, L. MALDI-TOF Mass Spectrometry in Clinical Analysis and Research. ACS Meas. Sci. Au 2022, 2, 385–404. [Google Scholar] [CrossRef]
- Samarah, L.Z.; Vertes, A. Mass Spectrometry Imaging of Biological Tissues by Laser Desorption Ionization from Silicon Nanopost Arrays. Methods Mol. Biol. 2021, 2437, 89–98. [Google Scholar] [CrossRef]
- Robichaud, G.; Garrard, K.P.; Barry, J.A.; Muddiman, D.C. MSiReader: An Open-Source Interface to View and Analyze High Resolving Power MS Imaging Files on Matlab Platform. J. Am. Soc. Mass Spectrom. 2013, 24, 718–721. [Google Scholar] [CrossRef]
- Parry, R.M.; Galhena, A.S.; Gamage, C.M.; Bennett, R.V.; Wang, M.D.; Fernández, F.M. OmniSpect: An Open MATLAB-Based Tool for Visualization and Analysis of Matrix-Assisted Laser Desorption/Ionization and Desorption Electrospray Ionization Mass Spectrometry Images. J. Am. Soc. Mass Spectrom. 2013, 24, 646–649. [Google Scholar] [CrossRef]
- Bemis, K.D.; Harry, A.; Eberlin, L.S.; Ferreira, C.; van de Ven, S.M.; Mallick, P.; Stolowitz, M.; Vitek, O. Cardinal: An R package for statistical analysis of mass spectrometry-based imaging experiments. Bioinformatics 2015, 31, 2418–2420. [Google Scholar] [CrossRef]
- De Raad, M.; de Rond, T.; Rübel, O.; Keasling, J.D.; Northen, T.R.; Bowen, B.P. OpenMSI Arrayed Analysis Toolkit: Analyzing Spatially Defined Samples Using Mass Spectrometry Imaging. Anal. Chem. 2017, 89, 5818–5823. [Google Scholar] [CrossRef]
- Rübel, O.; Greiner, A.; Cholia, S.; Louie, K.; Bethel, E.W.; Northen, T.R.; Bowen, B.P. OpenMSI: A High-Performance Web-Based Platform for Mass Spectrometry Imaging. Anal. Chem. 2013, 85, 10354–10361. [Google Scholar] [CrossRef] [PubMed]
- Paschke, C.; Leisner, A.; Hester, A.; Maass, K.; Guenther, S.; Bouschen, W.; Spengler, B. Mirion—A Software Package for Automatic Processing of Mass Spectrometric Images. J. Am. Soc. Mass Spectrom. 2013, 24, 1296–1306. [Google Scholar] [CrossRef] [PubMed]
- Veselkov, K.; Sleeman, J.; Claude, E.; Vissers, J.P.C.; Galea, D.; Mroz, A.; Laponogov, I.; Towers, M.; Tonge, R.; Mirnezami, R.; et al. BASIS: High-performance bioinformatics platform for processing of large-scale mass spectrometry imaging data in chemically augmented histology. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Aichler, M.; Walch, A. MALDI Imaging mass spectrometry: Current frontiers and perspectives in pathology research and practice. Lab. Investig. 2015, 95, 422–431. [Google Scholar] [CrossRef]
- Gonçalves, J.P.L.; Bollwein, C.; Schwamborn, K. Mass Spectrometry Imaging Spatial Tissue Analysis toward Personalized Medicine. Life 2022, 12, 1037. [Google Scholar] [CrossRef]
- Berghmans, E.; Boonen, K.; Maes, E.; Mertens, I.; Pauwels, P.; Baggerman, G. Implementation of MALDI Mass Spectrometry Imaging in Cancer Proteomics Research: Applications and Challenges. J. Pers. Med. 2020, 10, 54. [Google Scholar] [CrossRef]
- Mirnezami, R.; Spagou, K.; Vorkas, P.; Lewis, M.; Kinross, J.; Want, E.; Shion, H.; Goldin, R.; Darzi, A.; Takats, Z.; et al. Chemical mapping of the colorectal cancer microenvironment via MALDI imaging mass spectrometry (MALDI-MSI) reveals novel cancer-associated field effects. Mol. Oncol. 2013, 8, 39–49. [Google Scholar] [CrossRef]
- Gonçalves, J.P.L.; Bollwein, C.; Schlitter, A.M.; Kriegsmann, M.; Jacob, A.; Weichert, W.; Schwamborn, K. MALDI-MSI: A Powerful Approach to Understand Primary Pancreatic Ductal Adenocarcinoma and Metastases. Molecules 2022, 27, 4811. [Google Scholar] [CrossRef]
- Smith, A.; Galli, M.; Piga, I.; Denti, V.; Stella, M.; Chinello, C.; Fusco, N.; Leni, D.; Manzoni, M.; Roversi, G.; et al. Molecular signatures of medullary thyroid carcinoma by matrix-assisted laser desorption/ionisation mass spectrometry imaging. J. Proteom. 2019, 191, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Boyle, S.T.; Mittal, P.; Kaur, G.; Hoffmann, P.; Samuel, M.S.; Klingler-Hoffmann, M. Uncovering Tumor–Stroma Inter-Relationships Using MALDI Mass Spectrometry Imaging. J. Proteome Res. 2020, 19, 4093–4103. [Google Scholar] [CrossRef]
- Balluff, B.; Frese, C.K.; Maier, S.K.; Schöne, C.; Kuster, B.; Schmitt, M.; Aubele, M.; Höfler, H.; Deelder, A.M.; Heck, A.J.; et al. De novo discovery of phenotypic intratumour heterogeneity using imaging mass spectrometry. J. Pathol. 2014, 235, 3–13. [Google Scholar] [CrossRef]
- Capitoli, G.; Piga, I.; Galimberti, S.; Leni, D.; Pincelli, A.I.; Garancini, M.; Clerici, F.; Mahajneh, A.; Brambilla, V.; Smith, A.; et al. MALDI-MSI as a Complementary Diagnostic Tool in Cytopathology: A Pilot Study for the Characterization of Thyroid Nodules. Cancers 2019, 11, 1377. [Google Scholar] [CrossRef] [PubMed]
- Pertzborn, D.; Arolt, C.; Ernst, G.; Lechtenfeld, O.J.; Kaesler, J.; Pelzel, D.; Guntinas-Lichius, O.; von Eggeling, F.; Hoffmann, F. Multi-Class Cancer Subtyping in Salivary Gland Carcinomas with MALDI Imaging and Deep Learning. Cancers 2022, 14, 4342. [Google Scholar] [CrossRef]
- Föll, M.C.; Volkmann, V.; Enderle-Ammour, K.; Timme, S.; Wilhelm, K.; Guo, D.; Vitek, O.; Bronsert, P.; Schilling, O. Moving translational mass spectrometry imaging towards transparent and reproducible data analyses: A case study of an urothelial cancer cohort analyzed in the Galaxy framework. Clin. Proteom. 2022, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hutchens, T.W.; Yip, T.-T. New desorption strategies for the mass spectrometric analysis of macromolecules. Rapid Commun. Mass Spectrom. 1993, 7, 576–580. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Xuan, C.; Zhang, B.-B.; Liao, M.; Deng, K.-F.; He, M.; Zhao, J.-M. SELDI-TOF MS profiling of serum for detection of nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2009, 28, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, M.; Jennek, S.; Ludwig, S.; Klitzsch, A.; Kraft, F.; Melle, C.; Baniahmad, A. The tumor suppressor ING1b is a novel corepressor for the androgen receptor and induces cellular senescence in prostate cancer cells. J. Mol. Cell Biol. 2016, 8, 207–220. [Google Scholar] [CrossRef]
- Xue, A.; Gandy, R.C.; Chung, L.; Baxter, R.C.; Smith, R.C. Discovery of diagnostic biomarkers for pancreatic cancer in immunodepleted serum by SELDI-TOF MS. Pancreatology 2012, 12, 124–129. [Google Scholar] [CrossRef]
- Simsek, C.; Sonmez, O.; Yurdakul, A.S.; Ozmen, F.; Zengin, N.; Keyf, A.I.; Kubilay, D.; Gulbahar, O.; Karataylı, S.C.; Bozdayı, M.; et al. Importance of Serum SELDI-TOF-MS Analysis in the Diagnosis of Early Lung Cancer. Asian Pac. J. Cancer Prev. 2013, 14, 2037–2042. [Google Scholar] [CrossRef][Green Version]
- Zeidan, B.A.; Townsend, P.A. SELDI-TOF proteomic profiling of breast carcinomas identifies clinicopathologically relevant groups of patients similar to previously defined clusters from cDNA expression. Breast Cancer Res. 2008, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Solassol, J.; Du-Thanh, A.; Maudelonde, T.; Guillot, B. Serum Proteomic Profiling Reveals Potential Biomarkers for Cutaneous Malignant Melanoma. Int. J. Biol. Markers 2011, 26, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Gemoll, T.; Roblick, U.J.; Auer, G.; Jörnvall, H.; Habermann, J.K. SELDI-TOF serum proteomics and colorectal cancer: A current overview. Arch. Physiol. Biochem. 2010, 116, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Pillai, J.; Chincholkar, T.; Dixit, R.; Pandey, M. A systematic review of proteomic biomarkers in oral squamous cell cancer. World J. Surg. Oncol. 2021, 19, 315. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Luo, Z.; Tang, D.; Liu, L.; Yao, D.; Zhu, L.; Wang, Z. Identification of carboxyl terminal peptide of Fibrinogen as a potential serum biomarker for gastric cancer. Tumor Biol. 2015, 37, 6963–6970. [Google Scholar] [CrossRef] [PubMed]
- Husi, H.; Fernandes, M.; Skipworth, R.J.; Miller, J.; Cronshaw, A.D.; Fearon, K.C.H.; Ross, J.A. Identification of diagnostic upper gastrointestinal cancer tissue type-specific urinary biomarkers. Biomed. Rep. 2019, 10, 165–174. [Google Scholar] [CrossRef]
- Wu, J.; Ji, Y.; Zhao, L.; Ji, M.; Ye, Z.; Li, S. A Mass Spectrometric Analysis Method Based on PPCA and SVM for Early Detection of Ovarian Cancer. Comput. Math. Methods Med. 2016, 2016, 6169249. [Google Scholar] [CrossRef]
- Mu, A.K.-W.; Lim, B.-K.; Aminudin, N.; Hashim, O.H.; Shuib, A.S. Application of SELDI-TOF in N-glycopeptides profiling of the urine from patients with endometrial, ovarian and cervical cancer. Arch. Physiol. Biochem. 2016, 122, 111–116. [Google Scholar] [CrossRef]
- Schlichtemeier, S.M.; Nahm, C.B.; Xue, A.; Gill, A.J.; Smith, R.C.; Hugh, T.J. SELDI-TOF MS Analysis of Hepatocellular Carcinoma in an Australian Cohort. J. Surg. Res. 2019, 238, 127–136. [Google Scholar] [CrossRef]
- Nuerrula, Y.; Rexiati, M.; Liu, Q.; Wang, Y.-J. Differential expression and clinical significance of serum protein among patients with clear-cell renal cell carcinoma. Cancer Biomark. 2015, 15, 485–491. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, L. Anaphylatoxin C3a: A potential biomarker for esophageal cancer diagnosis. Mol. Clin. Oncol. 2017, 8, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P. Proteomic Protocol in Esophageal Adenocarcinoma. Breast Cancer 2018, 1756, 281–293. [Google Scholar] [CrossRef]
- Kumar, V.; Ray, S.; Ghantasala, S.; Srivastava, S. An Integrated Quantitative Proteomics Workflow for Cancer Biomarker Discovery and Validation in Plasma. Front. Oncol. 2020, 10, 543997. [Google Scholar] [CrossRef] [PubMed]
- Faria, S.S.; Morris, C.F.M.; Silva, A.R.; Fonseca, M.P.; Forget, P.; Castro, M.S.; Fontes, W. A Timely Shift from Shotgun to Targeted Proteomics and How It Can Be Groundbreaking for Cancer Research. Front. Oncol. 2017, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Whiteaker, J.R.; Zhao, L.; Abbatiello, S.E.; Burgess, M.; Kuhn, E.; Lin, C.; Pope, M.E.; Razavi, M.; Anderson, N.L.; Pearson, T.W.; et al. Evaluation of Large Scale Quantitative Proteomic Assay Development Using Peptide Affinity-Based Mass Spectrometry. Mol. Cell. Proteom. 2011, 10, M110.005645. [Google Scholar] [CrossRef]
- Kontostathi, G.; Makridakis, M.; Bitsika, V.; Tsolakos, N.; Vlahou, A.; Zoidakis, J. Development and Validation of Multiple Reaction Monitoring (MRM) Assays for Clinical Applications. Methods Mol. Biol. 2019, 1959, 205–223. [Google Scholar] [CrossRef]
- Zhao, Y.; Brasier, A.R. Applications of selected reaction monitoring (SRM)-mass spectrometry (MS) for quantitative measurement of signaling pathways. Methods 2013, 61, 313–322. [Google Scholar] [CrossRef][Green Version]
- Colangelo, C.M.; Chung, L.; Bruce, C.; Cheung, K.-H. Review of software tools for design and analysis of large scale MRM proteomic datasets. Methods 2013, 61, 287–298. [Google Scholar] [CrossRef]
- Sherwood, C.A.; Eastham, A.; Lee, L.W.; Risler, J.; Mirzaei, H.; Falkner, J.A.; Martin, D.B. Rapid Optimization of MRM-MS Instrument Parameters by Subtle Alteration of Precursor and Product m/z Targets. J. Proteome Res. 2009, 8, 3746–3751. [Google Scholar] [CrossRef]
- Yang, J.J.; Han, Y.; Mah, C.H.; Wanjaya, E.; Peng, B.; Xu, T.F.; Liu, M.; Huan, T.; Fang, M.L. Streamlined MRM method transfer between instruments assisted with HRMS matching and retention-time prediction. Anal. Chim. Acta 2019, 1100, 88–96. [Google Scholar] [CrossRef]
- Fukuda, T.; Nomura, M.; Kato, Y.; Tojo, H.; Fujii, K.; Nagao, T.; Bando, Y.; Fehniger, T.E.; Marko-Varga, G.; Nakamura, H.; et al. A selected reaction monitoring mass spectrometric assessment of biomarker candidates diagnosing large-cell neuroendocrine lung carcinoma by the scaling method using endogenous references. PLoS ONE 2017, 12, e0176219. [Google Scholar] [CrossRef] [PubMed]
- Hüttenhain, R.; Choi, M.; de la Fuente, L.M.; Oehl, K.; Chang, C.-Y.; Zimmermann, A.-K.; Malander, S.; Olsson, H.; Surinova, S.; Clough, T.; et al. A Targeted Mass Spectrometry Strategy for Developing Proteomic Biomarkers: A Case Study of Epithelial Ovarian Cancer. Mol. Cell. Proteom. 2019, 18, 1836–1850. [Google Scholar] [CrossRef] [PubMed]
- Duriez, E.; Masselon, C.D.; Mesmin, C.; Court, M.; Demeure, K.; Allory, Y.; Malats, N.; Matondo, M.; Radvanyi, F.; Garin, J.; et al. Large-Scale SRM Screen of Urothelial Bladder Cancer Candidate Biomarkers in Urine. J. Proteome Res. 2017, 16, 1617–1631. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Fillmore, T.L.; Sun, X.; Zhao, R.; Schepmoes, A.A.; Hossain, M.; Xie, F.; Wu, S.; Kim, J.-S.; Jones, N.; et al. Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc. Natl. Acad. Sci. USA 2012, 109, 15395–15400. [Google Scholar] [CrossRef]
- Shi, T.; Song, E.; Nie, S.; Rodland, K.D.; Liu, T.; Qian, W.-J.; Smith, R.D. Advances in targeted proteomics and applications to biomedical research. Proteomics 2016, 16, 2160–2182. [Google Scholar] [CrossRef]
- Shi, T.; Sun, X.; Gao, Y.; Fillmore, T.L.; Schepmoes, A.A.; Zhao, R.; He, J.; Moore, R.J.; Kagan, J.; Rodland, K.D.; et al. Targeted Quantification of Low ng/mL Level Proteins in Human Serum without Immunoaffinity Depletion. J. Proteome Res. 2013, 12, 3353–3361. [Google Scholar] [CrossRef]
- Khoo, A.; Liu, L.Y.; Nyalwidhe, J.O.; Semmes, O.J.; Vesprini, D.; Downes, M.R.; Boutros, P.C.; Liu, S.K.; Kislinger, T. Proteomic discovery of non-invasive biomarkers of localized prostate cancer using mass spectrometry. Nat. Rev. Urol. 2021, 18, 707–724. [Google Scholar] [CrossRef]
- Van Bentum, M.; Selbach, M. An Introduction to Advanced Targeted Acquisition Methods. Mol. Cell. Proteom. 2021, 20, 100165. [Google Scholar] [CrossRef]
- Park, J.; Oh, H.J.; Han, D.; Wang, J.I.; Park, I.A.; Ryu, H.S.; Kim, Y. Parallel Reaction Monitoring-Mass Spectrometry (PRM-MS)-Based Targeted Proteomic Surrogates for Intrinsic Subtypes in Breast Cancer: Comparative Analysis with Immunohistochemical Phenotypes. J. Proteome Res. 2019, 19, 2643–2653. [Google Scholar] [CrossRef]
- Doerr, A. Targeting with PRM. Nat. Chem. Biol. 2012, 9, 950. [Google Scholar] [CrossRef]
- Sherman, J.; McKay, M.J.; Ashman, K.; Molloy, M.P. How specific is my SRM?: The issue of precursor and product ion redundancy. Proteomics 2009, 9, 1120–1123. [Google Scholar] [CrossRef] [PubMed]
- Gallien, S.; Kim, S.Y.; Domon, B. Large-Scale Targeted Proteomics Using Internal Standard Triggered-Parallel Reaction Monitoring (IS-PRM) *. Mol. Cell. Proteom. 2015, 14, 1630–1644. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.C.; Russell, J.D.; Bailey, D.J.; Westphall, M.S.; Coon, J.J. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol. Cell. Proteom. 2012, 11, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-Independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef]
- Messner, C.B.; Demichev, V.; Nic Bloomfield, N.; Yu, J.S.L.; White, M.; Kreidl, M.; Egger, A.-S.; Freiwald, A.; Ivosev, G.; Wasim, F.; et al. Ultra-fast proteomics with Scanning SWATH. Nat. Biotechnol. 2021, 39, 846–854. [Google Scholar] [CrossRef]
- Krasny, L.; Bland, P.; Kogata, N.; Wai, P.; Howard, B.A.; Natrajan, R.C.; Huang, P.H. SWATH mass spectrometry as a tool for quantitative profiling of the matrisome. J. Proteom. 2018, 189, 11–22. [Google Scholar] [CrossRef]
- Thomas, S.N.; Friedrich, B.; Schnaubelt, M.; Chan, D.W.; Zhang, H.; Aebersold, R. Orthogonal Proteomic Platforms and Their Implications for the Stable Classification of High-Grade Serous Ovarian Cancer Subtypes. Iscience 2020, 23, 101079. [Google Scholar] [CrossRef]
- Yan, Z.; Zhou, Y.; Shan, Y.; Wu, Q.; Zhang, S.; Liang, Z.; Zhang, L.; Zhang, Y. Label-free quantification of differentially expressed proteins in mouse liver cancer cells with high and low metastasis rates by a SWATH acquisition method. Sci. China Chem. 2014, 57, 718–722. [Google Scholar] [CrossRef]
- Bouchal, P.; Schubert, O.T.; Faktor, J.; Capkova, L.; Imrichova, H.; Zoufalova, K.; Paralova, V.; Hrstka, R.; Liu, Y.; Ebhardt, H.A.; et al. Breast Cancer Classification Based on Proteotypes Obtained by SWATH Mass Spectrometry. Cell Rep. 2019, 28, 832–843. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, X.; Sang, Z.; Li, Z.; Liu, F.; Mao, J.; Yan, D.; Zhao, Y.; Wang, H.; Li, P.; et al. Quantitative proteomics by SWATH-MS reveals sophisticated metabolic reprogramming in hepatocellular carcinoma tissues. Sci. Rep. 2017, 7, 45913. [Google Scholar] [CrossRef] [PubMed]
- González-Fernández, M.J.; Fabrikov, D.; Ramos-Bueno, R.P.; Guil-Guerrero, J.L.; Ortea, I. SWATH Differential Abundance Proteomics and Cellular Assays Show In Vitro Anticancer Activity of Arachidonic Acid- and Docosahexaenoic Acid-Based Monoacylglycerols in HT-29 Colorectal Cancer Cells. Nutrients 2019, 11, 2984. [Google Scholar] [CrossRef] [PubMed]
- Eagle, G.L.; Herbert, J.M.J.; Zhuang, J.; Oates, M.; Khan, U.T.; Kitteringham, N.R.; Clarke, K.; Park, B.K.; Pettitt, A.R.; Jenkins, R.E.; et al. Assessing technical and biological variation in SWATH-MS-based proteomic analysis of chronic lymphocytic leukaemia cells. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Singh, A.N.; Sharma, N. Quantitative SWATH-Based Proteomic Profiling for Identification of Mechanism-Driven Diagnostic Biomarkers Conferring in the Progression of Metastatic Prostate Cancer. Front. Oncol. 2020, 10, 493. [Google Scholar] [CrossRef]
- Jiang, H.; English, A.M. Quantitative Analysis of the Yeast Proteome by Incorporation of Isotopically Labeled Leucine. J. Proteome Res. 2002, 1, 345–350. [Google Scholar] [CrossRef]
- Zhu, H.; Pan, S.; Gu, S.; Bradbury, E.M.; Chen, X. Amino acid residue specific stable isotope labeling for quantitative proteomics. Rapid Commun. Mass Spectrom. 2002, 16, 2115–2123. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Krüger, M.; Moser, M.; Ussar, S.; Thievessen, I.; Luber, C.A.; Forner, F.; Schmidt, S.; Zanivan, S.; Fässler, R.; Mann, M. SILAC Mouse for Quantitative Proteomics Uncovers Kindlin-3 as an Essential Factor for Red Blood Cell Function. Cell 2008, 134, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Konzer, A.; Ruhs, A.; Braun, T.; Krüger, M. Global Protein Quantification of Mouse Heart Tissue Based on the SILAC Mouse. Methods Mol. Biol. 2013, 1005, 39–52. [Google Scholar] [CrossRef]
- Westman-Brinkmalm, A.; Abramsson, A.; Pannee, J.; Gang, C.; Gustavsson, M.K.; von Otter, M.; Blennow, K.; Brinkmalm, G.; Heumann, H.; Zetterberg, H. SILAC zebrafish for quantitative analysis of protein turnover and tissue regeneration. J. Proteom. 2011, 75, 425–434. [Google Scholar] [CrossRef]
- Nolte, H.; Hölper, S.; Housley, M.P.; Islam, S.; Piller, T.; Konzer, A.; Stainier, D.Y.R.; Braun, T.; Krüger, M. Dynamics of zebrafish fin regeneration using a pulsed SILAC approach. Proteomics 2014, 15, 739–751. [Google Scholar] [CrossRef]
