
Optimized Proteome Reduction for Integrative Top–Down Proteomics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.1.1. Concentration and Purity Assessment of Isolated Protein Standards
2.1.2. Mammalian and Plant Proteome Sample Preparation
2.2. 1DE: SDS-PAGE
2.3. 2DE: IEF and SDS-PAGE
2.4. 1DE Image Analysis
2.5. 2DE Image Analysis
3. Results
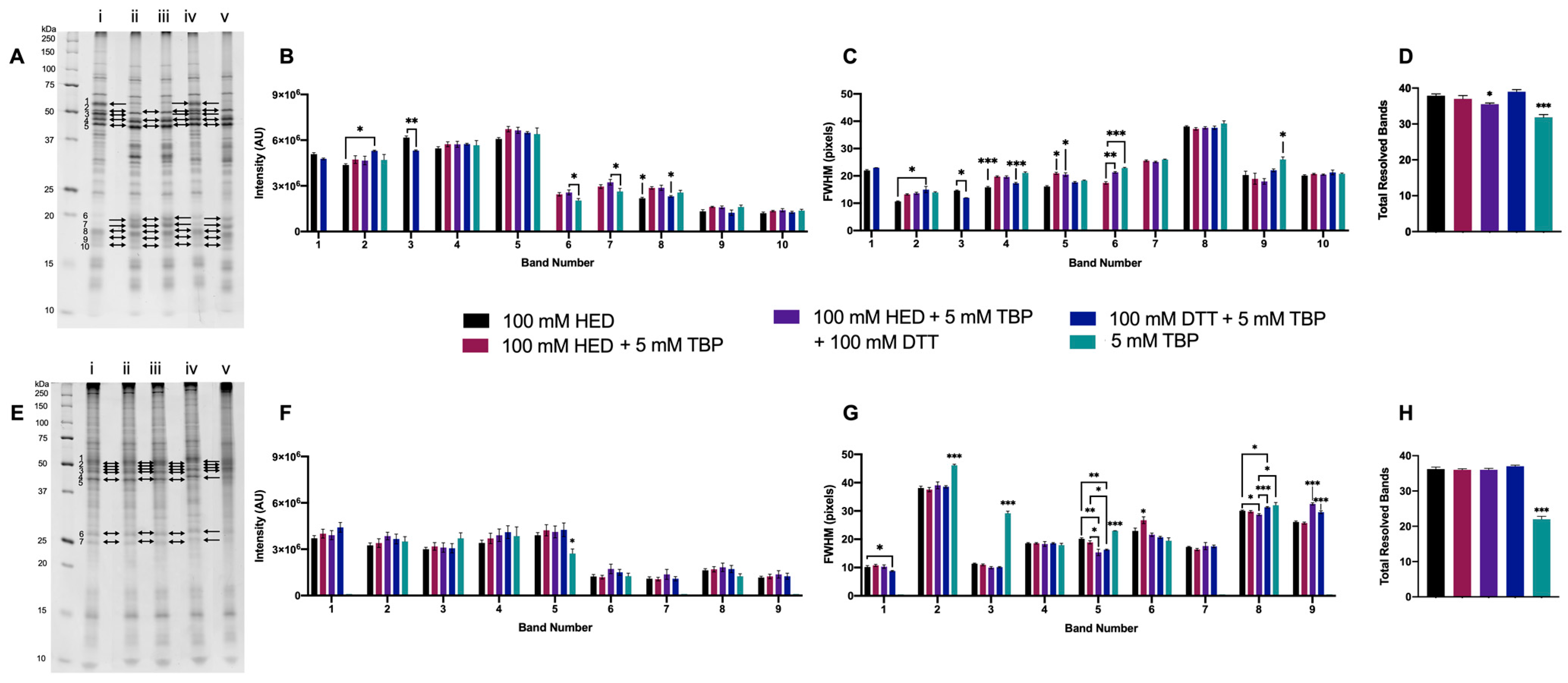
3.1. 1DE Experiments
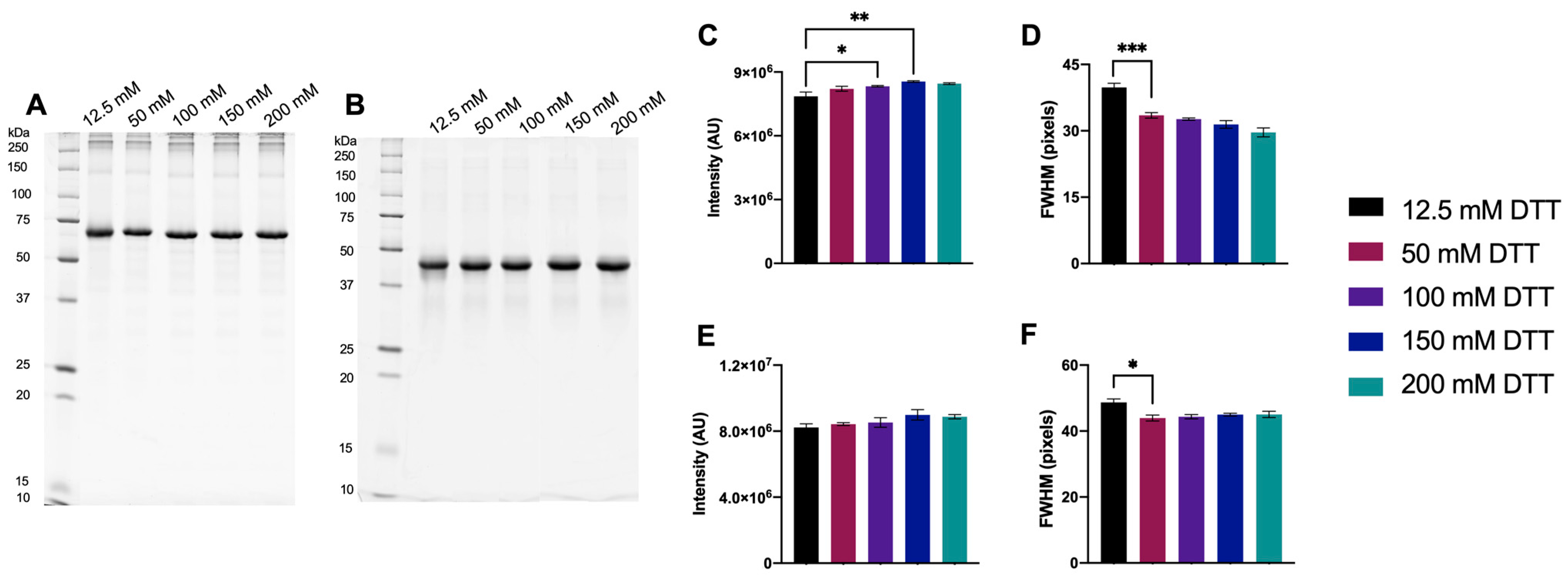
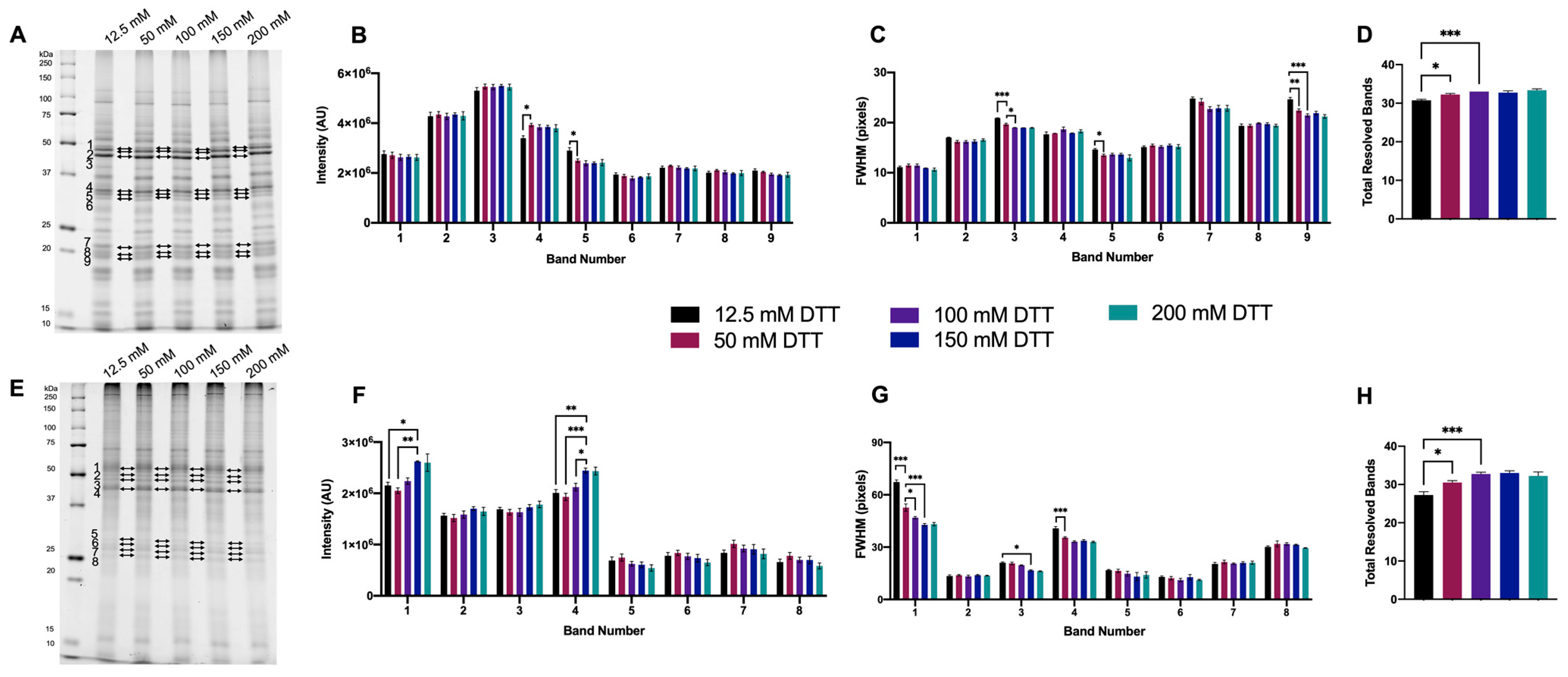
3.1.1. Initial Testing of DTT
3.1.2. Optimized DTT Concentration