- Looso, M.; Borchardt, T.; Krüger, M.; Braun, T. Advanced Identification of Proteins in Uncharacterized Proteomes by Pulsed In Vivo Stable Isotope Labeling-Based Mass Spectrometry. Mol. Cell. Proteom. 2010, 9, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Fredens, J.; Engholm-Keller, K.; Giessing, A.; Pultz, D.; Larsen, M.R.; Højrup, P.; Møller-Jensen, J.; Færgeman, N.J. Quantitative proteomics by amino acid labeling in C. elegans. Nat. Methods 2011, 8, 845–847. [Google Scholar] [CrossRef] [PubMed]
- Larance, M.; Bailly, A.P.; Pourkarimi, E.; Hay, R.T.; Buchanan, G.; Coulthurst, S.; Xirodimas, D.P.; Gartner, A.; Lamond, A.I. Stable-isotope labeling with amino acids in nematodes. Nat. Methods 2011, 8, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Macek, B.; Carpy, A.; Koch, A.; Bicho, C.C.; Borek, W.E.; Hauf, S.; Sawin, K.E. Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) Technology in Fission Yeast. Cold Spring Harb. Protoc. 2017, 2017, top079814. [Google Scholar] [CrossRef][Green Version]
- Geiger, T.; Cox, J.; Ostasiewicz, P.; Wisniewski, J.R.; Mann, M. Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 2010, 7, 383–385. [Google Scholar] [CrossRef]
- Beller, N.C.; Hummon, A.B. Advances in stable isotope labeling: Dynamic labeling for spatial and temporal proteomic analysis. Mol. Omics 2022, 18, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, T.W.; Rose, C.M.; Bailey, D.J.; Riley, N.M.; Molden, R.C.; Nestler, A.J.; Merrill, A.E.; Smith, L.M.; Hebert, A.S.; Westphall, M.S.; et al. Neutron-Encoded Mass Signatures for Quantitative Top-Down Proteomics. Anal. Chem. 2014, 86, 2314–2319. [Google Scholar] [CrossRef]
- Hebert, A.S.; Merrill, A.; Bailey, D.J.; Still, A.J.; Westphall, M.S.; Strieter, E.R.; Pagliarini, D.J.; Coon, J.J. Neutron-encoded mass signatures for multiplexed proteome quantification. Nat. Methods 2013, 10, 332–334. [Google Scholar] [CrossRef]
- Monetti, M.; Nagaraj, N.; Sharma, K.; Mann, M. Large-scale phosphosite quantification in tissues by a spike-in SILAC method. Nat. Methods 2011, 8, 655–658. [Google Scholar] [CrossRef]
- Zanivan, S.; Maione, F.; Hein, M.Y.; Hernández-Fernaud, J.R.; Ostasiewicz, P.; Giraudo, E.; Mann, M. SILAC-Based Proteomics of Human Primary Endothelial Cell Morphogenesis Unveils Tumor Angiogenic Markers. Mol. Cell. Proteom. 2013, 12, 3599–3611. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, Y.; Ye, Y.; Zhao, X.; Deng, S.; He, G.; Zhu, H.; Xu, N.; Liang, S. SILAC–based quantitative MS approach for real-time recording protein-mediated cell-cell interactions. Sci. Rep. 2018, 8, 8441. [Google Scholar] [CrossRef]
- Beller, N.C.; Lukowski, J.K.; Ludwig, K.R.; Hummon, A.B. Spatial Stable Isotopic Labeling by Amino Acids in Cell Culture: Pulse-Chase Labeling of Three-Dimensional Multicellular Spheroids for Global Proteome Analysis. Anal. Chem. 2021, 93, 15990–15999. [Google Scholar] [CrossRef] [PubMed]
- Snider, J.; Wang, D.; Bogenhagen, D.F.; Haley, J.D. Pulse SILAC Approaches to the Measurement of Cellular Dynamics. Adv. Exp. Med. Biol. 2019, 1140, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Fenyo, D.; Neubert, T.A. Evaluation of the Variation in Sample Preparation for Comparative Proteomics Using Stable Isotope Labeling by Amino Acids in Cell Culture. J. Proteome Res. 2009, 8, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.F.; Tang, F.; Yin, J.; Miao, W.; Wang, Y. Parallel-reaction monitoring revealed altered expression of a number of epitranscriptomic reader, writer, and eraser proteins accompanied with colorectal cancer metastasis. Proteomics 2022, e2200059. [Google Scholar] [CrossRef]
- Qi, T.F.; Miao, W.; Wang, Y. Targeted Profiling of Epitranscriptomic Reader, Writer, and Eraser Proteins Accompanied with Radioresistance in Breast Cancer Cells. Anal. Chem. 2022, 94, 1525–1530. [Google Scholar] [CrossRef]
- Zhang, Y.; Dreyer, B.; Govorukhina, N.; Heberle, A.M.; Končarević, S.; Krisp, C.; Opitz, C.A.; Pfänder, P.; Bischoff, R.; Schlüter, H.; et al. Comparative Assessment of Quantification Methods for Tumor Tissue Phosphoproteomics. Anal. Chem. 2022, 94, 10893–10906. [Google Scholar] [CrossRef]
- Griffith, A.A.; Callahan, K.P.; King, N.G.; Xiao, Q.; Su, X.; Salomon, A.R. SILAC Phosphoproteomics Reveals Unique Signaling Circuits in CAR-T Cells and the Inhibition of B Cell-Activating Phosphorylation in Target Cells. J. Proteome Res. 2022, 21, 395–409. [Google Scholar] [CrossRef]
- Capello, M.; Katayama, H.; Hanash, S.M. Proteomic Profiling of the Tumor Microenvironment. Methods Mol. Biol. 2022, 2435, 157–167. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Wu, W.-L.; Jang, C.-W.; Yen, Y.-C.; Wang, S.-H.; Tsai, F.-Y.; Shen, Y.-Y.; Chen, Y.-W. Interferon-stimulated gene 15 modulates cell migration by interacting with Rac1 and contributes to lymph node metastasis of oral squamous cell carcinoma cells. Oncogene 2019, 38, 4480–4495. [Google Scholar] [CrossRef]
- Zhang, X.; Nguyen, K.D.; Rudnick, P.A.; Roper, N.; Kawaler, E.; Maity, T.K.; Awasthi, S.; Gao, S.; Biswas, R.; Venugopalan, A.; et al. Quantitative Mass Spectrometry to Interrogate Proteomic Heterogeneity in Metastatic Lung Adenocarcinoma and Validate a Novel Somatic Mutation CDK12-G879V. Mol. Cell. Proteom. 2019, 18, 622–641. [Google Scholar] [CrossRef]
- Gygi, S.P.; Rist, B.; Gerber, S.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Colangelo, C.M.; Williams, K.R.; Dobrin, N.; Randall, N.W. Isotope-Coded Affinity Tags for Protein Quantification. Methods Mol. Biol. 2006, 328, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Shiio, Y.; Aebersold, R. Quantitative proteome analysis using isotope-coded affinity tags and mass spectrometry. Nat. Protoc. 2006, 1, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Yi, E.C.; Li, X.-J.; Cooke, K.; Lee, H.; Raught, B.; Page, A.; Aneliunas, V.; Hieter, P.; Goodlett, D.R.; Aebersold, R. Increased quantitative proteome coverage with13C/12C-based, acid-cleavable isotope-coded affinity tag reagent and modified data acquisition scheme. Proteomics 2005, 5, 380–387. [Google Scholar] [CrossRef]
- Xiao, Z.; Veenstra, T.D. Comparison of protein expression by isotope-coded affinity tag labeling. In Clinical Proteomics; Humana Press: Totowa, NJ, USA, 2008; Volume 428, pp. 181–192. [Google Scholar] [CrossRef]
- Von Haller, P.D.; Yi, E.; Donohoe, S.; Vaughn, K.; Keller, A.; Nesvizhskii, A.I.; Eng, J.; Li, X.-J.; Goodlett, D.R.; Aebersold, R.; et al. The Application of New Software Tools to Quantitative Protein Profiling via Isotope-Coded Affinity Tag (ICAT) and Tandem Mass Spectrometry. Mol. Cell. Proteom. 2003, 2, 428–442. [Google Scholar] [CrossRef]
- Kang, U.-B.; Ahn, Y.; Lee, J.W.; Kim, Y.-H.; Kim, J.; Yu, M.-H.; Noh, D.-Y.; Lee, C. Differential profiling of breast cancer plasma proteome by isotope-coded affinity tagging method reveals biotinidase as a breast cancer biomarker. BMC Cancer 2010, 10, 114. [Google Scholar] [CrossRef]
- Wdowiak, A.P.; Duong, M.N.; Joyce, R.D.; Boyatzis, A.E.; Walkey, M.C.; Nealon, G.L.; Arthur, P.G.; Piggott, M.J. Isotope-Coded Maleimide Affinity Tags for Proteomics Applications. Bioconjugate Chem. 2021, 32, 1652–1666. [Google Scholar] [CrossRef]
- Köcher, T.; Pichler, P.; Schutzbier, M.; Stingl, C.; Kaul, A.; Teucher, N.; Hasenfuss, G.; Penninger, J.M.; Mechtler, K. High Precision Quantitative Proteomics Using iTRAQ on an LTQ Orbitrap: A New Mass Spectrometric Method Combining the Benefits of All. J. Proteome Res. 2009, 8, 4743–4752. [Google Scholar] [CrossRef]
- Trinh, H.V.; Grossmann, J.; Gehrig, P.; Roschitzki, B.; Schlapbach, R.; Greber, U.F.; Hemmi, S. iTRAQ-Based and Label-Free Proteomics Approaches for Studies of Human Adenovirus Infections. Int. J. Proteom. 2013, 2013, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wiese, S.; Reidegeld, K.A.; Meyer, H.E.; Warscheid, B. Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. Proteomics 2007, 7, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Pichler, P.; Köcher, T.; Holzmann, J.; Mazanek, M.; Taus, T.; Ammerer, G.; Mechtler, K. Peptide Labeling with Isobaric Tags Yields Higher Identification Rates Using iTRAQ 4-Plex Compared to TMT 6-Plex and iTRAQ 8-Plex on LTQ Orbitrap. Anal. Chem. 2010, 82, 6549–6558. [Google Scholar] [CrossRef]
- Wang, Y.; Arthur, E.W.; Liu, N.; Li, X.; Xiang, W.; Maxwell, A.; Li, Z.; Zhou, Z. iTRAQ-Based Quantitative Proteomics Analysis of HeLa Cells Infected with Chlamydia muridarum TC0668 Mutant and Wild-Type Strains. Front. Microbiol. 2019, 10, 2553. [Google Scholar] [CrossRef]
- Zha, C.; Jiang, X.H.; Peng, S.F. iTRAQ-Based Quantitative Proteomic Analysis on S100 Calcium Binding Protein A2 in Metastasis of Laryngeal Cancer. PLoS ONE 2015, 10, e0122322. [Google Scholar] [CrossRef] [PubMed]
- Rehman, I.; Glen, A.; Gan, C.; Hamdy, F.; Eaton, C.; Cross, S.; Catto, J.; Wright, P. iTRAQ-Facilitated Proteomic Analysis of Human Prostate Cancer Cells Identifies Proteins Associated with Progression. Eur. Urol. Suppl. 2008, 7, 252. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.; Su, X. iTRAQ-based proteomics analysis of the therapeutic effects of combined anticancer bioactive peptides and oxaliplatin on gastric cancer cells. Oncol. Rep. 2020, 43, 201–217. [Google Scholar] [CrossRef]
- Xia, C.; Yang, F.; He, Z.; Cai, Y. iTRAQ-based quantitative proteomic analysis of the inhibition of cervical cancer cell invasion and migration by metformin. Biomed. Pharmacother. 2019, 123, 109762. [Google Scholar] [CrossRef]
- Boylan, K.L.; Andersen, J.D.; Anderson, L.B.; Higgins, L.; Skubitz, A.P. Quantitative proteomic analysis by iTRAQ® for the identification of candidate biomarkers in ovarian cancer serum. Proteome Sci. 2010, 8, 31. [Google Scholar] [CrossRef]
- Chen, C.-J.; Chou, C.-Y.; Shu, K.-H.; Chen, H.-C.; Wang, M.-C.; Chang, C.-C.; Hsu, B.-G.; Wu, M.-S.; Yang, Y.-L.; Liao, W.-L.; et al. Discovery of Novel Protein Biomarkers in Urine for Diagnosis of Urothelial Cancer Using iTRAQ Proteomics. J. Proteome Res. 2021, 20, 2953–2963. [Google Scholar] [CrossRef]
- Tonack, S.; Jenkinson, C.; Cox, T.; Elliott, V.; E Jenkins, R.; Kitteringham, N.R.; Greenhalf, W.; Shaw, V.; Michalski, C.W.; Friess, H.; et al. iTRAQ reveals candidate pancreatic cancer serum biomarkers: Influence of obstructive jaundice on their performance. Br. J. Cancer 2013, 108, 1846–1853. [Google Scholar] [CrossRef]
- Bąchor, R.; Waliczek, M.; Stefanowicz, P.; Szewczuk, Z. Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics. Molecules 2019, 24, 701. [Google Scholar] [CrossRef]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Dayon, L.; Hainard, A.; Licker, V.; Turck, N.; Kuhn, K.; Hochstrasser, D.F.; Burkhard, P.R.; Sanchez, J.-C. Relative Quantification of Proteins in Human Cerebrospinal Fluids by MS/MS Using 6-Plex Isobaric Tags. Anal. Chem. 2008, 80, 2921–2931. [Google Scholar] [CrossRef]
- Werner, T.; Sweetman, G.; Savitski, M.F.; Mathieson, T.; Bantscheff, M.; Savitski, M.M. Ion Coalescence of Neutron Encoded TMT 10-Plex Reporter Ions. Anal. Chem. 2014, 86, 3594–3601. [Google Scholar] [CrossRef] [PubMed]
- Specht, H.; Slavov, N. Optimizing Accuracy and Depth of Protein Quantification in Experiments Using Isobaric Carriers. J. Proteome Res. 2020, 20, 880–887. [Google Scholar] [CrossRef]
- Hamood, F.; Bayer, F.P.; Wilhelm, M.; Kuster, B.; The, M. SIMSI-Transfer: Software-Assisted Reduction of Missing Values in Phosphoproteomic and Proteomic Isobaric Labeling Data Using Tandem Mass Spectrum Clustering. Mol. Cell. Proteom. 2022, 21, 100238. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.D.; Paulo, J.A.; O’Brien, J.J.; Gygi, S.P. Proteome-Wide Evaluation of Two Common Protein Quantification Methods. J. Proteome Res. 2018, 17, 1934–1942. [Google Scholar] [CrossRef]
- Casey, T.M.; Khan, J.M.; Bringans, S.D.; Koudelka, T.; Takle, P.S.; Downs, R.A.; Livk, A.; Syme, R.A.; Tan, K.-C.; Lipscombe, R.J. Analysis of Reproducibility of Proteome Coverage and Quantitation Using Isobaric Mass Tags (iTRAQ and TMT). J. Proteome Res. 2016, 16, 384–392. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.J.; O’Connell, J.D.; Paulo, J.A.; Thakurta, S.; Rose, C.M.; Weekes, M.P.; Huttlin, E.L.; Gygi, S.P. Compositional Proteomics: Effects of Spatial Constraints on Protein Quantification Utilizing Isobaric Tags. J. Proteome Res. 2017, 17, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Zhang, M.; Li, T.; Qin, X. Serum Proteomic Analysis by Tandem Mass Tags (TMT) Based Quantitative Proteomics in Gastric Cancer Patients. Clin. Lab. 2018, 64, 855–866. [Google Scholar] [CrossRef]
- Brenes, A.; Hukelmann, J.; Bensaddek, D.; Lamond, A.I. Multibatch TMT Reveals False Positives, Batch Effects and Missing Values. Mol. Cell. Proteom. 2019, 18, 1967–1980. [Google Scholar] [CrossRef]
- Sanford, J.A.; Wang, Y.; Hansen, J.R.; Gritsenko, M.A.; Weitz, K.K.; Sagendorf, T.J.; Tognon, C.E.; Petyuk, V.A.; Qian, W.-J.; Liu, T.; et al. Evaluation of Differential Peptide Loading on Tandem Mass Tag-Based Proteomic and Phosphoproteomic Data Quality. J. Am. Soc. Mass Spectrom. 2021, 33, 17–30. [Google Scholar] [CrossRef]
- Tsai, C.-F.; Smith, J.S.; Krajewski, K.; Zhao, R.; Moghieb, A.M.; Nicora, C.D.; Xiong, X.; Moore, R.J.; Liu, T.; Smith, R.D.; et al. Tandem Mass Tag Labeling Facilitates Reversed-Phase Liquid Chromatography-Mass Spectrometry Analysis of Hydrophilic Phosphopeptides. Anal. Chem. 2019, 91, 11606–11613. [Google Scholar] [CrossRef] [PubMed]
- Aljawad, M.F.; Al Faisal, A.H.M.; Alqanbar, M.F.; A Wilmarth, P.; Hassan, B.Q. Tandem mass tag-based quantitative proteomic analysis of cervical cancer. Proteom. Clin. Appl. 2022, e2100105. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, H.; Wang, Y.; Gao, J.; Zhou, S.; Li, Y.; Han, S.; Li, X.; Li, J. Proteomic Analysis of Human Esophageal Cancer Using Tandem Mass Tag Quantifications. BioMed Res. Int. 2020, 2020, 5849323. [Google Scholar] [CrossRef]
- Slavov, N. Single-cell protein analysis by mass spectrometry. Curr. Opin. Chem. Biol. 2020, 60, 1–9. [Google Scholar] [CrossRef]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Q.; Huang, G.; Lin, B.-Y.; Lin, D.-Z.; Ma, Y.; Zhang, Z.; Chen, T.; Zhou, J. Tandem Mass Tag-Based Proteomic Analysis of Potential Biomarkers for Hepatocellular Carcinoma Differentiation. OncoTargets Ther. 2021, 14, 1007–1020. [Google Scholar] [CrossRef]
- Hsu, J.-L.; Huang, S.-Y.; Chow, N.-H.; Chen, S.-H. Stable-Isotope Dimethyl Labeling for Quantitative Proteomics. Anal. Chem. 2003, 75, 6843–6852. [Google Scholar] [CrossRef]
- Boersema, P.J.; Raijmakers, R.; Lemeer, S.; Mohammed, S.; Heck, A.J. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 2009, 4, 484–494. [Google Scholar] [CrossRef]
- Munoz, J.; Low, T.Y.; Kok, Y.J.; Chin, A.; Frese, C.; Ding, V.; Choo, A.; Heck, A.J.R. The quantitative proteomes of human-induced pluripotent stem cells and embryonic stem cells. Mol. Syst. Biol. 2011, 7, 550. [Google Scholar] [CrossRef] [PubMed]
- Khidekel, N.; Ficarro, S.B.; Clark, P.M.; Bryan, M.C.; Swaney, D.L.; E Rexach, J.; E Sun, Y.; Coon, J.J.; Peters, E.C.; Hsieh-Wilson, L.C. Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat. Chem. Biol. 2007, 3, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, F.; Liu, Z.; Qin, H.; Song, C.; Huang, J.; Bian, Y.; Wei, X.; Dong, J.; Zou, H. Five-plex isotope dimethyl labeling for quantitative proteomics. Chem. Commun. 2013, 50, 1708–1710. [Google Scholar] [CrossRef] [PubMed]
- Tashima, A.K.; Fricker, L.D. Quantitative Peptidomics with Five-Plex Reductive Methylation Labels. J. Am. Soc. Mass Spectrom. 2017, 29, 866–878. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Li, Y.; Zhao, L.; Yuan, S.; Wang, Z.; Li, B.; Chen, Q. Stable isotope dimethyl labeling combined with LTQ mass spectrometric detection, a quantitative proteomics technology used in liver cancer research. Biomed. Rep. 2013, 1, 549–554. [Google Scholar] [CrossRef][Green Version]
- Hao, L.; Johnson, J.; Lietz, C.B.; Buchberger, A.; Frost, D.; Kao, W.J.; Li, L. Mass Defect-Based N,N-Dimethyl Leucine Labels for Quantitative Proteomics and Amine Metabolomics of Pancreatic Cancer Cells. Anal. Chem. 2017, 89, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Castillo, M.J.; Reynolds, K.J.; Gomes, A.; Fenselau, C.; Yao, X. Quantitative Protein Analysis Using Enzymatic [18O]Water Labeling. Curr. Protoc. Protein Sci. 2014, 76, 23.4.1–23.4.9. [Google Scholar] [CrossRef]
- Klingler, D.; Hardt, M. Protease- and Acid-catalyzed Labeling Workflows Employing 18O-enriched Water. J. Vis. Exp. 2013, e3891. [Google Scholar] [CrossRef]
- Ye, X.; Luke, B.; Andresson, T.; Blonder, J. 18O Stable Isotope Labeling in MS-based Proteomics. Briefings Funct. Genom. Proteom. 2009, 8, 136–144. [Google Scholar] [CrossRef]
- Smith, J.R.; Olivier, M.; Greene, A.S. Relative quantification of peptide phosphorylation in a complex mixture using 18O labeling. Physiol. Genom. 2007, 31, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, K.; Staes, A.; Van Damme, J.; De Groot, S.; Hugelier, K.; Demol, H.; Martens, L.; Goethals, M.; Vandekerckhove, J. Global phosphoproteome analysis on human HepG2 hepatocytes using reversed-phase diagonal LC. Proteomics 2005, 5, 3589–3599. [Google Scholar] [CrossRef] [PubMed]
- White, C.A.; Oey, N.; Emili, A. Global Quantitative Proteomic Profiling through 18O-Labeling in Combination with MS/MS Spectra Analysis. J. Proteome Res. 2009, 8, 3653–3665. [Google Scholar] [CrossRef]
- Miyagi, M.; Rao, K.S. Proteolytic 18O-labeling strategies for quantitative proteomics. Mass Spectrom. Rev. 2006, 26, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Fenselau, C.; Yao, X. Proteolytic Labeling with 18O for Comparative Proteomics Studies. Anal. Chem. 2007, 359, 135–142. [Google Scholar] [CrossRef]
- Capelo, J.; Carreira, R.; Fernandes, L.; Lodeiro, C.; Santos, H.; Simal-Gandara, J. Latest developments in sample treatment for 18O-isotopic labeling for proteomics mass spectrometry-based approaches: A critical review. Talanta 2010, 80, 1476–1486. [Google Scholar] [CrossRef]
- Rao, K.C.S.; Miyagi, M. Recent Technological Developments in Proteolytic 18O Labeling. Curr. Proteom. 2011, 8, 39–46. [Google Scholar] [CrossRef]
- Heller, M.; Mattou, H.; Menzel, C.; Yao, X. Trypsin catalyzed 16O-to-18O exchange for comparative proteomics: Tandem mass spectrometry comparison using MALDI-TOF, ESI-QTOF, and ESI-ion trap mass spectrometers. J. Am. Soc. Mass Spectrom. 2003, 14, 704–718. [Google Scholar] [CrossRef]
- Johnson, K.L.; Muddiman, D.C. A method for calculating 16O/18O peptide ion ratios for the relative quantification of proteomes. J. Am. Soc. Mass Spectrom. 2004, 15, 437–445. [Google Scholar] [CrossRef]
- Qian, W.-J.; Monroe, M.E.; Liu, T.; Jacobs, J.M.; Anderson, G.A.; Shen, Y.; Moore, R.J.; Anderson, D.J.; Zhang, R.; Calvano, S.E.; et al. Quantitative Proteome Analysis of Human Plasma Following In Vivo Lipopolysaccharide Administration Using 16O/18O Labeling and the Accurate Mass and Time Tag Approach. Mol. Cell. Proteom. 2005, 4, 700–709. [Google Scholar] [CrossRef]
- Patwardhan, A.J.; Strittmatter, E.F.; Camp, D.G.; Smith, R.D.; Pallavicini, M.G. Quantitative proteome analysis of breast cancer cell lines using 18O-labeling and an accurate mass and time tag strategy. Proteomics 2006, 6, 2903–2915. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.-M.; Lee, C.-W.; Chang, K.-P.; Hao, S.-P.; Lee, H.-M.; Liang, Y.; Hsueh, C.; Yu, C.-J.; Lee, I.-N.; Chang, Y.-J.; et al. Enhanced Interferon Signaling Pathway in Oral Cancer Revealed by Quantitative Proteome Analysis of Microdissected Specimens Using 16O/18O Labeling and Integrated Two-dimensional LC-ESI-MALDI Tandem MS. Mol. Cell. Proteom. 2009, 8, 1453–1474. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.; Toy, D.P.; Hancock, W.S.; Sgroi, D.C.; Karger, B.L. Proteomic Analysis of Ductal Carcinoma of the Breast Using Laser Capture Microdissection, LC−MS, and 16O/18O Isotopic Labeling. J. Proteome Res. 2004, 3, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Ntai, I.; Toby, T.K.; LeDuc, R.D.; Kelleher, N.L. A Method for Label-Free, Differential Top-Down Proteomics. Methods Mol. Biol. 2016, 1410, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, F.; Landolfi, A.; Iacobucci, I.; Monaco, V.; Caterino, M.; Celentano, S.; Zuccato, C.; Cattaneo, E.; Monti, M. New label-free methods for protein relative quantification applied to the investigation of an animal model of Huntington Disease. PLoS ONE 2020, 15, e0238037. [Google Scholar] [CrossRef]
- Schilling, B.; Rardin, M.J.; MacLean, B.X.; Zawadzka, A.M.; Frewen, B.E.; Cusack, M.P.; Sorensen, D.J.; Bereman, M.S.; Jing, E.; Wu, C.C.; et al. Platform-Independent and Label-Free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline. Mol. Cell. Proteom. 2012, 11, 202–214. [Google Scholar] [CrossRef]
- Kudlicki, A. The Optimal Exponent Base for emPAI Is 6.5. PLoS ONE 2012, 7, e32339. [Google Scholar] [CrossRef]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially Modified Protein Abundance Index (emPAI) for Estimation of Absolute Protein Amount in Proteomics by the Number of Sequenced Peptides per Protein. Mol. Cell. Proteom. 2005, 4, 1265–1272. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Milac, T.I.; Randolph, T.W.; Wang, P. Analyzing LC-MS/MS data by spectral count and ion abundance: Two case studies. Stat. Its Interface 2012, 5, 75–87. [Google Scholar] [CrossRef]
- Old, W.M.; Meyer-Arendt, K.; Aveline-Wolf, L.; Pierce, K.G.; Mendoza, A.; Sevinsky, J.R.; Resing, K.A.; Ahn, N.G. Comparison of Label-Free Methods for Quantifying Human Proteins by Shotgun Proteomics. Mol. Cell. Proteom. 2005, 4, 1487–1502. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Gou, M.; Qi, M.; Xiang, W.; Ji, Z.; Wang, W.-J.; Zhao, S.-C.; Liu, Y. Label free quantitative proteomics reveals the role of miR-200b in androgen-independent prostate cancer cells. Clin. Proteom. 2018, 15, 8. [Google Scholar] [CrossRef]
- Pinto, G.; D’Acierno, M.; Illiano, A.; Petruk, G.; Ferraro, G.; Merlino, A.; Monti, D.M.; Godovac-Zimmermann, J.; Amoresano, A. Label-free quantitative proteomics of the MCF-7 cellular response to a ferritin–metallodrug complex. Mol. Omics 2020, 16, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Neilson, K.A.; Ali, N.A.; Muralidharan, S.; Mirzaei, M.; Mariani, M.; Assadourian, G.; Lee, A.; van Sluyter, S.C.; Haynes, P.A. Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 2011, 11, 535–553. [Google Scholar] [CrossRef] [PubMed]
- Levin, Y.; Schwarz, E.; Wang, L.; Leweke, F.M.; Bahn, S. Label-free LC-MS/MS quantitative proteomics for large-scale biomarker discovery in complex samples. J. Sep. Sci. 2007, 30, 2198–2203. [Google Scholar] [CrossRef]
- Nahnsen, S.; Bielow, C.; Reinert, K.; Kohlbacher, O. Tools for Label-Free Peptide Quantification. Mol. Cell. Proteom. 2013, 12, 549–556. [Google Scholar] [CrossRef]
- Chawade, A.; Sandin, M.; Teleman, J.; Malmström, J.; Levander, F. Data Processing Has Major Impact on the Outcome of Quantitative Label-Free LC-MS Analysis. J. Proteome Res. 2014, 14, 676–687. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Mendoza, L.; Shteynberg, D.; Farrah, T.; Lam, H.; Tasman, N.; Sun, Z.; Nilsson, E.; Pratt, B.; Prazen, B.; et al. A guided tour of the Trans-Proteomic Pipeline. Proteomics 2010, 10, 1150–1159. [Google Scholar] [CrossRef]
- Ma, B.; Zhang, K.; Hendrie, C.; Liang, C.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful software for peptidede novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2337–2342. [Google Scholar] [CrossRef]
- Piersma, S.R.; Knol, J.C.; de Reus, I.; Labots, M.; Sampadi, B.K.; Pham, T.V.; Ishihama, Y.; Verheul, H.M.; Jimenez, C.R. Feasibility of label-free phosphoproteomics and application to base-line signaling of colorectal cancer cell lines. J. Proteom. 2015, 127, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Stepath, M.; Zülch, B.; Maghnouj, A.; Schork, K.; Turewicz, M.; Eisenacher, M.; Hahn, S.; Sitek, B.; Bracht, T. Systematic Comparison of Label-Free, SILAC, and TMT Techniques to Study Early Adaption toward Inhibition of EGFR Signaling in the Colorectal Cancer Cell Line DiFi. J. Proteome Res. 2019, 19, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.D.P.; Moreno, F.B.M.B.; Souza, G.H.M.F.; Verde, S.M.M.L.; Moreira, R.D.A.; Monteiro-Moreira, A.C.D.O. Label-Free Proteome Analysis of Plasma from Patients with Breast Cancer: Stage-Specific Protein Expression. Front. Oncol. 2017, 7, 14. [Google Scholar] [CrossRef]
- Min, H.; Han, D.; Kim, Y.; Cho, J.Y.; Jin, J.; Kim, Y. Label-Free Quantitative Proteomics and N-terminal Analysis of Human Metastatic Lung Cancer Cells. Mol. Cells 2014, 37, 457–466. [Google Scholar] [CrossRef]
- Gautam, S.S.; Singh, R.P.; Karsauliya, K.; Sonker, A.K.; Reddy, P.J.; Mehrotra, D.; Gupta, S.; Singh, S.; Kumar, R.; Singh, S.P. Label-free plasma proteomics for the identification of the putative biomarkers of oral squamous cell carcinoma. J. Proteom. 2022, 259, 104541. [Google Scholar] [CrossRef] [PubMed]
- Melton, L. Proteomics in multiplex. Nature 2004, 429, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Berrade, L.; Garcia, A.E.; Camarero, J.A. Protein Microarrays: Novel Developments and Applications. Pharm. Res. 2010, 28, 1480–1499. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Schneiderhan-Marra, N.; Joos, T.O. Protein Microarrays for Personalized Medicine. Clin. Chem. 2010, 56, 376–387. [Google Scholar] [CrossRef]
- Akbani, R.; Becker, K.-F.; Carragher, N.; Goldstein, T.; de Koning, L.; Korf, U.; Liotta, L.; Mills, G.B.; Nishizuka, S.S.; Pawlak, M.; et al. Realizing the Promise of Reverse Phase Protein Arrays for Clinical, Translational and Basic Research: A Workshop Report. Mol. Cell. Proteom. 2014, 13, 1625–1643. [Google Scholar] [CrossRef]
- Petricoin, E.; Wulfkuhle, J.; Howard, M.; Pierobon, M.; Espina, V.; Luchini, A.; Liotta, L.A. RPPA: Origins, Transition to a Validated Clinical Research Tool and Next Generations of the Technology. Adv. Exp. Med. Biol. 2019, 1188, 1–19. [Google Scholar] [CrossRef]
- Partolina, M.; Thoms, H.C.; MacLeod, K.G.; Rodriguez-Blanco, G.; Clarke, M.N.; Venkatasubramani, A.V.; Beesoo, R.; Larionov, V.; Neergheen-Bhujun, V.S.; Serrels, B.; et al. Global histone modification fingerprinting in human cells using epigenetic reverse phase protein array. Cell Death Discov. 2017, 3, 16077. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, W.; Guo, H.; Fang, Y.; Stockman, S.E.; Bai, S.; Ng, P.K.-S.; Li, Y.; Yu, Q.; Lu, Y.; et al. AKT isoform-specific expression and activation across cancer lineages. BMC Cancer 2018, 18, 742. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Nagashio, R.; Jiang, S.-X.; Saito, K.; Tsuchiya, B.; Ryuge, S.; Katono, K.; Nakashima, H.; Fukuda, E.; Goshima, N.; et al. Calnexin is a novel sero-diagnostic marker for lung cancer. Lung Cancer 2015, 90, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Signore, M.; Alfonsi, R.; Federici, G.; Nanni, S.; Addario, A.; Bertuccini, L.; Aiello, A.; Di Pace, A.L.; Sperduti, I.; Muto, G.; et al. Diagnostic and prognostic potential of the proteomic profiling of serum-derived extracellular vesicles in prostate cancer. Cell Death Dis. 2021, 12, 636. [Google Scholar] [CrossRef]
- Vinik, Y.; Ortega, F.G.; Mills, G.B.; Lu, Y.; Jurkowicz, M.; Halperin, S.; Aharoni, M.; Gutman, M.; Lev, S. Proteomic analysis of circulating extracellular vesicles identifies potential markers of breast cancer progression, recurrence and response. Sci. Adv. 2020, 6, eaba5714. [Google Scholar] [CrossRef]
- Yanagita, K.; Nagashio, R.; Jiang, S.-X.; Kuchitsu, Y.; Hachimura, K.; Ichinoe, M.; Igawa, S.; Fukuda, E.; Goshima, N.; Satoh, Y.; et al. Cytoskeleton-Associated Protein 4 Is a Novel Serodiagnostic Marker for Lung Cancer. Am. J. Pathol. 2018, 188, 1328–1333. [Google Scholar] [CrossRef]
- Hellström, C.; Dodig-Crnković, T.; Hong, M.-G.; Schwenk, J.M.; Nilsson, P.; Sjöberg, R. High-Density Serum/Plasma Reverse Phase Protein Arrays. Methods Mol. Biol. 2017, 1619, 229–238. [Google Scholar] [CrossRef]
- Nettikadan, S.; Radke, K.; Johnson, J.; Xu, J.; Lynch, M.; Mosher, C.; Henderson, E. Detection and Quantification of Protein Biomarkers from Fewer than 10 Cells. Mol. Cell. Proteom. 2006, 5, 895–901. [Google Scholar] [CrossRef]
- Sanchez-Carbayo, M.; Socci, N.D.; Lozano, J.J.; Haab, B.B.; Cordon-Cardo, C. Profiling Bladder Cancer Using Targeted Antibody Arrays. Am. J. Pathol. 2006, 168, 93–103. [Google Scholar] [CrossRef]
- Puig-Costa, M.; Codina-Cazador, A.; Cortés-Pastoret, E.; Oliveras-Ferraros, C.; Cufí, S.; Flaquer, S.; Llopis-Puigmarti, F.; Pujol-Amado, E.; Corominas-Faja, B.; Cuyàs, E.; et al. Discovery and validation of an INflammatory PROtein-driven GAstric cancer Signature (INPROGAS) using antibody microarray-based oncoproteomics. Oncotarget 2014, 5, 1942–1954. [Google Scholar] [CrossRef][Green Version]
- Syu, G.-D.; Dunn, J.; Zhu, H. Developments and Applications of Functional Protein Microarrays. Mol. Cell. Proteom. 2020, 19, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Song, G.; Chen, D.; Li, Y.; Liu, S.; Hu, S.; Rosa, C.; Eichinger, D.; Pino, I.; Zhu, H.; et al. Identification of Serological Biomarkers for Early Diagnosis of Lung Cancer Using a Protein Array-Based Approach. Mol. Cell. Proteom. 2017, 16, 2069–2078. [Google Scholar] [CrossRef]
- Wilson, J.J.; Burgess, R.; Mao, Y.-Q.; Luo, S.; Tang, H.; Jones, V.S.; Weisheng, B.; Huang, R.-Y.; Chen, X.; Huang, R.-P. Antibody Arrays in Biomarker Discovery. Adv. Clin. Chem. 2015, 69, 255–324. [Google Scholar] [CrossRef] [PubMed]
- Chandra, H.; Srivastava, S. Cell-free synthesis-based protein microarrays and their applications. Proteomics 2009, 10, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, N.; Raphael, J.V.; Hainsworth, E.; Demirkan, G.; Fuentes, M.G.; Rolfs, A.; Hu, Y.; LaBaer, J. Next-generation high-density self-assembling functional protein arrays. Nat. Methods 2008, 5, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Spera, R.; Labaer, J.; Nicolini, C. MALDI-TOF characterization of NAPPA-generated proteins. Biol. Mass Spectrom. 2011, 46, 960–965. [Google Scholar] [CrossRef]
- Melton, L. On the trail of SNPs. Nature 2003, 422, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Houser, B. Bio-Rad’s Bio-Plex® suspension array system, xMAP technology overview. Arch. Physiol. Biochem. 2012, 118, 192–196. [Google Scholar] [CrossRef]
- Camp, R.L.; Neumeister, V.; Rimm, D.L. A Decade of Tissue Microarrays: Progress in the Discovery and Validation of Cancer Biomarkers. J. Clin. Oncol. 2008, 26, 5630–5637. [Google Scholar] [CrossRef] [PubMed]
- Kononen, J.; Bubendorf, L.; Kallioniemi, O.; Bärlund, M.; Schraml, P.; Leighton, S.; Torhorst, J.; Mihatsch, M.J.; Sauter, G.; Kallionimeni, O.-P. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat. Med. 1998, 4, 844–847. [Google Scholar] [CrossRef]
- Hwang, S.-I.; Thumar, J.; Lundgren, D.H.; Rezaul, K.; Mayya, V.; Wu, L.; Eng, J.; E Wright, M.; Han, D.K. Direct cancer tissue proteomics: A method to identify candidate cancer biomarkers from formalin-fixed paraffin-embedded archival tissues. Oncogene 2006, 26, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Voduc, D.; Kenney, C.; Nielsen, T.O. Tissue Microarrays in Clinical Oncology. Semin. Radiat. Oncol. 2008, 18, 89–97. [Google Scholar] [CrossRef]
- Nie, S.; Gurrea, M.; Zhu, J.; Thakolwiboon, S.; Heth, J.A.; Muraszko, K.M.; Fan, X.; Lubman, D.M. Tenascin-C: A Novel Candidate Marker for Cancer Stem Cells in Glioblastoma Identified by Tissue Microarrays. J. Proteome Res. 2014, 14, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Drev, P.; Grazio, S.F.; Bračko, M. Tissue Microarrays for Routine Diagnostic Assessment of HER2 Status in Breast Carcinoma. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A. Tissue microarray studies in bladder cancer. Scand. J. Urol. Nephrol. 2008, 42, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Engellau, J.; Åkerman, M.; Anderson, H.; Domanski, H.A.; Rambech, E.; Alvegård, T.A.; Nilbert, M. Tissue Microarray Technique in Soft Tissue Sarcoma: Immunohistochemical Ki-67 Expression in Malignant Fibrous Histiocytoma. Appl. Immunohistochem. Mol. Morphol. 2001, 9, 358–363. [Google Scholar] [CrossRef]
- Vlajnic, T.; Eppenberger-Castori, S.; Bubendorf, L. Protocols for Tissue Microarrays in Prostate Cancer Studies. Methods Mol. Biol. 2018, 1786, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Espejo, A.; Côté, J.; Bednarek, A.; Richard, S.; Bedford, M.T. A protein-domain microarray identifies novel protein–protein interactions. Biochem. J. 2002, 367, 697–702. [Google Scholar] [CrossRef]
- Kaushansky, A.; E Allen, J.; Gordus, A.; A Stiffler, M.; Karp, E.S.; Chang, B.H.; MacBeath, G. Quantifying protein–protein interactions in high throughput using protein domain microarrays. Nat. Protoc. 2010, 5, 773–790. [Google Scholar] [CrossRef]
- Chen, J.; Sagum, C.; Bedford, M.T. Protein domain microarrays as a platform to decipher signaling pathways and the histone code. Methods 2019, 184, 4–12. [Google Scholar] [CrossRef]
- Tian, W.; Wang, L.; Lei, H.; Sun, Y.; Xiao, Z. Antibody production and application for immunoassay development of environmental hormones: A review. Chem. Biol. Technol. Agric. 2018, 5, 5. [Google Scholar] [CrossRef]
- Hou, J.-Y.; Liu, T.-C.; Lin, G.-F.; Li, Z.-X.; Zou, L.-P.; Li, M.; Wu, Y.-S. Development of an immunomagnetic bead-based time-resolved fluorescence immunoassay for rapid determination of levels of carcinoembryonic antigen in human serum. Anal. Chim. Acta 2012, 734, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Cao, Y.; Xu, Y.; Li, G. Colorimetric Immunoassay for Detection of Tumor Markers. Int. J. Mol. Sci. 2010, 11, 5077–5094. [Google Scholar] [CrossRef] [PubMed]
- Ladd, J.; Taylor, A.D.; Piliarik, M.; Homola, J.; Jiang, S. Label-free detection of cancer biomarker candidates using surface plasmon resonance imaging. Anal. Bioanal. Chem. 2008, 393, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Gil Rosa, B.; Akingbade, O.E.; Guo, X.; Gonzalez-Macia, L.; Crone, M.A.; Cameron, L.P.; Freemont, P.; Choy, K.-L.; Güder, F.; Yeatman, E.; et al. Multiplexed immunosensors for point-of-care diagnostic applications. Biosens. Bioelectron. 2022, 203, 114050. [Google Scholar] [CrossRef]
- Dixit, C.K.; Kadimisetty, K.; Otieno, B.A.; Tang, C.; Malla, S.; Krause, C.E.; Rusling, J.F. Electrochemistry-based approaches to low cost, high sensitivity, automated, multiplexed protein immunoassays for cancer diagnostics. Analyst 2015, 141, 536–547. [Google Scholar] [CrossRef]
- Zheng, W.; Zhou, S.; Xu, J.; Liu, Y.; Huang, P.; Liu, Y.; Chen, X. Tumor Marker Detection: Ultrasensitive Luminescent In Vitro Detection for Tumor Markers Based on Inorganic Lanthanide Nano-Bioprobes. Adv. Sci. 2016, 3, 1600197. [Google Scholar] [CrossRef]
- Sardesai, N.P.; Kadimisetty, K.; Faria, R.C.; Rusling, J.F. A microfluidic electrochemiluminescent device for detecting cancer biomarker proteins. Anal. Bioanal. Chem. 2013, 405, 3831–3838. [Google Scholar] [CrossRef]
- Sharafeldin, M.; Kadimisetty, K.; Bhalerao, K.S.; Chen, T.; Rusling, J.F. 3D-Printed Immunosensor Arrays for Cancer Diagnostics. Sensors 2020, 20, 4514. [Google Scholar] [CrossRef]
- Dhanapala, L.; Krause, C.; Jones, A.; Rusling, J. Printed Electrodes in Microfluidic Arrays for Cancer Biomarker Protein Detection. Biosensors 2020, 10, 115. [Google Scholar] [CrossRef]
- Kadimisetty, K.; Malla, S.; Bhalerao, K.S.; Mosa, I.M.; Bhakta, S.; Lee, N.H.; Rusling, J.F. Automated 3D-Printed Microfluidic Array for Rapid Nanomaterial-Enhanced Detection of Multiple Proteins. Anal. Chem. 2018, 90, 7569–7577. [Google Scholar] [CrossRef]
- Prince, E.; Kheiri, S.; Wang, Y.; Xu, F.; Cruickshank, J.; Topolskaia, V.; Tao, H.; Young, E.W.K.; McGuigan, A.P.; Cescon, D.W.; et al. Microfluidic Arrays of Breast Tumor Spheroids for Drug Screening and Personalized Cancer Therapies. Adv. Healthc. Mater. 2021, 11, 2101085. [Google Scholar] [CrossRef]
- Anderson, K.S.; Ramachandran, N.; Wong, J.; Raphael, J.V.; Hainsworth, E.; Demirkan, G.; Cramer, D.; Aronzon, D.; Hodi, F.S.; Harris, L.; et al. Application of Protein Microarrays for Multiplexed Detection of Antibodies to Tumor Antigens in Breast Cancer. J. Proteome Res. 2008, 7, 1490–1499. [Google Scholar] [CrossRef]
- Munge, B.S.; Stracensky, T.; Gamez, K.; DiBiase, D.; Rusling, J.F. Multiplex Immunosensor Arrays for Electrochemical Detection of Cancer Biomarker Proteins. Electroanalysis 2016, 28, 2644–2658. [Google Scholar] [CrossRef]
- Roulhac, P.L.; Ward, J.M.; Thompson, J.W.; Soderblom, E.J.; Silva, M.; Moseley, M.A.; Jarvis, E.D. Microproteomics: Quantitative Proteomic Profiling of Small Numbers of Laser-Captured Cells. Cold Spring Harb. Protoc. 2011, 2011, pdb.prot5573. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Espina, V.; Wulfkuhle, J.D.; Calvert, V.S.; VanMeter, A.; Zhou, W.; Coukos, G.; Geho, D.H.; Petricoin, E.F.; Liotta, L.A. Laser-capture microdissection. Nat. Protoc. 2006, 1, 586–603. [Google Scholar] [CrossRef]
- Shen, S.; Li, J.; Huo, S.; Ma, M.; Zhu, X.; Rasam, S.; Duan, X.; Qu, M.; Titus, M.A.; Qu, J. Parallel, High-Quality Proteomic and Targeted Metabolomic Quantification Using Laser Capture Microdissected Tissues. Anal. Chem. 2021, 93, 8711–8718. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Yang, S.; Tian, Y.; Zhang, W.; Zhou, B.; Bu, L.; Huo, S. Analysis of the expression protein profiles of lung squamous carcinoma cell using shot-gun proteomics strategy. Med. Oncol. 2008, 26, 215–221. [Google Scholar] [CrossRef]
- Zhang, Y.; Ye, Y.; Shen, D.; Jiang, K.; Zhang, H.; Sun, W.; Zhang, J.; Xu, F.; Cui, Z.; Wang, S. Identification of transgelin-2 as a biomarker of colorectal cancer by laser capture microdissection and quantitative proteome analysis. Cancer Sci. 2010, 101, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Johann, N.J.; Mukherjee, S.; Prieto, D.A.; Veenstra, T.D.; Blonder, J. Profiling Solid Tumor Heterogeneity by LCM and Biological MS of Fresh-Frozen Tissue Sections. Methods Mol. Biol. 2011, 755, 95–106. [Google Scholar] [CrossRef]
- Liotta, L.A.; Pappalardoa, P.A.; Carpino, A.; Haymonda, A.; Howarda, M.; Espina, V. Laser Capture Proteomics: Spatial tissue molecular profiling from the bench to personalized medicine. Expert Rev. Proteom. 2021, 18, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Staunton, L.; Tonry, C.; Lis, R.; Finn, S.; Leary, J.O.; Loda, M.; Bowden, M.; Pennington, S. Profiling the tumor microenvironment proteome in prostate cancer using laser capture microdissection coupled to LC-MS. A technical report. EuPA Open Proteom. 2016, 10, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, D.; Li, Y.; Tang, Z.; Cao, Z.C.; Chen, H.W.; Mallikaratchy, P.; Sefah, K.; Yang, C.J.; Tan, W. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc. Natl. Acad. Sci. USA 2006, 103, 11838–11843. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, H.; Han, D.; Peng, B.; Zhang, H.; Zhang, L.; Li, J.; Liu, J.; Cui, C.; Fang, S.; et al. Elucidation and Structural Modeling of CD71 as a Molecular Target for Cell-Specific Aptamer Binding. J. Am. Chem. Soc. 2019, 141, 10760–10769. [Google Scholar] [CrossRef]
- Li, H.; Vanarsa, K.; Zhang, T.; Soomro, S.; Cicalese, P.A.; Duran, V.; Dasari, S.; Lee, K.H.; Pedroza, C.; Kisiel, J.B.; et al. Comprehensive aptamer-based screen of 1317 proteins uncovers improved stool protein markers of colorectal cancer. J. Gastroenterol. 2021, 56, 659–672. [Google Scholar] [CrossRef]
- Ostroff, R.M.; Bigbee, W.L.; Franklin, W.; Gold, L.; Mehan, M.; Miller, Y.E.; Pass, H.I.; Rom, W.; Siegfried, J.M.; Stewart, A.; et al. Unlocking Biomarker Discovery: Large Scale Application of Aptamer Proteomic Technology for Early Detection of Lung Cancer. PLoS ONE 2010, 5, e15003. [Google Scholar] [CrossRef]
- Jung, Y.J.; Katilius, E.; Ostroff, R.M.; Kim, Y.; Seok, M.; Lee, S.; Jang, S.; Kim, W.S.; Choi, C.-M. Development of a Protein Biomarker Panel to Detect Non–Small-Cell Lung Cancer in Korea. Clin. Lung Cancer 2016, 18, e99–e107. [Google Scholar] [CrossRef]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef]
- Lai, X.; Wang, M.; McElyea, S.D.; Sherman, S.; House, M.; Korc, M. A microRNA signature in circulating exosomes is superior to exosomal glypican-1 levels for diagnosing pancreatic cancer. Cancer Lett. 2017, 393, 86–93. [Google Scholar] [CrossRef]
- Moon, P.-G.; Lee, J.-E.; Cho, Y.-E.; Lee, S.J.; Jung, J.H.; Chae, Y.S.; Bae, H.-I.; Kim, Y.-B.; Kim, I.-S.; Park, H.Y.; et al. Identification of Developmental Endothelial Locus-1 on Circulating Extracellular Vesicles as a Novel Biomarker for Early Breast Cancer Detection. Clin. Cancer Res. 2016, 22, 1757–1766. [Google Scholar] [CrossRef]
- Sandfeld-Paulsen, B.; Aggerholm-Pedersen, N.; Baek, R.; Jakobsen, K.R.; Meldgaard, P.; Folkersen, B.H.; Rasmussen, T.R.; Varming, K.; Jørgensen, M.M.; Sorensen, B.S. Exosomal proteins as prognostic biomarkers in non-small cell lung cancer. Mol. Oncol. 2016, 10, 1595–1602. [Google Scholar] [CrossRef] [PubMed]
- Sandfeld-Paulsen, B.; Jakobsen, K.R.; Bæk, R.; Folkersen, B.H.; Rasmussen, T.R.; Meldgaard, P.; Varming, K.; Jørgensen, M.M.; Sorensen, B.S. Exosomal Proteins as Diagnostic Biomarkers in Lung Cancer. J. Thorac. Oncol. 2016, 11, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, K.R.; Paulsen, B.S.; Bæk, R.; Varming, K.; Sorensen, B.; Jørgensen, M.M. Exosomal proteins as potential diagnostic markers in advanced non-small cell lung carcinoma. J. Extracell. Vesicles 2015, 4, 26659. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Kosaka, N.; Konishi, Y.; Ohta, H.; Okamoto, H.; Sonoda, H.; Nonaka, R.; Yamamoto, H.; Ishii, H.; Mori, M.; et al. Ultra-sensitive liquid biopsy of circulating extracellular vesicles using ExoScreen. Nat. Commun. 2014, 5, 3591. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, S.B.; Lundberg, M.; Villablanca, A.; Christensen, S.L.T.; Belling, K.C.; Nielsen, B.S.; Knowles, M.; Gee, N.; Nielsen, H.J.; Brünner, N.; et al. Detection of serological biomarkers by proximity extension assay for detection of colorectal neoplasias in symptomatic individuals. J. Transl. Med. 2013, 11, 253. [Google Scholar] [CrossRef] [PubMed]
- Eltahir, M.; Isaksson, J.; Mattsson, J.; Kärre, K.; Botling, J.; Lord, M.; Mangsbo, S.; Micke, P. Plasma Proteomic Analysis in Non-Small Cell Lung Cancer Patients Treated with PD-1/PD-L1 Blockade. Cancers 2021, 13, 3116. [Google Scholar] [CrossRef]
- Berggrund, M.; Enroth, S.; Lundberg, M.; Assarsson, E.; Stålberg, K.; Lindquist, D.; Hallmans, G.; Grankvist, K.; Olovsson, M.; Gyllensten, U. Identification of Candidate Plasma Protein Biomarkers for Cervical Cancer Using the Multiplex Proximity Extension Assay. Mol. Cell. Proteom. 2019, 18, 735–743. [Google Scholar] [CrossRef]
- Enroth, S.; Berggrund, M.; Lycke, M.; Lundberg, M.; Assarsson, E.; Olovsson, M.; Stålberg, K.; Sundfeldt, K.; Gyllensten, U. A two-step strategy for identification of plasma protein biomarkers for endometrial and ovarian cancer. Clin. Proteom. 2018, 15, 38. [Google Scholar] [CrossRef]
- Enblad, G.; Karlsson, H.; Gammelgård, G.; Wenthe, J.; Lövgren, T.; Amini, R.M.; Wikstrom, K.I.; Essand, M.; Savoldo, B.; Hallböök, H.; et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin. Cancer Res. 2018, 24, 6185–6194. [Google Scholar] [CrossRef]
- Liu, S.; Shen, M.; Hsu, E.-C.; Zhang, C.A.; Garcia-Marques, F.; Nolley, R.; Koul, K.; Rice, M.A.; Aslan, M.; Pitteri, S.J.; et al. Discovery of PTN as a serum-based biomarker of pro-metastatic prostate cancer. Br. J. Cancer 2020, 124, 896–900. [Google Scholar] [CrossRef]
- Guzman, N.A.; Guzman, D.E. An emerging micro-scale immuno-analytical diagnostic tool to see the unseen. Holding promise for precision medicine and P4 medicine. J. Chromatogr. B 2015, 1021, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Guzman, N.A.; Blanc, T.; Phillips, T.M. Immunoaffinity capillary electrophoresis as a powerful strategy for the quantification of low-abundance biomarkers, drugs, and metabolites in biological matrices. Electrophoresis 2008, 29, 3259–3278. [Google Scholar] [CrossRef] [PubMed]
- Guzman, N.A.; Guzman, D.E. Immunoaffinity Capillary Electrophoresis in the Era of Proteoforms, Liquid Biopsy and Preventive Medicine: A Potential Impact in the Diagnosis and Monitoring of Disease Progression. Biomolecules 2021, 11, 1443. [Google Scholar] [CrossRef]
- Guzman, N.A.; Guzman, D.E. A Two-Dimensional Affinity Capture and Separation Mini-Platform for the Isolation, Enrichment, and Quantification of Biomarkers and Its Potential Use for Liquid Biopsy. Biomedicines 2020, 8, 255. [Google Scholar] [CrossRef]
- Phillips, T.M.; Wellner, E.F. Analysis of Inflammatory Mediators in Newborn Dried Blood Spot Samples by Chip-Based Immunoaffinity Capillary Electrophoresis. Methods Mol. Biol. 2019, 1972, 185–198. [Google Scholar] [CrossRef]
- Guzman, N.; Guzman, D. From a Central Laboratory to the Bedside: A Point-of-Care Instrument for Monitoring Wellness and Disease Using Two-Dimensional Immunoaffinity Capillary Electrophoresis Technology Analysis of IgE Response in Nippostrongylus Brasiliensis-Infected Mice View Project Miniaturized Immunoaffinity Capillary Electrophoresis View Project. 2018. Available online: https://www.researchgate.net/publication/324484712 (accessed on 14 October 2022).
- A Home-Based Portable Instrument to Monitor Wellness and Disease—Atlas of Science. Available online: https://atlasofscience.org/a-home-based-portable-instrument-to-monitor-wellness-and-disease (accessed on 14 October 2022).
- Koziol, J.A.; Imai, H.; Dai, L.; Zhang, J.-Y.; Tan, E.M. Early detection of hepatocellular carcinoma using autoantibody profiles from a panel of tumor-associated antigens. Cancer Immunol. Immunother. 2018, 67, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.; Choquet-Kastylevsky, G.; Joubert-Caron, R. Cancer Immunomics Using Autoantibody Signatures for Biomarker Discovery. Mol. Cell. Proteom. 2007, 6, 1115–1122. [Google Scholar] [CrossRef]
- Mintoo, M.; Chakravarty, A.; Tilvawala, R. N-Terminomics Strategies for Protease Substrates Profiling. Molecules 2021, 26, 4699. [Google Scholar] [CrossRef]
- Starr, A.E.; Bellac, C.L.; Dufour, A.; Goebeler, V.; Overall, C.M. Biochemical Characterization and N-terminomics Analysis of Leukolysin, the Membrane-type 6 Matrix Metalloprotease (MMP25). J. Biol. Chem. 2012, 287, 13382–13395. [Google Scholar] [CrossRef]
- Alcaraz, L.B.; Mallavialle, A.; David, T.; Derocq, D.; Delolme, F.; Dieryckx, C.; Mollevi, C.; Boissière-Michot, F.; Simony-Lafontaine, J.; Du Manoir, S.; et al. A 9-kDa matricellular SPARC fragment released by cathepsin D exhibits pro-tumor activity in the triple-negative breast cancer microenvironment. Theranostics 2021, 11, 6173–6192. [Google Scholar] [CrossRef]
- Bennett, T.A.; Montesinos, P.; Moscardo, F.; Martinez-Cuadron, D.; Martinez, J.; Sierra, J.; García, R.; de Oteyza, J.P.; Fernandez, P.; Serrano, J.; et al. Pharmacological Profiles of Acute Myeloid Leukemia Treatments in Patient Samples by Automated Flow Cytometry: A Bridge to Individualized Medicine. Clin. Lymphoma Myeloma Leuk. 2013, 14, 305–318. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pemovska, T.; Kontro, M.; Yadav, B.; Edgren, H.; Eldfors, S.; Szwajda, A.; Almusa, H.; Bespalov, M.M.; Ellonen, P.; Elonen, E.; et al. Individualized Systems Medicine Strategy to Tailor Treatments for Patients with Chemorefractory Acute Myeloid Leukemia. Cancer Discov. 2013, 3, 1416–1429. [Google Scholar] [CrossRef] [PubMed]
- Unger, F.T.; Lange, N.; Krüger, J.; Compton, C.; Moore, H.; Agrawal, L.; Juhl, H.; David, K.A. Nanoproteomic analysis of ischemia-dependent changes in signaling protein phosphorylation in colorectal normal and cancer tissue. J. Transl. Med. 2016, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Riley, N.M.; Bertozzi, C.R.; Pitteri, S.J. A Pragmatic Guide to Enrichment Strategies for Mass Spectrometry–Based Glycoproteomics. Mol. Cell. Proteom. 2021, 20, 100029. [Google Scholar] [CrossRef] [PubMed]
- Beaudette, P.; Popp, O.; Dittmar, G. Proteomic techniques to probe the ubiquitin landscape. Proteomics 2015, 16, 273–287. [Google Scholar] [CrossRef]
- Diallo, I.; Seve, M.; Cunin, V.; Minassian, F.; Poisson, J.-F.; Michelland, S.; Bourgoin-Voillard, S. Current trends in protein acetylation analysis. Expert Rev. Proteom. 2018, 16, 139–159. [Google Scholar] [CrossRef]
- Li, L.; Wu, R.; Yan, G.; Gao, M.; Deng, C.; Zhang, X. A novel method to isolate protein N-terminal peptides from proteome samples using sulfydryl tagging and gold-nanoparticle-based depletion. Anal. Bioanal. Chem. 2015, 408, 441–448. [Google Scholar] [CrossRef]
- Low, T.Y.; Mohtar, M.A.; Lee, P.Y.; Omar, N.; Zhou, H.; Ye, M. Widening the bottleneck of phosphoproteomics: Evolving strategies for phosphopeptide enrichment. Mass Spectrom. Rev. 2020, 40, 309–333. [Google Scholar] [CrossRef]
- Mohammed, S.; Heck, A. Strong cation exchange (SCX) based analytical methods for the targeted analysis of protein post-translational modifications. Curr. Opin. Biotechnol. 2011, 22, 9–16. [Google Scholar] [CrossRef]
- Darling, A.L.; Uversky, V.N. Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front. Genet. 2018, 9, 158. [Google Scholar] [CrossRef]
- Monti, M.; Orrù, S.; Pagnozzi, D.; Pucci, P. Functional proteomics. Clin. Chim. Acta 2005, 357, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Turiák, L.; Sugár, S.; Ács, A.; Tóth, G.; Gömöry, T.; Telekes, A.; Vékey, K.; Drahos, L. Site-specific N-glycosylation of HeLa cell glycoproteins. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Cufaro, M.C.; Pieragostino, D.; Lanuti, P.; Rossi, C.; Cicalini, I.; Federici, L.; De Laurenzi, V.; Del Boccio, P. Extracellular Vesicles and Their Potential Use in Monitoring Cancer Progression and Therapy: The Contribution of Proteomics. J. Oncol. 2019, 2019, 1639854. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Valladares, M.; Bruserud, O.; Selheim, F. The Implementation of Mass Spectrometry-Based Proteomics Workflows in Clinical Routines of Acute Myeloid Leukemia: Applicability and Perspectives. Int. J. Mol. Sci. 2020, 21, 6830. [Google Scholar] [CrossRef] [PubMed]
- Berenguer, C.V.; Pereira, F.; Pereira, J.A.M.; Câmara, J.S. Volatilomics: An Emerging and Promising Avenue for the Detection of Potential Prostate Cancer Biomarkers. Cancers 2022, 14, 3982. [Google Scholar] [CrossRef]
- Samaržija, I. Post-Translational Modifications That Drive Prostate Cancer Progression. Biomolecules 2021, 11, 247. [Google Scholar] [CrossRef]
- Abyadeh, M.; Meyfour, A.; Gupta, V.; Moghaddam, M.Z.; Fitzhenry, M.J.; Shahbazian, S.; Salekdeh, G.H.; Mirzaei, M. Recent Advances of Functional Proteomics in Gastrointestinal Cancers—A Path towards the Identification of Candidate Diagnostic, Prognostic, and Therapeutic Molecular Biomarkers. Int. J. Mol. Sci. 2020, 21, 8532. [Google Scholar] [CrossRef]
- Li, Y.; Kong, X.; Wang, Z.; Xuan, L. Recent advances of transcriptomics and proteomics in triple-negative breast cancer prognosis assessment. J. Cell. Mol. Med. 2022, 26, 1351–1362. [Google Scholar] [CrossRef]
- Kim, Y.; Han, K.-H. Epidemiology and surveillance of hepatocellular carcinoma. Liver Cancer 2012, 1, 2–14. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Caldwell, S.; Park, S.H. The epidemiology of hepatocellular cancer: From the perspectives of public health problem to tumor biology. J. Gastroenterol. 2009, 44, 96–101. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, W.S.; Steggerda, J.; Yang, J.D.; Kuo, A.; Sundaram, V.; Lu, S.C. Current status of hepatocellular carcinoma detection: Screening strategies and novel biomarkers. Ther. Adv. Med. Oncol. 2019, 11, 1758835919869120. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Hatano, E.; Tada, M.; Kawabata, Y.; Tamagawa, S.; Kurimoto, A.; Iwama, H.; Toriguchi, K.; Sueoka, H.; Iida, K.; et al. Enhanced patterns on intraoperative contrast-enhanced ultrasonography predict outcomes after curative liver resection in patients with hepatocellular carcinoma. Surg. Today 2020, 51, 764–776. [Google Scholar] [CrossRef]
- Ludwig, D.R.; Fraum, T.J.; Cannella, R.; Tsai, R.; Naeem, M.; LeBlanc, M.; Salter, A.; Tsung, A.; Fleckenstein, J.; Shetty, A.S.; et al. Expanding the Liver Imaging Reporting and Data System (LI-RADS) v2018 diagnostic population: Performance and reliability of LI-RADS for distinguishing hepatocellular carcinoma (HCC) from non-HCC primary liver carcinoma in patients who do not meet strict LI-RADS high-risk criteria. HPB 2019, 21, 1697–1706. [Google Scholar] [CrossRef]
- Osho, A.; Rich, N.E.; Singal, A.G. Role of imaging in management of hepatocellular carcinoma: Surveillance, diagnosis and treatment response. Hepatoma Res. 2020, 6, 55. [Google Scholar] [CrossRef]
- Cao, J.; Shen, C.; Wang, H.; Shen, H.; Chen, Y.; Nie, A.; Yan, G.; Lu, H.; Liu, Y.; Yang, P. Identification of N-Glycosylation Sites on Secreted Proteins of Human Hepatocellular Carcinoma Cells with a Complementary Proteomics Approach. J. Proteome Res. 2009, 8, 662–672. [Google Scholar] [CrossRef]
- Song, C.; Wang, F.; Ye, M.; Cheng, K.; Chen, R.; Zhu, J.; Tan, Y.; Wang, H.; Figeys, D.; Zou, H. Improvement of the Quantification Accuracy and Throughput for Phosphoproteome Analysis by a Pseudo Triplex Stable Isotope Dimethyl Labeling Approach. Anal. Chem. 2011, 83, 7755–7762. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Wang, J.; Jia, H.-L.; Zhu, W.-W.; Lu, L.; Ye, Q.-H.; Nelson, P.J.; Qin, Y.; Gao, D.-M.; Zhou, H.-J.; et al. Core fucosylated glycan-dependent inhibitory effect of QSOX1-S on invasion and metastasis of hepatocellular carcinoma. Cell Death Discov. 2019, 5, 84. [Google Scholar] [CrossRef]
- Jiang, B.; Huang, J.; Yu, Z.; Wu, M.; Liu, M.; Yao, J.; Zhao, H.; Yan, G.; Ying, W.; Cao, W.; et al. A multi-parallel N-glycopeptide enrichment strategy for high-throughput and in-depth mapping of the N-glycoproteome in metastatic human hepatocellular carcinoma cell lines. Talanta 2019, 199, 254–261. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Chien, K.-Y.; Wu, C.-C.; Chang, W.-Y.; Chu, L.J.; Chen, M.-C.; Yeh, C.-T.; Yu, J.-S. Super-SILAC mix coupled with SIM/AIMS assays for targeted verification of phosphopeptides discovered in a large-scale phosphoproteome analysis of hepatocellular carcinoma. J. Proteom. 2017, 157, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Block, T.M.; Comunale, M.A.; Lowman, M.; Steel, L.F.; Romano, P.R.; Fimmel, C.; Tennant, B.C.; London, W.T.; Evans, A.A.; Blumberg, B.S.; et al. Use of targeted glycoproteomics to identify serum glycoproteins that correlate with liver cancer in woodchucks and humans. Proc. Natl. Acad. Sci. USA 2005, 102, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, Y.; Chui, J.; Guo, K.; Shun, Q.; Lu, W.; Jin, H.; Wei, L.; Yang, P. Investigation on glycosylation patterns of proteins from human liver cancer cell lines based on the multiplexed proteomics technology. Arch. Biochem. Biophys. 2007, 459, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Cheng, J.; Tsai, H.; Young, K.; Hsieh, S.; Ho, C. Plasma proteome plus site-specific N-glycoprofiling for hepatobiliary carcinomas. J. Pathol. Clin. Res. 2019, 5, 199–212. [Google Scholar] [CrossRef]
- Sun, Z.; Sun, D.; Wang, F.; Cheng, K.; Zhang, Z.; Xu, B.; Ye, M.; Wang, L.; Zou, H. Differential analysis of N-glycoproteome between hepatocellular carcinoma and normal human liver tissues by combination of multiple protease digestion and solid phase based labeling. Clin. Proteom. 2014, 11, 26. [Google Scholar] [CrossRef]
- Ang, I.L.; Poon, T.C.W.; Lai, P.B.S.; Chan, A.T.C.; Ngai, S.-M.; Hui, A.Y.; Johnson, P.J.; Sung, J.J.Y. Study of Serum Haptoglobin and Its Glycoforms in the Diagnosis of Hepatocellular Carcinoma: A Glycoproteomic Approach. J. Proteome Res. 2006, 5, 2691–2700. [Google Scholar] [CrossRef]
- Gao, Q.; Zhu, H.; Dong, L.; Shi, W.; Chen, R.; Song, Z.; Huang, C.; Li, J.; Dong, X.; Zhou, Y.; et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 2019, 179, 1240. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Arnold, M.W. Colon Cancer: The Road Traveled. Surg. Oncol. Clin. N. Am. 2018, 27, 15–18. [Google Scholar] [CrossRef]
- Mody, K.; Bekaii-Saab, T. Clinical Trials and Progress in Metastatic Colon Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 349–365. [Google Scholar] [CrossRef]
- Fatemi, S.R.; Pourhoseingholi, M.A.; Asadi, F.; Vahedi, M.; Pasha, S.; Alizadeh, L.; Zali, M.R. Recurrence and Five Year Survival in Colorectal Cancer Patients after Surgery. Iran. J. Cancer Prev. 2015, 8, e3439. [Google Scholar] [CrossRef] [PubMed]
- Ratto, C.; Parello, A.; Donisi, L.; Litta, F. Colon, Rectum and Anus: Anatomic, Physiologic and Diagnostic Bases for Disease Management; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar] [CrossRef]
- Siebenhüner, A.R.; Güller, U.; Warschkow, R. Population-based SEER analysis of survival in colorectal cancer patients with or without resection of lung and liver metastases. BMC Cancer 2020, 20, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A. Controversies in the pathological assessment of colorectal cancer. World J. Gastroenterol. 2014, 20, 9850–9861. [Google Scholar] [CrossRef] [PubMed]
- Kirana, C.; Shi, H.; Laing, E.; Hood, K.; Miller, R.; Bethwaite, P.; Keating, J.; Jordan, T.W.; Hayes, M.; Stubbs, R. Cathepsin D Expression in Colorectal Cancer: From Proteomic Discovery through Validation Using Western Blotting, Immunohistochemistry, and Tissue Microarrays. Int. J. Proteom. 2012, 2012, 1–10. [Google Scholar] [CrossRef]
- Ku, X.; Xu, Y.; Cai, C.; Yang, Y.; Cui, L.; Yan, W. In-Depth Characterization of Mass Spectrometry-Based Proteomic Profiles Revealed Novel Signature Proteins Associated with Liver Metastatic Colorectal Cancers. Anal. Cell. Pathol. 2019, 2019, 7653230. [Google Scholar] [CrossRef]
- Liu, X.; Xu, D.; Liu, Z.; Li, Y.; Zhang, C.; Gong, Y.; Jiang, Y.; Xing, B. THBS1 facilitates colorectal liver metastasis through enhancing epithelial–mesenchymal transition. Clin. Transl. Oncol. 2020, 22, 1730–1740. [Google Scholar] [CrossRef]
- Shen, Z.; Wang, B.; Luo, J.; Jiang, K.; Zhang, H.; Mustonen, H.; Puolakkainen, P.; Zhu, J.; Ye, Y.; Wang, S. Global-scale profiling of differential expressed lysine acetylated proteins in colorectal cancer tumors and paired liver metastases. J. Proteom. 2016, 142, 24–32. [Google Scholar] [CrossRef]
- Van Huizen, N.A.; Braak, R.R.C.V.D.; Doukas, M.; Dekker, L.J.; Ijzermans, J.N.; Luider, T.M. Up-regulation of collagen proteins in colorectal liver metastasis compared with normal liver tissue. J. Biol. Chem. 2019, 294, 281–289. [Google Scholar] [CrossRef]
- Van Huizen, N.A.; Burgers, P.C.; Saintmont, F.; Brocorens, P.; Gerbaux, P.; Stingl, C.; Dekker, L.J.M.; Ijzermans, J.N.; Luider, T.M. Identification of 4-Hydroxyproline at the Xaa Position in Collagen by Mass Spectrometry. J. Proteome Res. 2019, 18, 2045–2051. [Google Scholar] [CrossRef]
- Fahrner, M.; Bronsert, P.; Fichtner-Feigl, S.; Jud, A.; Schilling, O. Proteome biology of primary colorectal carcinoma and corresponding liver metastases. Neoplasia 2021, 23, 1240–1251. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Whittaker, C.A.; Carr, S.A.; Tanabe, K.K.; Hynes, R.O. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014, 14, 518. [Google Scholar] [CrossRef] [PubMed]
- Van Huizen, N.A.; Burgers, P.C.; Van Rosmalen, J.; Doukas, M.; Ijzermans, J.N.M.; Luider, T.M. Down-Regulation of Collagen Hydroxylation in Colorectal Liver Metastasis. Front. Oncol. 2020, 10, 557737. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-K.; Song, M.-J.; Jung, Y.; Lee, W.-S.; Jang, H.H. Proteomic Analysis of Primary Colon Cancer and Synchronous Solitary Liver Metastasis. Cancer Genom. Proteom. 2019, 16, 583–592. [Google Scholar] [CrossRef]
- Voß, H.; Wurlitzer, M.; Smit, D.J.; Ewald, F.; Alawi, M.; Spohn, M.; Indenbirken, D.; Omidi, M.; David, K.; Juhl, H.; et al. Differential regulation of extracellular matrix proteins in three recurrent liver metastases of a single patient with colorectal cancer. Clin. Exp. Metastasis 2020, 37, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Yuzhalin, A.E.; Gordon-Weeks, A.N.; Tognoli, M.L.; Jones, K.; Markelc, B.; Konietzny, R.; Fischer, R.; Muth, A.; O’Neill, E.; Thompson, P.R.; et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat. Commun. 2018, 9, 4783. [Google Scholar] [CrossRef]
- Yang, Q.; Bavi, P.; Wang, J.Y.; Roehrl, M.H. Immuno-proteomic discovery of tumor tissue autoantigens identifies olfactomedin 4, CD11b, and integrin alpha-2 as markers of colorectal cancer with liver metastases. J. Proteom. 2017, 168, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Kirana, C.; Peng, L.; Miller, R.; Keating, J.P.; Glenn, C.; Shi, H.; Jordan, T.W.; Maddern, G.J.; Stubbs, R.S. Combination of laser microdissection, 2D-DIGE and MALDI-TOF MS to identify protein biomarkers to predict colorectal cancer spread. Clin. Proteom. 2019, 16, 3. [Google Scholar] [CrossRef]
- Michal, S.; Tal, G.-L.; Gali, P.; Miki, G.; Elana, B.; Baroch, B.; Hanoch, K.; Irit, B.A.; Riad, H. Characterization of Biomarkers in Colorectal Cancer Liver Metastases as a Prognostic Tool. J. Pers. Med. 2021, 11, 1059. [Google Scholar] [CrossRef]
- Turtoi, A.; Blomme, A.; Debois, D.; Somja, J.; Delvaux, D.; Patsos, G.; Di Valentin, E.; Peulen, O.; Mutijima, E.N.; De Pauw, E.; et al. Organized proteomic heterogeneity in colorectal cancer liver metastases and implications for therapies. Hepatology 2013, 59, 924–934. [Google Scholar] [CrossRef]
- Yang, W.; Shi, J.; Zhou, Y.; Liu, T.; Li, J.; Hong, F.; Zhang, K.; Liu, N. Co-expression Network Analysis Identified Key Proteins in Association with Hepatic Metastatic Colorectal Cancer. Proteom. Clin. Appl. 2019, 13, 1900017. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, Y.; Xu, L.; Zhan, S.; Xiao, Y.; Gao, Y.; Wu, B.; Ge, W. Protein content and functional characteristics of serum-purified exosomes from patients with colorectal cancer revealed by quantitative proteomics. Int. J. Cancer 2016, 140, 900–913. [Google Scholar] [CrossRef]
- Shiromizu, T.; Kume, H.; Ishida, M.; Adachi, J.; Kano, M.; Matsubara, H.; Tomonaga, T. Quantitation of putative colorectal cancer biomarker candidates in serum extracellular vesicles by targeted proteomics. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.-S.; Park, J.O.; Jang, S.C.; Yoon, Y.J.; Jung, J.W.; Choi, D.-Y.; Kim, J.-W.; Kang, J.S.; Park, J.; Hwang, D.; et al. Proteomic analysis of microvesicles derived from human colorectal cancer ascites. Proteomics 2011, 11, 2745–2751. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.; Schiffer, C.A. Acute myeloid leukaemia in adults. Lancet 2013, 381, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, E.; Berven, F.S.; Bartaula-Brevik, S.; Stokowy, T.; Hovland, R.; Vaudel, M.; Døskeland, S.O.; McCormack, E.; Batth, T.S.; Olsen, J.V.; et al. Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia. Cancers 2020, 12, 709. [Google Scholar] [CrossRef]
- Mariani, S.; Trisolini, S.M.; Minotti, C.; Breccia, M.; Cartoni, C.; De Propris, M.S.; Loglisci, G.; Latagliata, R.; Limongi, M.Z.; Testi, A.M.; et al. Very late acute myeloid leukemia relapse: Clinical features, treatment and outcome. Leuk. Lymphoma 2020, 62, 1022–1025. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Ramos, N.R.; Mo, C.C.; Karp, J.E.; Hourigan, C.S. Current Approaches in the Treatment of Relapsed and Refractory Acute Myeloid Leukemia. J. Clin. Med. 2015, 4, 665–695. [Google Scholar] [CrossRef]
- Forthun, R.B.; Aasebø, E.; Rasinger, J.D.; Bedringaas, S.L.; Berven, F.; Selheim, F.; Bruserud, O.; Gjertsen, B.T. Phosphoprotein DIGE profiles reflect blast differentiation, cytogenetic risk stratification, FLT3/NPM1 mutations and therapy response in acute myeloid leukaemia. J. Proteom. 2018, 173, 32–41. [Google Scholar] [CrossRef]
- Foss, E.J.; Radulovic, D.; Stirewalt, D.L.; Radich, J.; Sala-Torra, O.; Pogosova-Agadjanyan, E.L.; Hengel, S.M.; Loeb, K.R.; Deeg, H.J.; Meshinchi, S.; et al. Proteomic Classification of Acute Leukemias by Alignment-Based Quantitation of LC–MS/MS Data Sets. J. Proteome Res. 2012, 11, 5005–5010. [Google Scholar] [CrossRef]
- Aasebø, E.; Forthun, R.B.; Berven, F.; Selheim, F.; Hernandez-Valladares, M. Global Cell Proteome Profiling, Phospho-Signaling and Quantitative Proteomics for Identification of New Biomarkers in Acute Myeloid Leukemia Patients. Curr. Pharm. Biotechnol. 2015, 17, 52–70. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Helmy, M.; Cavalli, F.M.G.; Jin, L.; St-Germain, J.; Karisch, R.; Taylor, P.; Minden, M.D.; Taylor, M.D.; Neel, B.G.; et al. Integrated analysis of proteome, phosphotyrosine-proteome, tyrosine-kinome, and tyrosine-phosphatome in acute myeloid leukemia. Proteomics 2017, 17, 1600361. [Google Scholar] [CrossRef] [PubMed]
- Nepstad, I.; Hatfield, K.J.; Aasebø, E.; Hernandez-Valladares, M.; Brenner, A.K.; Bartaula-Brevik, S.; Berven, F.; Selheim, F.; Skavland, J.; Gjertsen, B.T.; et al. Two acute myeloid leukemia patient subsets are identified based on the constitutive PI3K-Akt-mTOR signaling of their leukemic cells; a functional, proteomic, and transcriptomic comparison. Expert Opin. Ther. Targets 2018, 22, 639–653. [Google Scholar] [CrossRef] [PubMed]
- De Boer, B.; Prick, J.; Pruis, M.G.; Keane, P.; Imperato, M.R.; Jaques, J.; Brouwers-Vos, A.Z.; Hogeling, S.M.; Woolthuis, C.M.; Nijk, M.T.; et al. Prospective Isolation and Characterization of Genetically and Functionally Distinct AML Subclones. Cancer Cell 2018, 34, 674–689. [Google Scholar] [CrossRef]
- Reikvam, H.; Aasebø, E.; Brenner, A.K.; Bartaula-Brevik, S.; Grønningsæter, I.S.; Forthun, R.B.; Hovland, R.; Bruserud, Ø. High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype. J. Clin. Med. 2019, 8, 970. [Google Scholar] [CrossRef]
- Aasebø, E.; Hernandez-Valladares, M.; Selheim, F.; Berven, F.S.; Brenner, A.K.; Bruserud, O. Proteomic Profiling of Primary Human Acute Myeloid Leukemia Cells Does Not Reflect Their Constitutive Release of Soluble Mediators. Proteomes 2018, 7, 1. [Google Scholar] [CrossRef]
- Grønningsæter, I.S.; Reikvam, H.; Aasebø, E.; Bartaula-Brevik, S.; Tvedt, T.H.; Bruserud, O.; Hatfield, K.J. Targeting Cellular Metabolism in Acute Myeloid Leukemia and the Role of Patient Heterogeneity. Cells 2020, 9, 1155. [Google Scholar] [CrossRef]
- Raffel, S.; Klimmeck, D.; Falcone, M.; Demir, A.; Pouya, A.; Zeisberger, P.; Lutz, C.; Tinelli, M.; Bischel, O.; Bullinger, L.; et al. Quantitative proteomics reveals specific metabolic features of acute myeloid leukemia stem cells. Blood 2020, 136, 1507–1519. [Google Scholar] [CrossRef]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]
- Brenner, A.K.; Aasebø, E.; Hernandez-Valladares, M.; Selheim, F.; Berven, F.; Grønningsæter, I.-S.; Bartaula-Brevik, S.; Bruserud, O. The Capacity of Long-Term In Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome. Cancers 2019, 11, 73. [Google Scholar] [CrossRef]
- Aasebø, E.; Berven, F.S.; Hovland, R.; Døskeland, S.O.; Bruserud, O.; Selheim, F.; Hernandez-Valladares, M. The Progression of Acute Myeloid Leukemia from First Diagnosis to Chemoresistant Relapse: A Comparison of Proteomic and Phosphoproteomic Profiles. Cancers 2020, 12, 1466. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, B.; Munje, C.R.; Rastogi, N.; Williamson, A.J.K.; Taylor, S.; Hole, P.S.; Hodges, M.; Doyle, M.; Baker, S.; Gilkes, A.F.; et al. Integrated nuclear proteomics and transcriptomics identifies S100A4 as a therapeutic target in acute myeloid leukemia. Leukemia 2019, 34, 427–440. [Google Scholar] [CrossRef]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Aasebø, E.; Hernandez-Valladares, M.; Hagen, K.M.; Rye, K.P.; Berven, F.S.; Selheim, F.; Reikvam, H.; et al. Effects of insulin and pathway inhibitors on the PI3K-Akt-mTOR phosphorylation profile in acute myeloid leukemia cells. Signal Transduct. Target. Ther. 2019, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.R.; Rücker-Braun, E.; Heidrich, K.; von Bonin, M.; Stölzel, F.; Thiede, C.; Middeke, J.M.; Ehninger, G.; Bornhäuser, M.; Schetelig, J.; et al. Pilot Study on Mass Spectrometry–Based Analysis of the Proteome of CD34+CD123+ Progenitor Cells for the Identification of Potential Targets for Immunotherapy in Acute Myeloid Leukemia. Proteomes 2018, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Braglia, L.; Zavatti, M.; Vinceti, M.; Martelli, A.M.; Marmiroli, S. Deregulated PTEN/PI3K/AKT/mTOR signaling in prostate cancer: Still a potential druggable target? Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2020, 1867, 118731. [Google Scholar] [CrossRef]
- Canesin, G.; Krzyzanowska, A.; Hellsten, R.; Bjartell, A. Cytokines and Janus kinase/signal transducer and activator of transcription signaling in prostate cancer: Overview and therapeutic opportunities. Curr. Opin. Endocr. Metab. Res. 2020, 10, 36–42. [Google Scholar] [CrossRef]
- Uo, T.; Sprenger, C.C.; Plymate, S.R. Androgen Receptor Signaling and Metabolic and Cellular Plasticity During Progression to Castration Resistant Prostate Cancer. Front. Oncol. 2020, 10, 580617. [Google Scholar] [CrossRef]
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2020, 77, 38–52. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Urbanucci, A.; Martin, S.E.; Khan, A.; Mathelier, A.; Thiede, B.; Walker, S.; Mills, I.G. High OGT activity is essential for MYC-driven proliferation of prostate cancer cells. Theranostics 2019, 9, 2183–2197. [Google Scholar] [CrossRef]
- McCann, J.J.; Vasilevskaya, I.A.; Neupane, N.P.; Shafi, A.A.; McNair, C.; Dylgjeri, E.; Mandigo, A.C.; Schiewer, M.J.; Schrecengost, R.S.; Gallagher, P.; et al. USP22 Functions as an Oncogenic Driver in Prostate Cancer by Regulating Cell Proliferation and DNA Repair. Cancer Res 2020, 80, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.M.; Paull, E.O.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Mariscal, J.; Vagner, T.; Kim, M.; Zhou, B.; Chin, A.; Zandian, M.; Freeman, M.R.; You, S.; Zijlstra, A.; Yang, W.; et al. Comprehensive palmitoyl-proteomic analysis identifies distinct protein signatures for large and small cancer-derived extracellular vesicles. J. Extracell. Vesicles 2020, 9, 1764192. [Google Scholar] [CrossRef]
- Nguyen, E.V.; Pereira, B.A.; Lawrence, M.G.; Ma, X.; Rebello, R.J.; Chan, H.; Niranjan, B.; Wu, Y.; Ellem, S.; Guan, X.; et al. Proteomic Profiling of Human Prostate Cancer-Associated Fibroblasts (CAF) Reveals LOXL2-Dependent Regulation of the Tumor Microenvironment. Mol. Cell. Proteom. 2019, 18, 1410–1427. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Liu, M.; Lai, S.; Hou, H.; Diao, T.; Zhang, D.; Wang, M.; Zhang, Y.; Wang, J. Androgen upregulates the palmitoylation of eIF3L in human prostate LNCaP cells. OncoTargets Ther. 2019, 12, 4451–4459. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.S.; Zhang, L.; Berger, A.; Lawrence, M.G.; Song, J.; Niranjan, B.; Davies, R.G.; Lister, N.L.; Sandhu, S.; Rubin, M.; et al. Characterization of the ERG-Regulated Kinome in Prostate Cancer Identifies TNIK as a Potential Therapeutic Target. Neoplasia 2019, 21, 389–400. [Google Scholar] [CrossRef]
- Zhao, H.; Pflug, B.R.; Lai, X.; Wang, M. Pyruvate dehydrogenase alpha 1 as a target of omega-3 polyunsaturated fatty acids in human prostate cancer through a global phosphoproteomic analysis. Proteomics 2016, 16, 2419–2431. [Google Scholar] [CrossRef]
- Faltermeier, C.M.; Drake, J.M.; Clark, P.M.; Smith, B.A.; Zong, Y.; Volpe, C.; Mathis, C.; Morrissey, C.; Castor, B.; Huang, J.; et al. Functional screen identifies kinases driving prostate cancer visceral and bone metastasis. Proc. Natl. Acad. Sci. USA 2015, 113, 172–181. [Google Scholar] [CrossRef]
- Wen, D.; Xu, Z.; Xia, L.; Liu, X.; Tu, Y.; Lei, H.; Wang, W.; Wang, T.; Song, L.; Ma, C.; et al. Important Role of SUMOylation of Spliceosome Factors in Prostate Cancer Cells. J. Proteome Res. 2014, 13, 3571–3582. [Google Scholar] [CrossRef]
- Jiang, N.; Hjorth-Jensen, K.; Hekmat, O.; Iglesias-Gato, D.; Kruse, T.; Wang, C.; Wei, W.; Ke, B.; Yan, B.; Niu, Y.; et al. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene 2014, 34, 2764–2776. [Google Scholar] [CrossRef]
- Toughiri, R.; Li, X.; Du, Q.; Bieberich, C.J. Phosphorylation of NuMA by Aurora-A kinase in PC-3 prostate cancer cells affects proliferation, survival, and interphase NuMA localization. J. Cell. Biochem. 2013, 114, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, J.; Zou, L.; Cui, J.; Su, F.; Jin, J.; Xiao, F.; Liu, M.; Zhao, G. Palmitoylome profiling indicates that androgens regulate the palmitoylation of α-tubulin in prostate cancer-derived LNCaP cells and supernatants. Oncol. Rep. 2019, 42, 2788–2796. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Luan, X.; Zhang, Y.; Robbe-Masselot, C.; Brockhausen, I.; Gao, Y. The expression and functional analysis of the sialyl-T antigen in prostate cancer. Glycoconj. J. 2020, 37, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.J.; Schnaubelt, M.; Hoti, N.; Hu, Y.; Zhou, Y.; Gooya, M.; Zhang, H. Impact of Increased FUT8 Expression on the Extracellular Vesicle Proteome in Prostate Cancer Cells. J. Proteome Res. 2020, 19, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Theurillat, J.-P.P.; Udeshi, N.D.; Errington, W.J.; Svinkina, T.; Baca, S.C.; Pop, M.; Wild, P.J.; Blattner, M.; Groner, A.C.; Rubin, M.A.; et al. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 2014, 346, 85–89. [Google Scholar] [CrossRef]
- Drake, J.M.; Graham, N.A.; Stoyanova, T.; Sedghi, A.; Goldstein, A.S.; Cai, H.; Smith, D.A.; Zhang, H.; Komisopoulou, E.; Huang, J.; et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc. Natl. Acad. Sci. USA 2012, 109, 1643–1648. [Google Scholar] [CrossRef]
- Li, F.; Glinskii, O.V.; Mooney, B.P.; Rittenhouse-Olson, K.; Pienta, K.J.; Glinsky, V.V. Cell surface Thomsen-Friedenreich proteome profiling of metastatic prostate cancer cells reveals potential link with cancer stem cell-like phenotype. Oncotarget 2017, 8, 98598–98608. [Google Scholar] [CrossRef]
- Ino, Y.; Arakawa, N.; Ishiguro, H.; Uemura, H.; Kubota, Y.; Hirano, H.; Toda, T. Phosphoproteome analysis demonstrates the potential role of THRAP3 phosphorylation in androgen-independent prostate cancer cell growth. Proteomics 2016, 16, 1069–1078. [Google Scholar] [CrossRef]
- Gulati, T.; Huang, C.; Caramia, F.; Raghu, D.; Paul, P.J.; Goode, R.J.; Keam, S.P.; Williams, S.G.; Haupt, S.; Kleifeld, O.; et al. Proteotranscriptomic Measurements of E6-Associated Protein (E6AP) Targets in DU145 Prostate Cancer Cells. Mol. Cell. Proteom. 2018, 17, 1170–1183. [Google Scholar] [CrossRef]
- Gao, Y.; Ha, Y.-S.; Kwon, T.G.; Cho, Y.-C.; Lee, S.; Lee, J.N. Characterization of Kinase Expression Related to Increased Migration of PC-3M Cells Using Global Comparative Phosphoproteome Analysis. Cancer Genom. Proteom. 2020, 17, 543–553. [Google Scholar] [CrossRef]
- Höti, N.; Lih, T.-S.; Pan, J.; Zhou, Y.; Yang, G.; Deng, A.; Chen, L.; Dong, M.; Yang, R.-B.; Tu, C.-F.; et al. A Comprehensive Analysis of FUT8 Overexpressing Prostate Cancer Cells Reveals the Role of EGFR in Castration Resistance. Cancers 2020, 12, 468. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Yang, W.; Steen, H.; Freeman, M.R.; Hemler, M.E. Antioxidant functions of DHHC3 suppress anti-cancer drug activities. Cell. Mol. Life Sci. 2020, 78, 2341–2353. [Google Scholar] [CrossRef]
- Hoti, N.; Yang, S.; Hu, Y.; Shah, P.; Haffner, M.C.; Zhang, H. Overexpression of α (1,6) fucosyltransferase in the development of castration-resistant prostate cancer cells. Prostate Cancer Prostatic Dis. 2018, 21, 137–146. [Google Scholar] [CrossRef]
- Lee, B.Y.; Hochgräfe, F.; Lin, H.-M.; Castillo, L.; Wu, J.; Raftery, M.J.; Shreeve, S.M.; Horvath, L.G.; Daly, R.J. Phosphoproteomic Profiling Identifies Focal Adhesion Kinase as a Mediator of Docetaxel Resistance in Castrate-Resistant Prostate Cancer. Mol. Cancer Ther. 2014, 13, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Sheikhpour, M.; Ahangari, G.; Sadeghizadeh, M.; Deezagi, A. A Novel Report of Apoptosis in Human Lung Carcinoma Cells Using Selective Agonist of D2-Like Dopamine Receptors: A New Approach for the Treatment of Human Non-Small Cell Lung Cancer. Int. J. Immunopathol. Pharmacol. 2013, 26, 393–402. [Google Scholar] [CrossRef]
- Bronte, G.; Rizzo, S.; La Paglia, L.; Adamo, V.; Siragusa, S.; Ficorella, C.; Santini, D.; Bazan, V.; Colucci, G.; Gebbia, N.; et al. Driver mutations and differential sensitivity to targeted therapies: A new approach to the treatment of lung adenocarcinoma. Cancer Treat. Rev. 2010, 36, S21–S29. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.M.; VanderMeer, R. Delays in the diagnosis of lung cancer. J. Thorac. Dis. 2011, 3, 183–188. [Google Scholar] [CrossRef]
- Strimbu, K.; Tavel, J.A. What Are Biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466. [Google Scholar] [CrossRef]
- An, T.; Qin, S.; Sun, D.; Huang, Y.; Hu, Y.; Li, S.; Zhang, H.; Li, B.; Situ, B.; Lie, L.; et al. Unique Protein Profiles of Extracellular Vesicles as Diagnostic Biomarkers for Early and Advanced Non-Small Cell Lung Cancer. Proteomics 2019, 19, e1800160. [Google Scholar] [CrossRef]
- Geary, B.; Walker, M.J.; Snow, J.T.; Lee, D.C.H.; Pernemalm, M.; Maleki-Dizaji, S.; Azadbakht, N.; Apostolidou, S.; Barnes, J.; Krysiak, P.; et al. Identification of a Biomarker Panel for Early Detection of Lung Cancer Patients. J. Proteome Res. 2019, 18, 3369–3382. [Google Scholar] [CrossRef]
- Li, W.; Zheng, H.; Qin, H.; Liu, G.; Ke, L.; Li, Y.; Li, N.; Zhong, X. Exploration of differentially expressed plasma proteins in patients with lung adenocarcinoma using iTRAQ-coupled 2D LC-MS/MS. Clin. Respir. J. 2018, 12, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Sabrkhany, S.; Kuijpers, M.J.E.; Knol, J.C.; Olde Damink, S.W.M.; Dingemans, A.C.; Verheul, H.M.; Piersma, S.R.; Pham, T.V.; Griffioen, A.W.; Oude Egbrink, M.G.A.; et al. Exploration of the platelet proteome in patients with early-stage cancer. J. Proteom. 2018, 177, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Kong, Y.; Wang, X.; Li, W.; Chen, S.; Wang, L.; Wang, C.; Zhang, Q. LC-MS/MS-Based Quantitative Proteomics Analysis of Different Stages of Non-Small-Cell Lung Cancer. BioMed Res. Int. 2021, 2021, 5561569. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Bin Kim, W.; Davis, A.A.; Park, L.C.; Anker, J.F.; Simon, N.I.; Rhee, K.; Song, J.; Cho, A.; Chang, S.; et al. Mass spectrometry-based serum proteomic signature as a potential biomarker for survival in patients with non-small cell lung cancer receiving immunotherapy. Transl. Lung Cancer Res. 2020, 9, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Hummelink, K.; Hurkmans, D.P.; Niemeijer, A.-L.N.; Monkhorst, K.; Roder, J.; Oliveira, C.; Roder, H.; Aerts, J.G.; Smit, E.F. A Serum Protein Classifier Identifying Patients with Advanced Non–Small Cell Lung Cancer Who Derive Clinical Benefit from Treatment with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2020, 26, 5188–5197. [Google Scholar] [CrossRef]
- Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 15 August 2021).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Haque, R.; Ahmed, S.A.; Inzhakova, G.; Shi, J.; Avila, C.; Polikoff, J.; Bernstein, L.; Enger, S.M.; Press, M.F. Impact of Breast Cancer Subtypes and Treatment on Survival: An Analysis Spanning Two Decades. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1848–1855. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13 Pt 1, 4429–4434. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Hass, R. Breast Carcinoma: From Initial Tumor Cell Detachment to Settlement at Secondary Sites. BioMed Res. Int. 2017, 2017, 8534371. [Google Scholar] [CrossRef]
- Velloso, F.J.; Bianco, A.F.R.; O Farias, J.; Torres, N.E.; Ferruzo, P.Y.; Anschau, V.; Jesus-Ferreira, H.C.; Chang, T.H.-T.; Sogayar, M.; Zerbini, L.F.; et al. The crossroads of breast cancer progression: Insights into the modulation of major signaling pathways. OncoTargets Ther. 2017, 10, 5491–5524. [Google Scholar] [CrossRef]
- Brenton, J.D.; Carey, L.A.; Ahmed, A.A.; Caldas, C. Molecular Classification and Molecular Forecasting of Breast Cancer: Ready for Clinical Application? J. Clin. Oncol. 2005, 23, 7350–7360. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929. [Google Scholar] [PubMed]
- Abramson, V.G.; Lehmann, B.D.; Ballinger, T.J.; Pietenpol, J.A. Subtyping of triple-negative breast cancer: Implications for therapy. Cancer 2015, 121, 8–16. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Whelan, S.A.; Lu, M.; Shen, D.; Chung, D.U.; Saxton, R.E.; Faull, K.F.; Whitelegge, J.P.; Chang, H.R. Proteomic-Based Biosignatures in Breast Cancer Classification and Prediction of Therapeutic Response. Int. J. Proteom. 2011, 2011, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Campone, M.; Valo, I.; Jézéquel, P.; Moreau, M.; Boissard, A.; Campion, L.; Loussouarn, D.; Verriele, V.; Coqueret, O.; Guette, C. Prediction of Recurrence and Survival for Triple-Negative Breast Cancer (TNBC) by a Protein Signature in Tissue Samples. Mol. Cell. Proteom. 2015, 14, 2936–2946. [Google Scholar] [CrossRef]
- Suman, S.; Basak, T.; Gupta, P.; Mishra, S.; Kumar, V.; Sengupta, S.; Shukla, Y. Quantitative proteomics revealed novel proteins associated with molecular subtypes of breast cancer. J. Proteom. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Sun, T.; Aceto, N.; Meerbrey, K.L.; Kessler, J.D.; Zhou, C.; Migliaccio, I.; Nguyen, D.X.; Pavlova, N.N.; Botero, M.; Huang, J.; et al. Activation of Multiple Proto-Oncogenic Tyrosine Kinases in Breast Cancer via Loss of the PTPN12 Phosphatase. Cell 2011, 144, 703–718. [Google Scholar] [CrossRef]
- Semaan, S.M.; Wang, X.; Stewart, P.A.; Marshall, A.G.; Sang, Q.-X.A. Differential phosphopeptide expression in a benign breast tissue, and triple-negative primary and metastatic breast cancer tissues from the same African-American woman by LC-LTQ/FT-ICR mass spectrometry. Biochem. Biophys. Res. Commun. 2011, 412, 127–131. [Google Scholar] [CrossRef]
- Lawrence, R.T.; Perez, E.; Hernández, D.; Miller, C.P.; Haas, K.M.; Irie, H.Y.; Lee, S.-I.; Blau, C.A.; Villén, J. The Proteomic Landscape of Triple-Negative Breast Cancer. Cell Rep. 2015, 11, 630–644. [Google Scholar] [CrossRef]
- Liu, N.Q.; Stingl, C.; Look, M.P.; Smid, M.; Braakman, R.B.; De Marchi, T.; Sieuwerts, A.M.; Span, P.; Sweep, F.; Linderholm, B.; et al. Comparative Proteome Analysis Revealing an 11-Protein Signature for Aggressive Triple-Negative Breast Cancer. J. Natl. Cancer Inst. 2014, 106, djt376. [Google Scholar] [CrossRef]
- Mittal, L.; Camarillo, I.G.; Varadarajan, G.S.; Srinivasan, H.; Aryal, U.K.; Sundararajan, R. High-throughput, Label-Free Quantitative Proteomic Studies of the Anticancer Effects of Electrical Pulses with Turmeric Silver Nanoparticles: An in vitro Model Study. Sci. Rep. 2020, 10, 7258. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.Q.; De Marchi, T.; Timmermans, A.M.; Beekhof, R.; Trapman-Jansen, A.M.; Foekens, R.; Look, M.P.; van Deurzen, C.H.M.; Span, P.; Sweep, F.; et al. Ferritin Heavy Chain in Triple Negative Breast Cancer: A Favorable Prognostic Marker that Relates to a Cluster of Differentiation 8 Positive (CD8+) Effector T-cell Response. Mol. Cell. Proteom. 2014, 13, 1814–1827. [Google Scholar] [CrossRef]
- Wu, X.; Zahari, M.S.; Ma, B.; Liu, R.; Renuse, S.; Sahasrabuddhe, N.A.; Chen, L.; Chaerkady, R.; Kim, M.-S.; Zhong, J.; et al. Global phosphotyrosine survey in triple-negative breast cancer reveals activation of multiple tyrosine kinase signaling pathways. Oncotarget 2015, 6, 29143–29160. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Albrechtsen, R.; Kronqvist, P.; Cox, J.; Mann, M.; Geiger, T. Proteomic maps of breast cancer subtypes. Nat. Commun. 2016, 7, 10259. [Google Scholar] [CrossRef]
- Koh, E.-Y.; You, J.-E.; Jung, S.-H.; Kim, P.-H. Biological Functions and Identification of Novel Biomarker Expressed on the Surface of Breast Cancer-Derived Cancer Stem Cells via Proteomic Analysis. Mol. Cells 2020, 43, 384–396. [Google Scholar] [CrossRef]
- Rezaul, K.; Thumar, J.K.; Lundgren, D.H.; Eng, J.K.; Claffey, K.P.; Wilson, L.; Han, D.K. Differential Protein Expression Profiles in Estrogen Receptor-Positive and -Negative Breast Cancer Tissues Using Label-Free Quantitative Proteomics. Genes Cancer 2010, 1, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Pozo, A.; Ferrer, N.I.; Ciruelos, E.; López-Vacas, R.; Martínez, F.G.; Espinosa, E.; Vara, J.F. Shotgun proteomics of archival triple-negative breast cancer samples. Proteom. Clin. Appl. 2013, 7, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Imielinski, M.B.; Rejtar, T.; Richardson, E.A.; Thakur, D.; Sgroi, D.C.; Karger, B.L. In Situ Proteomic Analysis of Human Breast Cancer Epithelial Cells Using Laser Capture Microdissection: Annotation by Protein Set Enrichment Analysis and Gene Ontology. Mol. Cell. Proteom. 2010, 9, 2529–2544. [Google Scholar] [CrossRef]
- De Marchi, T.; Liu, N.Q.; Stingl, C.; Timmermans, M.A.; Smid, M.; Look, M.P.; Tjoa, M.; Braakman, R.B.; Opdam, M.; Linn, S.C.; et al. 4-protein signature predicting tamoxifen treatment outcome in recurrent breast cancer. Mol. Oncol. 2015, 10, 24–39. [Google Scholar] [CrossRef]
- Do, M.; Kim, H.; Yeo, I.; Lee, J.; Park, I.A.; Ryu, H.S.; Kim, Y. Clinical Application of Multiple Reaction Monitoring-Mass Spectrometry to Human Epidermal Growth Factor Receptor 2 Measurements as a Potential Diagnostic Tool for Breast Cancer Therapy. Clin. Chem. 2020, 66, 1339–1348. [Google Scholar] [CrossRef]
- Xu, Y.; Zhuo, J.; Duan, Y.; Shi, B.; Chen, X.; Zhang, X.; Xiao, L.; Lou, J.; Huang, R.; Zhang, Q.; et al. Construction of protein profile classification model and screening of proteomic signature of acute leukemia. Int. J. Clin. Exp. Pathol. 2014, 7, 5569–5581. [Google Scholar] [PubMed]
- Aivado, M.; Spentzos, D.; Germing, U.; Alterovitz, G.; Meng, X.-Y.; Grall, F.; Giagounidis, A.A.N.; Klement, G.; Steidl, U.; Otu, H.H.; et al. Serum proteome profiling detects myelodysplastic syndromes and identifies CXC chemokine ligands 4 and 7 as markers for advanced disease. Proc. Natl. Acad. Sci. USA 2007, 104, 1307–1312. [Google Scholar] [CrossRef]
- Braoudaki, M.; Tzortzatou-Stathopoulou, F.; Anagnostopoulos, A.K.; Papathanassiou, C.; Vougas, K.; Karamolegou, K.; Tsangaris, G.T. Proteomic analysis of childhood de novo acute myeloid leukemia and myelodysplastic syndrome/AML: Correlation to molecular and cytogenetic analyses. Amino Acids 2010, 40, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Kornblau, S.M.; Tibes, R.; Qiu, Y.H.; Chen, W.; Kantarjian, H.M.; Andreeff, M.; Coombes, K.R.; Mills, G.B. Functional proteomic profiling of AML predicts response and survival. Blood 2009, 113, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Hoff, F.W.; Hu, C.W.; Qiu, Y.; Ligeralde, A.; Yoo, S.-Y.; Scheurer, M.E.; de Bont, E.S.; Qutub, A.A.; Kornblau, S.M.; Horton, T.M. Recurrent Patterns of Protein Expression Signatures in Pediatric Acute Lymphoblastic Leukemia: Recognition and Therapeutic Guidance. Mol. Cancer Res. 2018, 16, 1263–1274. [Google Scholar] [CrossRef]
- Hoff, F.W.; Hu, C.W.; Qiu, Y.; Ligeralde, A.; Yoo, S.-Y.; Mahmud, H.; de Bont, E.S.; Qutub, A.A.; Horton, T.M.; Kornblau, S.M. Recognition of Recurrent Protein Expression Patterns in Pediatric Acute Myeloid Leukemia Identified New Therapeutic Targets. Mol. Cancer Res. 2018, 16, 1275–1286. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Zhang, N.; Qiu, Y.H.; Post, S.; Creighton, C.J.; Cortes, J.; Coombes, K.R.; Kornblau, S.M. Loss of TRIM62 Expression Is an Independent Adverse Prognostic Factor in Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2014, 15, 115–127. [Google Scholar] [CrossRef]
- Butler, J.S.; Qiu, Y.H.; Zhang, N.; Yoo, S.-Y.; Coombes, K.R.; Dent, S.Y.R.; Kornblau, S.M. Low expression of ASH2L protein correlates with a favorable outcome in acute myeloid leukemia. Leuk. Lymphoma 2016, 58, 1207–1218. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Singh, N.; Qiu, Y.; Chen, W.; Zhang, N.; Coombes, K.R. Highly Phosphorylated FOXO3A Is an Adverse Prognostic Factor in Acute Myeloid Leukemia. Clin. Cancer Res. 2010, 16, 1865–1874. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Qiu, Y.H.; Zhang, N.; Singh, N.; Faderl, S.; Ferrajoli, A.; York, H.; Qutub, A.; Coombes, K.; Watson, D.K. Abnormal expression of FLI1 protein is an adverse prognostic factor in acute myeloid leukemia. Blood 2011, 118, 5604–5612. [Google Scholar] [CrossRef]
- Pierce, A.; Whetton, A.D.; Meyer, S.; Ravandi-Kashani, F.; Borthakur, G.; Coombes, K.R.; Zhang, N.; Kornblau, S. Transglutaminase 2 expression in acute myeloid leukemia: Association with adhesion molecule expression and leukemic blast motility. Proteomics 2013, 13, 2216–2224. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Hu, C.W.; Qiu, Y.; Ruvolo, V.R.; Go, R.L.; Hubner, S.E.; Coombes, K.R.; Andreeff, M.; Qutub, A.A.; Kornblau, S.M. LGALS3 is connected to CD74 in a previously unknown protein network that is associated with poor survival in patients with AML. EBioMedicine 2019, 44, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, Y.; Yao, Y.; Fang, Z.; Miao, Q.R.; Ye, M. Quantitative proteomic and phosphoproteomic studies reveal novel 5-fluorouracil resistant targets in hepatocellular carcinoma. J. Proteom. 2019, 208, 103501. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-T.; Liao, L.-Z.; Lu, C.-H.; Huang, Y.-H.; Lin, Y.-K.; Lin, J.-H.; Chow, L.-P. Quantitative phosphoproteomic analysis identifies the potential therapeutic target EphA2 for overcoming sorafenib resistance in hepatocellular carcinoma cells. Exp. Mol. Med. 2020, 52, 497–513. [Google Scholar] [CrossRef]
- Melas, I.N.; Lauffenburger, U.A.; Alexopoulos, L.G. Identification of signaling pathways related to drug efficacy in hepatocellular carcinoma via integration of phosphoproteomic, genomic and clinical data. In Proceedings of the 13th IEEE International Conference on BioInformatics and BioEngineering, Chania, Greece, 10–13 November 2013. [Google Scholar] [CrossRef]
- Yu, L.; Shen, J.; Mannoor, K.; Guarnera, M.; Jiang, F. Identification of ENO1 as a Potential Sputum Biomarker for Early-Stage Lung Cancer by Shotgun Proteomics. Clin. Lung Cancer 2014, 15, 372–378. [Google Scholar] [CrossRef]
- Füzéry, A.K.; Levin, J.; Chan, M.M.; Chan, D.W. Translation of proteomic biomarkers into FDA approved cancer diagnostics: Issues and challenges. Clin. Proteom. 2013, 10, 13. [Google Scholar] [CrossRef]
- Sukari, A.; Nagasaka, M.; Wakeling, E. EGFR -Mutant Non–Small Cell Lung Cancer in the Era of Precision Medicine: Importance of Germline EGFR T790M Testing. J. Natl. Compr. Cancer Netw. 2017, 15, 1188–1192. [Google Scholar] [CrossRef]
- Jänne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L.; et al. AZD9291 in EGFR Inhibitor–Resistant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Zhang, Z.; Bast, R.; Yu, Y.; Li, J.; Sokoll, L.J.; Rai, A.J.; Rosenzweig, J.M.; Cameron, B.; Wang, Y.Y.; Meng, X.-Y.; et al. Three Biomarkers Identified from Serum Proteomic Analysis for the Detection of Early Stage Ovarian Cancer. Cancer Res. 2004, 64, 5882–5890. [Google Scholar] [CrossRef]
- Bhawal, R.; Oberg, A.L.; Zhang, S.; Kohli, M. Challenges and Opportunities in Clinical Applications of Blood-Based Proteomics in Cancer. Cancers 2020, 12, 2428. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Gies, A.; Weigl, K.; Tikk, K.; Benner, A.; Schrotz-King, P.; Borchers, C.H.; Brenner, H. Evaluation and Validation of Plasma Proteins Using Two Different Protein Detection Methods for Early Detection of Colorectal Cancer. Cancers 2019, 11, 1426. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Alborn, W.E.; Slebos, R.J.C.; Liebler, D.C. Comparison of Protein Immunoprecipitation-Multiple Reaction Monitoring with ELISA for Assay of Biomarker Candidates in Plasma. J. Proteome Res. 2013, 12, 5996–6003. [Google Scholar] [CrossRef] [PubMed]
- Larkin, S.E.T.; E Johnston, H.; Jackson, T.R.; Jamieson, D.G.; I Roumeliotis, T.; I Mockridge, C.; Michael, A.; Manousopoulou, A.; Papachristou, E.K.; Brown, M.D.; et al. Detection of candidate biomarkers of prostate cancer progression in serum: A depletion-free 3D LC/MS quantitative proteomics pilot study. Br. J. Cancer 2016, 115, 1078–1086. [Google Scholar] [CrossRef]
- Tsaur, I.; Thurn, K.; Juengel, E.; Gust, K.M.; Borgmann, H.; Mager, R.; Bartsch, G.; Oppermann, E.; Ackermann, H.; Nelson, K.; et al. sE-cadherin serves as a diagnostic and predictive parameter in prostate cancer patients. J. Exp. Clin. Cancer Res. 2015, 34, 43. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, F.; Wang, F.; Li, C.; Meng, G.; Duan, H.; Ma, Q.; Zhang, W. Serum peptidome profiling analysis for the identification of potential biomarkers in cervical intraepithelial neoplasia patients. Biochem. Biophys. Res. Commun. 2015, 465, 476–480. [Google Scholar] [CrossRef]
- De la Rosa, A.B.; Lugo-Melchor, O.; Briones-Cerecero, E.; Chagolla-López, A.; De León-Rodríguez, A.; Santos, L.; Vázquez-Ortiz, G.; Salcedo, M. Analysis of human serum from women affected by cervical lesions. J. Exp. Ther. Oncol. 2008, 7, 65–72. [Google Scholar]
- Dytfeld, D.; Luczak, M.; Wrobel, T.; Usnarska-Zubkiewicz, L.; Brzezniakiewicz, K.; Jamroziak, K.; Giannopoulos, K.; Przybylowicz-Chalecka, A.; Ratajczak, B.; Czerwinska-Rybak, J.; et al. Comparative proteomic profiling of refractory/relapsed multiple myeloma reveals biomarkers involved in resistance to bortezomib-based therapy. Oncotarget 2016, 7, 56726–56736. [Google Scholar] [CrossRef]
- Harshman, S.W.; Canella, A.; Ciarlariello, P.D.; Agarwal, K.; Branson, O.E.; Rocci, A.; Cordero, H.; Phelps, M.A.; Hade, E.M.; Dubovsky, J.A.; et al. Proteomic characterization of circulating extracellular vesicles identifies novel serum myeloma associated markers. J. Proteom. 2016, 136, 89–98. [Google Scholar] [CrossRef]
- Zhang, H.-T.; Tian, E.-B.; Chen, Y.-L.; Deng, H.-T.; Wang, Q.-T. Proteomic Analysis for Finding Serum Pathogenic Factors and Potential Biomarkers in Multiple Myeloma. Chin. Med. J. 2015, 128, 1108–1113. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- International Society of Hematology. FDA Approves New Drug for Chronic Lymphocytic Leukemia in Patients with a Specific Chromosomal Abnormality. Available online: https://ishworld.org/1/news/35/fda-approves-new-drug-for-chronic-lymphocytic-leukemia-in-patients-with-a-specif (accessed on 1 October 2022).
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Vakoc, C.R. Targeting Cancer Cells with BET Bromodomain Inhibitors. Cold Spring Harb. Perspect. Med. 2017, 7, a026674. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, G.; Belkina, A.C.; Denis, G.V. Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today: Technol. 2016, 19, 45–50. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 133. [Google Scholar] [CrossRef]
- Bai, L.; Smith, D.C.; Wang, S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol. Ther. 2014, 144, 82–95. [Google Scholar] [CrossRef]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.A.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.G.; Vergara, D.; et al. A HGF/cMET Autocrine Loop Is Operative in Multiple Myeloma Bone Marrow Endothelial Cells and May Represent a Novel Therapeutic Target. Clin. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef]
- Armstrong, H.K.; Gillis, J.L.; Johnson, I.R.D.; Nassar, Z.D.; Moldovan, M.; Levrier, C.; Sadowski, M.C.; Chin, M.Y.; Guns, E.S.T.; Tarulli, G.; et al. Dysregulated fibronectin trafficking by Hsp90 inhibition restricts prostate cancer cell invasion. Sci. Rep. 2018, 8, 2090. [Google Scholar] [CrossRef]
- Roolf, C.; Dybowski, N.; Sekora, A.; Mueller, S.; Knuebel, G.; Tebbe, A.; Escobar, H.M.; Godl, K.; Junghanss, C.; Schaab, C. Phosphoproteome Analysis Reveals Differential Mode of Action of Sorafenib in Wildtype and Mutated FLT3 Acute Myeloid Leukemia (AML) Cells. Mol. Cell. Proteom. 2017, 16, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.C.; Fahrmann, J.F.; Celiktas, M.; Aguilar, M.; Marini, K.D.; Jolly, M.K.; Katayama, H.; Wang, H.; Murage, E.N.; Dennison, J.B.; et al. MCAM Mediates Chemoresistance in Small-Cell Lung Cancer via the PI3K/AKT/SOX2 Signaling Pathway. Cancer Res 2017, 77, 4414–4425. [Google Scholar] [CrossRef] [PubMed]
- Lanning, B.R.; Whitby, L.R.; Dix, M.M.; Douhan, J.; Gilbert, A.M.; Hett, E.C.; Johnson, T.O.; Joslyn, C.M.; Kath, J.C.; Niessen, S.; et al. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat. Chem. Biol. 2014, 10, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Deane, F.M.; Lin, A.J.S.; Hains, P.G.; Pilgrim, S.L.; Robinson, P.J.; McCluskey, A. FD5180, a Novel Protein Kinase Affinity Probe, and the Effect of Bead Loading on Protein Kinase Identification. ACS Omega 2017, 2, 3828–3838. [Google Scholar] [CrossRef]
- Parker, C.G.; Galmozzi, A.; Wang, Y.; Correia, B.E.; Sasaki, K.; Joslyn, C.M.; Kim, A.S.; Cavallaro, C.L.; Lawrence, R.M.; Johnson, S.R.; et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 2017, 168, 527–541. [Google Scholar] [CrossRef]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.-A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef]
- Médard, G.; Pachl, F.; Ruprecht, B.; Klaeger, S.; Heinzlmeir, S.; Helm, D.; Qiao, H.; Ku, X.; Wilhelm, M.; Kuehne, T.; et al. Optimized Chemical Proteomics Assay for Kinase Inhibitor Profiling. J. Proteome Res. 2015, 14, 1574–1586. [Google Scholar] [CrossRef]
- Wong, G.Y.M.; Diakos, C.; Hugh, T.J.; Molloy, M.P. Proteomic Profiling and Biomarker Discovery in Colorectal Liver Metastases. Int. J. Mol. Sci. 2022, 23, 6091. [Google Scholar] [CrossRef]
- Schaffer, L.V.; Millikin, R.J.; Miller, R.M.; Anderson, L.C.; Fellers, R.T.; Ge, Y.; Kelleher, N.L.; LeDuc, R.; Liu, X.; Payne, S.H.; et al. Identification and Quantification of Proteoforms by Mass Spectrometry. Proteomics 2019, 19, e1800361. [Google Scholar] [CrossRef]
- Zhang, H.; Ge, Y. Comprehensive Analysis of Protein Modifications by Top-Down Mass Spectrometry. Circ. Cardiovasc. Genet. 2011, 4, 711. [Google Scholar] [CrossRef]
- Ntai, I.; Fornelli, L.; DeHart, C.J.; Hutton, J.E.; Doubleday, P.F.; LeDuc, R.D.; van Nispen, A.J.; Fellers, R.T.; Whiteley, G.; Boja, E.S.; et al. Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk. Proc. Natl. Acad. Sci. USA 2018, 115, 4140–4145. [Google Scholar] [CrossRef]
- Pandeswari, P.B.; Sabareesh, V. Middle-down approach: A choice to sequence and characterize proteins/proteomes by mass spectrometry. RSC Adv. 2019, 9, 313–344. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Le Large, T.Y.S.; El Hassouni, B.; Funel, N.; Kok, B.; Piersma, S.R.; Pham, T.V.; Olive, K.; Kazemier, G.; Van Laarhoven, H.W.; Jimenez, C.R.; et al. Proteomic analysis of gemcitabine-resistant pancreatic cancer cells reveals that microtubule-associated protein 2 upregulation associates with taxane treatment. Ther. Adv. Med. Oncol. 2019, 11, 1758835919841233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Maity, T.K.; Ross, K.E.; Qi, Y.; Cultraro, C.M.; Bahta, M.; Pitts, S.; Keswani, M.; Gao, S.; Nguyen, K.D.P.; et al. Alterations in the Global Proteome and Phosphoproteome in Third Generation EGFR TKI Resistance Reveal Drug Targets to Circumvent Resistance. Cancer Res 2021, 81, 3051–3066. [Google Scholar] [CrossRef] [PubMed]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and Their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Brandi, J.; Dando, I.; Pozza, E.D.; Biondani, G.; Jenkins, R.; Elliott, V.; Park, K.; Fanelli, G.; Zolla, L.; Costello, E.; et al. Proteomic analysis of pancreatic cancer stem cells: Functional role of fatty acid synthesis and mevalonate pathways. J. Proteom. 2017, 150, 310–322. [Google Scholar] [CrossRef]
- Finkernagel, F.; Reinartz, S.; Schuldner, M.; Malz, A.; Jansen, J.M.; Wagner, U.; Worzfeld, T.; Graumann, J.; Von Strandmann, E.P.; Müller, R. Dual-platform affinity proteomics identifies links between the recurrence of ovarian carcinoma and proteins released into the tumor microenvironment. Theranostics 2019, 9, 6601–6617. [Google Scholar] [CrossRef]
- Brunner, A.; Thielert, M.; Vasilopoulou, C.; Ammar, C.; Coscia, F.; Mund, A.; Hoerning, O.B.; Bache, N.; Apalategui, A.; Lubeck, M.; et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Biol. 2022, 18, e10798. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef]
- Sun, Y.V.; Hu, Y.-J. Integrative Analysis of Multi-Omics Data for Discovery and Functional Studies of Complex Human Diseases. Adv. Genet. 2016, 93, 147–190. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ma, D.; Zhao, S.; Suo, C.; Shi, J.; Xue, M.-Z.; Ruan, M.; Wang, H.; Zhao, J.; Li, Q.; et al. Multi-Omics Profiling Reveals Distinct Microenvironment Characterization and Suggests Immune Escape Mechanisms of Triple-Negative Breast Cancer. Clin. Cancer Res. 2019, 25, 5002–5014. [Google Scholar] [CrossRef] [PubMed]




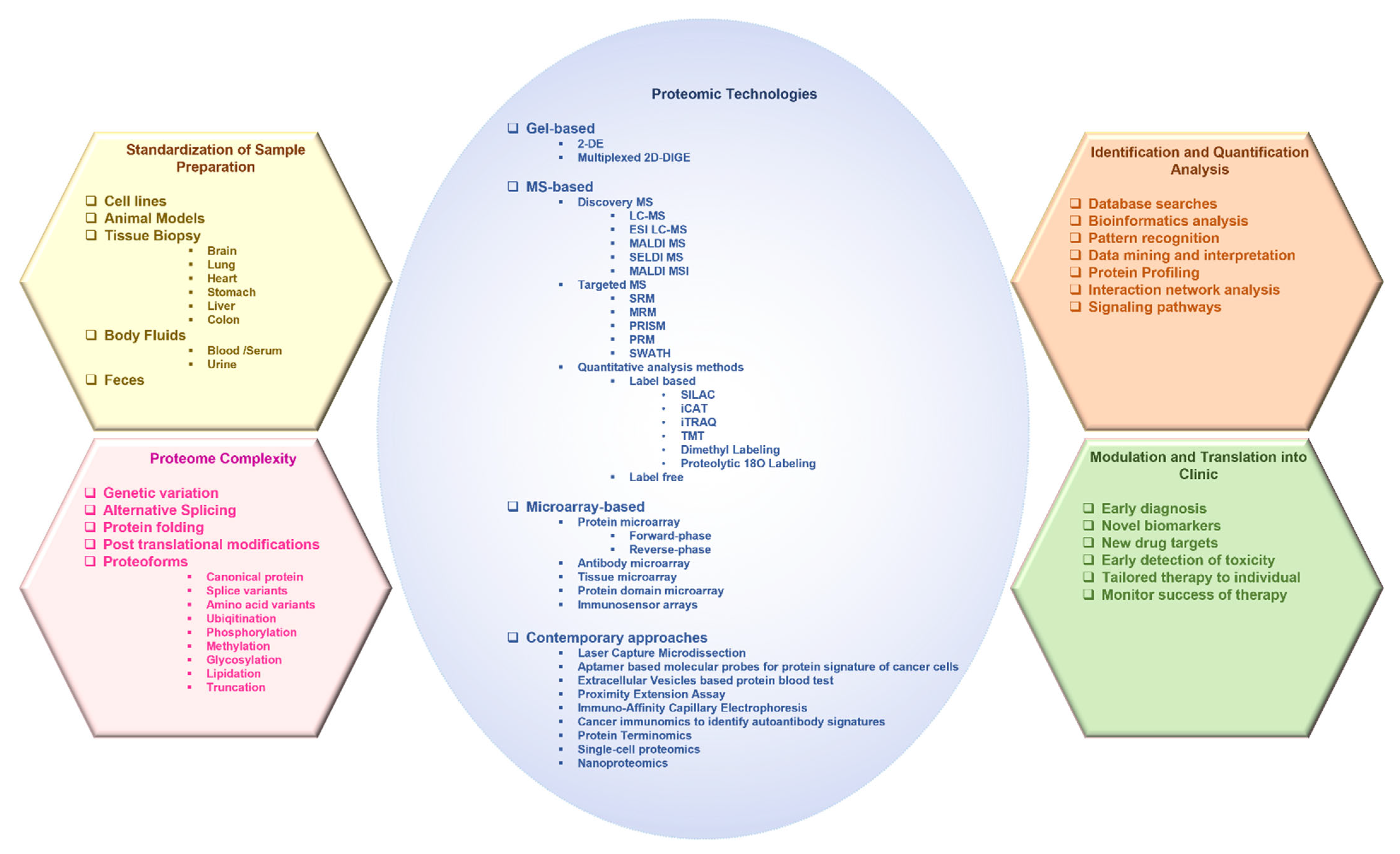{kind=link}
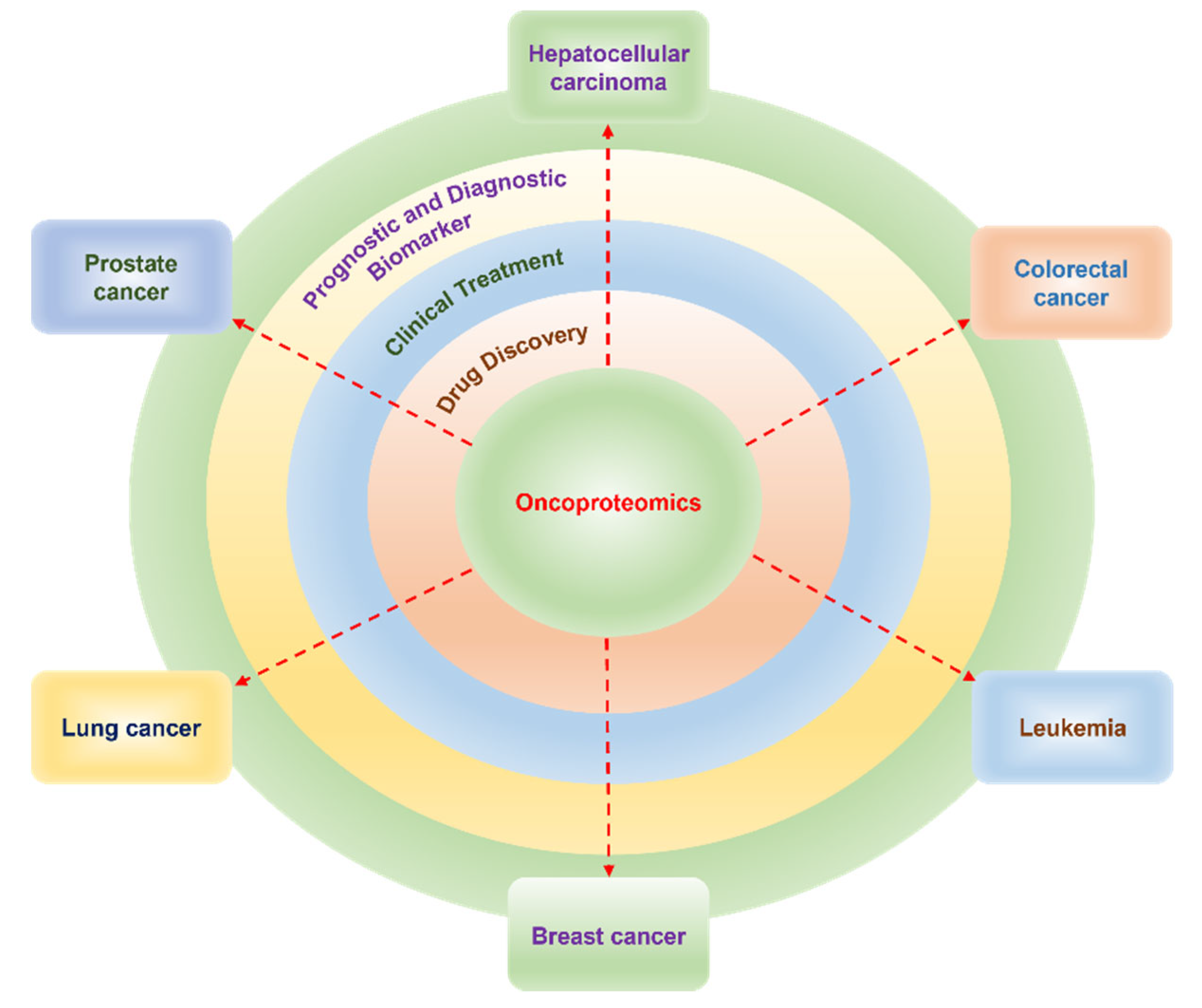{kind=link}
{kind=link}
| Reference | Proteomics Techniques | Biospecimen | Key Findings |
|---|---|---|---|
| Cao et al. [373] | Glycopeptide enrichment methods: hydrophilic affinity (HA) and hydrazide chemistry (HC) were used to complement LC-MS/MS | Human HCC cells | A total of 300 glycosylation sites within 194 glycoproteins were identified |
| Song et al. [374] | Pseudo triplex dimethyl labeling approach coupled with online RP-SCX-RP LC-MS/MS | Human HCC and normal liver tissues | A total of 1934 phosphopeptides from 1033 phosphoproteins were identified |
| Zhang et al. [375] | Lectin affinity chromatography (LAC)-nLC-ESI-MS/MS | Human HCC serum samples | A biomarker for postoperative recurrence of Quiescin Sulfhydryl Oxidase 1 (QSOX1) was identified |
| Jiang et al. [376] | A multi-parallel enrichment strategy based on the optimized ZIC-HILIC enrichment method assisted by a filter-coated 96-well plate MALDI-TOF MS | Three human HCC cell lines | A total of 5466 N-glycosites in 2383 glycoproteins were identified |
| Lin et al. [377] | Dimethyl labeling coupled with online 3DSCX-TiO2/RP LC-MS/MS and super-SILAC mix coupled with SIM/AIMS | Human HCC tissue | A total of 7868 phosphopeptides were identified |
| Block et al. [378] | LAC-2DE-HPLC-MS/MS | Animal models (woodchucks) of HCC | Golgi protein 73 (GP73) was identified as a diagnostic biomarker |
| Zhou et al. [379] | The 2-DE was followed by the fluorescence staining of glycoprotein and MALDI-TOF-MS/MS | Three human HCC cell lines | A total of 80 glycoproteins were identified |
| Chang et al. [380] | LC-MS/MS | Human HCC plasma samples | Indicators of HCC tumor grade C3 with mannan endo-1,4-beta-mannosidase (Man5), Man6, or Man7 glycoform at asparagine 85 were identified |
| Sun et al. [381] | Hydrazine chemistry and multiple protease digestion-dimethyl labeling-SCX-RP LC-MS/MS | Human HCC and healthy liver tissues | 2329 N-glycosites on 1052N-glycoproteins were identified |
| Ang et al. [382] | LAC-2DE- MALDI-MS/MS | HCC patient serum samples | A diagnostic biomarker (haptoglobin) Hp was identified |
| Gao Q. et al. [383] | Nano-LC-MS/MS | Human tissue | Solute carrier family 10 members (1SLC10A1), pyrroline-5-carboxylate reductase 2 (PYCR2), and alcohol dehydrogenase 1A (Class I) (ADH1A) were identified |
| Reference | Proteomics Techniques | Biospecimen | Key Findings |
|---|---|---|---|
| Kirana et al. [391] | 2D-DIGE, MALDI-TOF MS | Fresh frozen tissue | Overexpression of cathepsin D (CTSD) in cells from the main tumor body showed a significant correlation with subsequent distant metastasis and shorter cancer-specific survival |
| Ku et al. [392] | TMT labeling, nano-LC-MS/MS | Fresh frozen tissue | Filamin A-interacting protein 1-like (FILIP1L) and plasminogen (PLG) upregulated in CRLM |
| Liu et al. [393] | TMT-labeling, LC-MS/MS | Fresh frozen tissue | fibronectin (FN1), metallo proteinase inhibitor 1 (TIMP1), thrombospondin-1 (THBS1), and periostin (POSTN) upregulated in CRLM |
| Shen et al. [394] | Acetylated peptide enrichment, TMT labeling, LC-MS/MS | Fresh frozen tissue | Acetylated histones, such as HIST2H3AK19Ac and H2BLK121Ac, changed while acetylated non-histones, such as tropomyosin beta chain (TPM2), K152Ac and alcohol dehydrogenase 1B (ADH1B), K331Ac altered in CRLM |
| van Huizen et al. [395] | Label-free nano-LC-MS/MS | Formalin-fixed paraffin-embedded tissue | Four collagen types, COL10A1, COL12A1 (the most abundant), COL14A1, and COL15A1 were upregulated in CRLM, while six non-collagen colon-specific proteins, cadherin-17 (CDH17), protein phosphatase-1 regulatory subunit-1B (PPP1R1B/DARP-32), keratin, type 1 cytoskeletal 20 (KRT20), carcinoembryonic antigen-related cell-adhesion molecule 5 (CEACAM5), cell-surface AA33 antigen (GPA33), and mucin-13 (MUC13), were upregulated in CRLM |
| van Huizen et al. [396] | Nano-LC-ESI-ETD-HCD | Formalin-fixed paraffin-embedded tissue | A lower ratio of 4xHyp at position 584 of the collagenalpha-2(I) chain (COL1A2) was found in CRLM |
| Fahrner M et al. [397] | Label-free LC-MS/MS | Formalin-fixed paraffin-embedded tissue | Metabolic proteins such as pyruvate carboxylase (PC) and fructose-bisphosphate aldolase B (ALDOB), and fructose-1,6-bisphosphatase 1 (FBP1) were upregulated in CRLM. Immune system proteins were enriched such as complement components C1, C4, C5, and C9 in CRLM. Structural proteins were depleted, such as desmin (DES), synemin (SYNM), and filamin-C (FLNC) in CRLM |
| Naba et al. [398] | ECM enrichment, off-gel electrophoresis, LC-MS/MS | Fresh frozen tissue | Hemopexin (HPX), osteopontin/secreted phosphoprotein 1 (SPP1), cartilage oligomeric matrix protein (COMP), insulin-like growth factor-binding protein complex acid labile subunit (IGFALS), fibronectin type III domain-containing protein1 (FNDC1), bone morphogenetic protein 1 (BMP1), and complement C1q tumor necrosis factor-related protein 5 (C1QTNF5). Extracellular matrix protein signatures are potential tissue or serological biomarkers |
| van Huizen et al. [399] | Label-free nano-LC-MS/MS | Formalin-fixed paraffin-embedded tissue | Hydroxylation of collagen was significantly lowered in CRLM and primary CRC as compared with a normal colon. Eleven peptides with a specific number of hydroxylation were downregulated in CRLM as compared with normal liver tissue |
| Kim et al. [400] | 2-DE, MALDI-TOF MS | Fresh frozen tissue | Serpin family A member 1 (SERPINA1), apolipoprotein AI (APOA1), intelectin 1 (ITLN1), desmin (DES), diazepam-binding inhibitor (DBI), succinate dehydrogenase complex flavoprotein subunit A (SDHA), and carbonic anhydrase 1 (CA1) were upregulated in CRLM |
| Voß et al. [401] | Label-free LC-MS/MS | Fresh frozen tissue | Fifty-six extracellular matrix-associated proteins including tenascin C (TNC), nidogen-1 (NID1), fibulin-1 (FBLN1), and vitronectin (VTN) were upregulated |
| Yuzhalin et al. [402] | Extra Cellular Matrix enrichment, label-free, nano-LC-MS/MS | Fresh frozen tissue | Increased level of citrullinated proteins in CRLM as compared with normal liver peptidyl arginine deiminase 4 (PAD4)-driven citrullination of the extracellular matrix is essential for CRLM growth Other upregulated proteins include versican (VCAN), metalloproteinase inhibitor 1 precursor (T1MP1), latent-transforming growth factor beta-binding protein (LTBP) 1–3, epithelial discoidin domain-containing receptor 1 (DDR1), and protein S100-A10 (S100A10) |
| Yang et al. [403] | 1D and 2-DE, nano-LC-MS/MS | Fresh frozen tissue | Olfactomedin 4 (OLFM4), CD11b/integrin alpha m (ITGAM), and integrin alpha-2 (ITGA2) significantly upregulated in primary CRC and CRLM |
| Kirana C et al. [404] | 2-DE, MALDI-TOF MS | Fresh frozen tissue | HLA class I histocompatibility antigen B alpha chain (HLAB), A disintegrin, and metalloproteinase with thrombospondin motifs 2(ADAMTS2), latent-transforming growth factor beta-binding protein 3 (LTBP3), protein jagged-2 (JAG2), and nucleoside diphosphate kinase B (NME2) were upregulated in tumor cells and associated with CRC progression by invasion, metastasis, and CRC-specific survival |
| Michal et al. [405] | Label-free LC-MS/MS | Formalin-fixed paraffin-embedded tissue | Upregulation of matrix metalloproteinase 7 (MMP7) and dehydropeptidase 1 (DPEP1) in the poor-prognosis group. Downregulation of lysyl oxidase-like 1 (LOXL1) in the poor-prognosis group. A third of differentially expressed proteins were associated with the extracellular matrix |
| Turtoi et al. [406] | MALDI-MS imaging, nano-UPLC-qTOF MS | Formalin-fixed paraffin-embedded tissue | The latent-transforming growth factor beta-binding protein 2 (LTBP2) and transforming growth factor-beta-induced protein ig-h3 (TGFBI) were upregulated in CRLM and were absent in normal tissues |
| Yang et al. [407] | Label-free nano-LC-MS/MS | Fresh frozen tissue | Nine key proteins were identified in CRLM: heat shock protein family D member 1 (HSPD1), eukaryotic translation elongation factor 1 gamma, heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1), fibrinogen beta chain (FGB), Talin 1 (TLN 1), adaptor-related protein complex 2 subunit alpha-2 (AP2A2), serrated RNA effector molecule homolog (SRRT), apolipoproteinC3 (APOC3), and phosphoglucomutase 5 (PGM5). The fibrinogen α chain was reported as a key biomarker for CRLM |
| Chen et al. [408] | HPLC-MS/MS (orbitrap fusion) | Exosomes purified from the serum of CRC and normal patients | Identified metalloproteinase-9, galectin-3 binding protein, and the insulin-like growth factor |
| Shiromizu et al. [409] | LC-MS/MS (Q exactive) | Exosomes purified from the serum of CRC and normal patients | Identified mucin-5B, matrixmetalloproteinase-9, and transferrin receptor protein 1 |
| Choi et al. [410] | LC-ESI-MS/MS (LTQ) | Microvesicles derived from CRC patient ascites | Identified the G-protein-coupled receptor E5, galectin-3, epithelial cell adhesion molecule, aminopeptidase N, and trophoblast glycoprotein |
| Reference | Proteomics Techniques | Biospecimen | Key Findings |
|---|---|---|---|
| Nepstad et al. [420] | Super-SILAC DDA LC-MS/MS | AML cells derived from patients | Higher phosphorylation of transcription regulators decreased cytokine release and increased integrin expression on cells from acute myeloid leukemia (AML) patients with high constitutive activation of the PI3K-AKT-mTOR signaling pathway |
| Boer et al. [421] | Label-free DDA, LC-MS/MS | Peripheral blood and bone marrow cells from AML patients | Differential expression of leukemia-enriched plasma membrane proteins on distinct AML subclones. Some of the proteins (e.g., interleukin 3 receptor subunit alpha (IL3RA), IL2RA, T cell immunoglobulin and mucin-domain containing-3 (TIM3), and cluster of differentiation 44 (CD44), CD96, CD47, CD32, CD99, and CLEC12A have been previously identified by other non-MS-based technologies |
| Reikvam et al. [422] | Label-free DDA, LC-MS/MS | Leukemic cells from the peripheral blood of AML patients | Patient subsets with high constitutive cytokine release levels show high expression of proteins involved in intracellular signaling interacting with integrins, ras-related C3 botulinum toxin substrate 1 (RAC1), and spleen associated tyrosine kinase (SYK). AML cells with low cytokine release showed high expression of transcriptional regulators |
| Aasebo et al. [423] | Label-free DDA, LC-MS/MS | Circulating AML blast from the peripheral blood of AML patients | The constitutive release of mediators from primary AML differs from the intracellular protein levels |
| Tong et al. [419] | Label-free DDA, LC-MS/MS | Cell suspension AML patients and control | The study showed the connection between the protein tyrosine kinase and protein tyrosine phosphatase, and its effect on protein-phosphotyrosine signaling networks |
| Forthun et al. [416] | Label-free DDA, SRM, LC-MS/MS | Leukemic cells from AML patients | Phosphoprotein profiles revealed blast differentiation and cytogenic risk stratification |
| Grønningsæter et al. [424] | Label-free DDA, LC-MS/MS | AML cells derived from AML patients | Strong antiproliferative and proapoptotic effects of metabolic pathways inhibitors were observed on the cells of AML patients |
| Raffel et al. [425] | TMT DDA LC-MS/MS | AML cells from the bone marrow aspirations of AML patients | The expression of cell adhesion molecules, proteins of the oxidative phosphorylation process, and spliceosome factors were characterized in leukemia stem cells (LSCs) |
| Raffel et al. [426] | TMT DDA LC-MS/MS | Patient AML bone marrow, cord blood, and healthy mobilized peripheral blood samples | BCAA transaminase 1 (BCAT1) was enriched in leukemia stem cells (LSCs) and linked with a branched-chain amino acid (BCAA) metabolism to epigenomic and post-translational hypoxia-inducible factor 1-α (HIF1α) regulation via α-ketoglutarate (αKG)-dependent dioxygenase |
| Aesebo et al. [412] | Super-SILAC DDA LC-MS/MS | Primary cells from AML patients | High expression of RNA processing proteins, low expression of vacuolar-type ATPase (V-ATPase) proteins, and higher activity of casein kinase 2 (CSK2) and cyclin-dependent kinases (CDKs) could help predict chemo-resistant AML relapse |
| Brenner et al. [427] | Super-SILAC DDA LC-MS/MS | AML cells derived from the peripheral blood of patients | Transcription factors and proteins involved in mRNA splicing were highly expressed in AML cells with self-renewal capacity |
| Aesebo et al. [428] | Super-SILAC DDA LC-MS/MS | Primary cells from the peripheral blood of AML patients | Higher expression of mitochondrial ribosomal subunit proteins, RNA processing proteins, DNA repair proteins, and high activity of CDKs at AML relapse |
| Alanazi et al. [429] | iTRAQ DDA LC-MS/MS | Peripheral blood and bone marrow cells from AML patients | Over-expression of nuclear S100A4 in AML cells. Nuclear S100A4 is crucial for AML survival |
| Nepstad et al. [430] | Super-SILAC DDA LC-MS/MS | AML cells derived from patients | Enhanced phosphorylation and activation of PI3K-AKT-mTOR pathway by insulin was coupled to reduced antiproliferative effects of metabolic inhibitors in AML patient subsets |
| Schmidt et al. [431] | TMT DDA LC-MS/MS | Leukemic progenitors of AML patients | Protein modification and cytoskeleton reorganization proteins showed an altered abundance in the proteome of leukemic progenitor cells |
| Reference | Proteomic Techniques | Biospecimen | Key Findings |
|---|---|---|---|
| Itkonen et al. [437] | RPPA, O-GlcNAc chromatin consensus motif imposed by O-GlcNAc transferase (OGT) used as a bait; combination with MYC chromatin immunoprecipitation (ChIP)-MS | Prostate cancer cells | O-GlcNAc transferase (OGT) is an essential mediator in androgen-independency, which is the major mechanism of PCa progression |
| McCann et al. [438] | LC-MS/MS | Overexpression or depletion of ubiquitin specific peptidase 22 (USP22) in PCa cells and analysis of the ubiquitylome | Depletion of USP22 sensitizes cells to genotoxic insult; analysis of the USP22-sensitive ubiquitylome identified the nucleotide excision repair protein, xeroderma pigmentosum C (XPC), as a critical mediator of the USP22-mediated response to genotoxic insult |
| Drake et al. [439] | LC-MS/MS | Phosphoproteome of treated naïve and metastatic CRPC tissue samples integrated with genomic and transcriptomic data | Six major signaling pathways with phosphorylation of several key residues were significantly enriched in CRPC tumors; clinically relevant information (kinase target potential based on patient-specific networks) potentially suitable for patient stratification and targeted therapies in late-stage PCa is provided |
| Mariscal et al. [440] | LC-MS/MS | Palmitoyl proteome analysis of large and small cancer-derived PCa extracellular vesicles (EVs) | STEAP1, STEAP2 metalloreductase, and ABCC4 were identified as PCa-specific palmitoyl-proteins abundant in both EV populations; their localization in EVs was reduced upon inhibition of palmitoylation in the producing cells |
| Nguyen et al. [441] | LC-MS/MS | Human prostate cancer (PCa)-associated fibroblasts | (Phospho) proteomic profiling of PCa-associated fibroblasts-derived lysyl oxidase-like 2 (LOXL2) is an important mediator of intercellular communication within the prostate tumor microenvironment |
| Cui et al. [442] | Nano-LC-MS/MS | Proteomic experiments using a clickable palmitate probe (Alk-C16) between three individual pairs of androgen-treated and non-treated LNCaP cells | Androgen treatment significantly increased the palmitoylation level of eIF3L, which may be used as a biomarker for the diagnosis of early-stage PCa |
| Lee et al. [443] | MS | DU145 and RWPE1cells | Characterization of the ERG-regulated kinome. TNIK is suggested as a potential therapeutic target |
| Zhao et al. [444] | High-resolution MS/MS | Analysis of global phosphoproteomic changes induced by fish oil in human PCa | Pyruvate dehydrogenase α-1 is a target of omega-3 polyunsaturated fatty acids in human PCa |
| Faltermeier et al. [445] | MS-based phosphoproteomics dataset | Phosphoproteomics data from a mouse model of PCa progression. Integrated with gene expression analysis and literature mining | A total of 125 wild-type kinases implicated in human PCa metastasis were selected for screening for in vivo metastatic ability; the RAF family, MERTK, and NTRK2 kinases drive PCa bone and visceral metastasis, and are highly expressed in human metastatic PCa tissues, potentially representing important therapeutic targets |
| Wen et al. [446] | SILAC-MS | Quantitative proteomics to identify SUMOylated proteins in SUMO stably transfected PC-3 cells | More than 900 putative target proteins of SUMO were identified; mutation of newly identified SUMO modification sites of ubiquitin specific peptidase 39 (USP39) further promotes the proliferation-enhancing effect of USP39 on PCa cells |
| Jiang et al. [447] | LC MS/MS | Quantitative proteomic approach to compare protein phosphorylation in orthotopic xenograft tumors grown in either intact or castrated mice | Changes in phosphorylation of Yes1 associated transcriptional regulator (YAP1) and P21 (RAC1) activated kinase 2 (PAK2) and their elevated levels in CRPC were identified. YAP2 and PAK2 regulate cell colony formation and invasion in androgen-independent cells. PAK2 influences cell proliferation and mitotic timing. Pharmacologic inhibitors of PAK2 and YAP1 were able to inhibit the growth of androgen-independent PC-3 xenografts. |
| Toughiri et al. [448] | LC-MS/MS | Proteome analysis of Aurora-A substrates using small molecule inhibitor and reverse in-gel kinase assay in PC-3 cells | The nuclear mitotic apparatus (NuMA) becomes hypo-phosphorylated in vivo upon Aurora-A inhibition; mutation of three of these phospho-sites significantly diminishes cell proliferation and increases the rate of apoptosis. |
| Li et al. [449] | Nano LC-MS/MS | LNCaP cells were metabolically labeled with Alk-C16, a palmitate probe, and treated with R1881, an androgen, or DMSO, after which palmitoylome profiling was performed | Androgen treatment significantly increased the palmitoylation level of α-tubulin and Ras-related protein Rab-7a (Rab7a), which are essential for cell proliferation; in the supernatant of LNCaP cells, the palmitoylation level of α-tubulin was also increased following androgen treatment, which may represent a biomarker for early-stage PCa |
| Bai et al. [450] | MALDI-TOF-MS analysis | Proteomics analysis to determine the O-glycan profiles of PCa cells metastasized to bone (PC-3), brain (DU145), lymph node (LNCaP), and vertebra (VCaP) in comparison to immortalized RWPE-1 cells derived from normal prostatic tissue. | PCa cells exhibit an elevation of simple/short O-glycans, with a reduction of complex O-glycans, increased O-glycan sialylation, and decreased fucosylation. Core 1 sialylation is increased in all PCa cells. The expression of sialyl-3T antigen, which is the product of ST3Gal-I is increased. ST3Gal-I is associated with PC-3 cell proliferation, migration, and apoptosis. Downregulation of ST3Gal-I reduces the tumor size in the xenograft mouse model. |
| Clark et al. [451] | Nano-ESI-LC-MS/MS | EV-derived glycoproteins upon overexpression of FUT8 in PCa cells | A reduced number of vesicles secreted by PCa cells. Increase in the abundance of proteins associated with cell motility and PCa metastasis. Altered glycans on select EV-derived glycoproteins |
| Theurillat et al. [452] | SILAC-MS | Changes in the ubiquitin landscape induced by prostate cancer-associated mutations of speckle-type POZ protein (SPOP) in immortalized prostate epithelial cells expressing endogenous SPOP | DEK proto-oncogene and tripartite motif containing 24 (TRIM24) are effector substrates consistently upregulated by SPOP mutants with decreases in ubiquitination and proteasomal degradation resulting from heteromeric complexes of wild-type and mutant SPOP protein; DEK stabilization promotes prostate epithelial cell invasion |
| Drake et al. [453] | MS | Phosphotyrosine peptide enrichment and quantitative mass spectrometry (MS) in oncogene (non-tyrosin kinase)-driven mouse model of PCa progression | Elevated tyrosine kinase signaling (EGFR, EPHA2, JAK2, ABL1, and steroid receptor coactivator (SRC) tyrosine kinase activation) was observed |
| Li et al. [454] | LTQ Orbitrap LC-MS/MS | Cell surface Thomsen–Friedenreich (TF) antigen proteome profiling of metastatic PCa cells | A cluster of differentiation 44 (CD44), CD49f, CD133, CD59, CD138, EphA2, α2 integrin, β1 integrin, transferrin receptor, and profilin express TF antigen. TF antigen-positive prostate cancer cells form significantly more and larger prostaspheres under both non-differentiating and differentiating conditions and express higher levels of stem cell markers. |
| Ino et al. [455] | MS | Comparative phosphoproteome analysis of a PCa cell line LNCaP, and an LNCaP-derived androgen-independent cell line LNCaP-AI | The phosphorylation level of THRAP3 was significantly lowered in LNCaP-AI cells; the nonphosphorylatable mutant form of THRAP3 and the phosphorylation-mimic form differ significantly in protein binding repertoire; many of the differentially interacting proteins were identified as being involved in RNA splicing and processing |
| Gulati et al. [456] | SILAC-MS | Knockdown of E6-associated protein (E6AP) in DU145 cells and analysis of a proteome | Clusterin is a novel target of E6AP; the concomitant knockdown of clusterin and E6AP partially restores cell growth |
| Gao et al. [457] | LC-MS | Highly aggressive PC-3 and PC-3M cells | Compared phosphoproteomics of differentially expressed kinases. PAK2, STE20-like kinase (SLK), mammalian STE20-like protein kinase 4 (MST4), mitogen-activated protein kinase 2 (MAP2K2), and A-Raf proto-oncogene, serine/threonine kinase (ARAF) were kinases that were potentially associated with increased migration in PC-3M cells |
| Hoti et al. [458] | LC-MS/MS | Comprehensive proteomic approaches of alpha (1,6) fucosyltransferase (FUT8) overexpressing PCa cells | EGFR and its downstream signaling were upregulated; cell survival was increased in androgen-depleted conditions |
| Sharma et al. [459] | MS | Palmitoyl proteome analysis of breast, PCa cell lines and ±DHHC3 ablation | Putative substrates include 22–28 antioxidant/redox-regulatory proteins and ablation of protein acyltransferase DHHC3 elevated oxidative stress. DHHC3 ablation in combination with chemotherapeutic drug treatment elevated oxidative stress with a greater than additive effect, and enhanced the anti-growth effects of the chemotherapeutic agents. DHHC3 ablation synergized with poly-ADP ribose polymerase (PARP) inhibitor PJ-34, to decrease cell proliferation and increase oxidative stress |
| Hoti et al. [460] | iTRAQ MS and LC- MS\MS | Proteomics of androgen-dependent and androgen-resistant LAPC4 cells | Alpha (1,6) fucosyltransferase (FUT8) was significantly overexpressed in the androgen-resistant LAPC4 cells; an overexpression of FUT8 might be responsible for the decreased PSA expression in prostate cancer specimens |
| Lee et al. [461] | LC-MS/MS in combination with SILAC | Phosphoproteomics of metastatic docetaxel-resistant PCa cell lines (DU145-Rx and PC-3-Rx) | Increased phosphorylation of focal adhesion kinase (FAK) mediates chemoresistance in CRPC |
| Reference | Proteomics Techniques | Biospecimen | Key Findings |
|---|---|---|---|
| An. et al. [466] | LC-ESI MS/MS | Serum of lung cancer patients | Thirty-two different proteins were identified |
| Geary et al. [467] | sequential windowed Acquisition of all theoretical fragment ion MS | Serum of lung cancer patients | Eleven different proteins were identified |
| Li. et al. [468] | iTRAQ-2DE-LC MS/MS | Plasma of lung cancer patients | Multiple inositol polyphosphate phosphatase 1, thyroxine-binding globulin, mannan-binding lectin serine protease 1, cathelicidin antimicrobial peptide, carnosine dipeptidase 1, fibrinogen-like protein 1, ADAMTS-like protein 4, and haptoglobin were identified |
| Sabrkhany et al. [469] | nLC-MS/MS | Plasma of lung cancer patients | Forty-nine different proteins were identified |
| Zhou et al. [470] | LC-MS/MS | Serum of NSCLC patients | Elongation factor 1, alpha 2, proteasome subunit alpha type, and spermatogenesis-associated protein were identified |
| Chae et al. [471] | VeriStrat test MALDI-TOF MS | Serum of NSCLC patients | The VS-Good group demonstrated significantly higher progression-free survival (PFS) and overall survival (OS) compared to the VS-Poor group among overall NSCLC patients, regardless of treatment |
| Muller et al. [472] | MS | Serum of patients with advanced NSCLC treated with nivolumab | A total of 274 MS protein signatures were associated with progression-free survival (PFS) and overall survival (OS) in patients |
| Reference | Proteomic Techniques | Biospecimen | Key Findings |
|---|---|---|---|
| He et al. [482] | Label-free LC-MS/MS | Breast cancer tissue | Heat shock protein (HSP) 70 kDa-8, periostin, RhoA, actinin alpha 4, cathepsin D, preproprotein, annexin 1, and aldehyde dehydrogenase 1 family member A1 (ALDH1A1), G3BP stress aranule assembly factor 1 (G3BP) were upregulated and Thymosin-β4, transketolase, and transferrin were downregulated as prognostic biomarkers and drug targets |
| Campone et al. [483] | iTRAQ labeling MALDI-MS/MS | Breast cancer tissue | Desmoplakin (DP), thrombospondin-1 (TPS1), and tryptophanyl-tRNA synthetase (TrpRS) were upregulated as prognostic biomarkers or drug targets |
| Suman et al. [484] | iTRAQ labeling MALDI-MS/MS | Breast cancer tissue and serum | Alpha-2-macroglobulin (A2M) was upregulated, and complement component 4 binding protein alpha (C4BPA) was downregulated as a prognostic biomarker |
| Sun et al. [485] | Dimethyl labeling LC-MS/MS | Breast cancer cell lines | Protein tyrosine phosphatase non-receptor type 12 (PTPN12) was downregulated as a prognostic biomarker |
| Semaan et al. [486] | Label-free LC-LTQ/FT-ICR MS | Breast cancer tissue | Tripartite motif containing 28 (TRIM28), HSP90-alpha, heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1), clathrin heavy chain (CLTC), and myosin-9, heparin binding growth factor (HDGF) phosphorylated and HSP90, Abl interactor 1 (AB1), PTRF1 isoform 1 of polymerase I and transcript release factor, AHNAK nucleoprotein, and SEPT2 dephosphorylated were identified as drug targets |
| Lawrence et al. [487] | iBAQ (absolute quantitation) LC-MS/MS | Cell lines and tumors | NF-κB was upregulated as a prognostic biomarker |
| Liu et al. [488] | Label-free nLC-MS/MS) | Breast cancer tissue | UMP-CMP kinase (CMPK1), apoptosis-inducing factor 1, mitochondrial (AIFM1), ferritin heavy chain (FTH1), echinoderm microtubule-associated protein-like 4 (EML4), neutral alpha glucosidase AB (GANAB), catenin alpha-1 (CTNNA1), AP-1 complex subunit gamma-1 (AP1G1), syntaxin-12 (STX12), AP-1 complex subunit mu-1 (AP1M1), and F-actin capping protein subunit beta (CAPZB) proteins were upregulated and C-1-tetrahydrofolate synthase cytoplasmic (MTHFD1) was downregulated as a prognostic biomarker |
| Mittal et al. [489] | Label-free quantification LC-MS/MS | Breast cancer cell lines | Enolase 1 (ENO1) was upregulated as a prognostic biomarker |
| Liu et al. [490] | Label free nLC-MS/MS | Breast cancer tissue | Ferritin heavy chain 1 (FTH1) was upregulated as a prognostic biomarker |
| Wu et al. [491] | SILAC MS | Breast cancer cell lines | AXL receptor tyrosine kinase was upregulated as a prognostic biomarker |
| Tyanova et al. [492] | Super-SILAC LC-MS/MS | Cell lines and breast cancer tumors | Minichromosome maintenance complex component 5 (MCM5), stathmin 1 (STMN1), glutaminase (GLS), RNA terminal phosphate cyclaselike 1 (RCL1), chromosome 9 open reading frame 114 (C9ORF114), and ENO1 were upregulated and anterior gradient 2 (AGR2), melanophilin (MLPH), HID1 domain-containing (HID1), centralized master bidders list (CMBL), and forkhead box A1 (FOXA1) were downregulated as prognostic biomarkers |
| Shenoy et al. [17] | In-solution digestion and LC -MS/MS | Patient’s tissue and breast cancer cell lines | Pyrroline-5-carboxylate reductase 1 (PYCR1) was identified as a biomarker |
| Koh et al. [493] | In-solution digestion and LC -MS/MS | Breast CSCs and breast cancer cell lines | CD66c was identified as a biomarker |
| Proteomic Biomarker | Biospecimen | Cancer | Reference |
|---|---|---|---|
| Alfa-Feto-Protein-L3 | Serum | HCC | [515] |
| Epidermal growth factor receptor (EGFR)-T790M | Tumor tissue | Lung cancer | [516,517] |
| Cancer antigen 125 (CA125), prealbumin, apolipoprotein A1, beta-2-microglobulin, and transferrin | Serum | Ovarian cancer | [518] |
| Thrombospondin 1 (THBS1), bromodomain and WD repeat domain containing 3 (BRWD3), epidermal growth factor receptor (EGFR), and complement factor H related 3 (CFHR3) | Plasma | Breast cancer | [519] |
| MBL-associated serine protease 1 (MASP1), osteopontin (OPN), paraoxonase 3 (PON3), and transferrin receptor (TFRC) | Plasma | Colorectal cancer | [520,521] |
| sE-cadherin, TSR1 ribosome maturation factor (TSR1), serum amyloid A (SAA), kallikrein related peptidase 3 (KLK3) | Serum | Prostate cancer | [522,523] |
| Peptide profiling | Serum | Cervical cancer | [524,525] |
| Serum amyloid A2 (SAA2), kallikrein B1 (KLKB1), apolipoprotein A1 (APOA1), and cluster of differentiation-44 (CD44) | Serum | Multiple Myeloma | [526,527,528] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Punetha, A.; Kotiya, D. Advancements in Oncoproteomics Technologies: Treading toward Translation into Clinical Practice. Proteomes 2023, 11, 2. https://doi.org/10.3390/proteomes11010002
Punetha A, Kotiya D. Advancements in Oncoproteomics Technologies: Treading toward Translation into Clinical Practice. Proteomes. 2023; 11(1):2. https://doi.org/10.3390/proteomes11010002
Chicago/Turabian StylePunetha, Ankita, and Deepak Kotiya. 2023. "Advancements in Oncoproteomics Technologies: Treading toward Translation into Clinical Practice" Proteomes 11, no. 1: 2. https://doi.org/10.3390/proteomes11010002
APA StylePunetha, A., & Kotiya, D. (2023). Advancements in Oncoproteomics Technologies: Treading toward Translation into Clinical Practice. Proteomes, 11(1), 2. https://doi.org/10.3390/proteomes11010002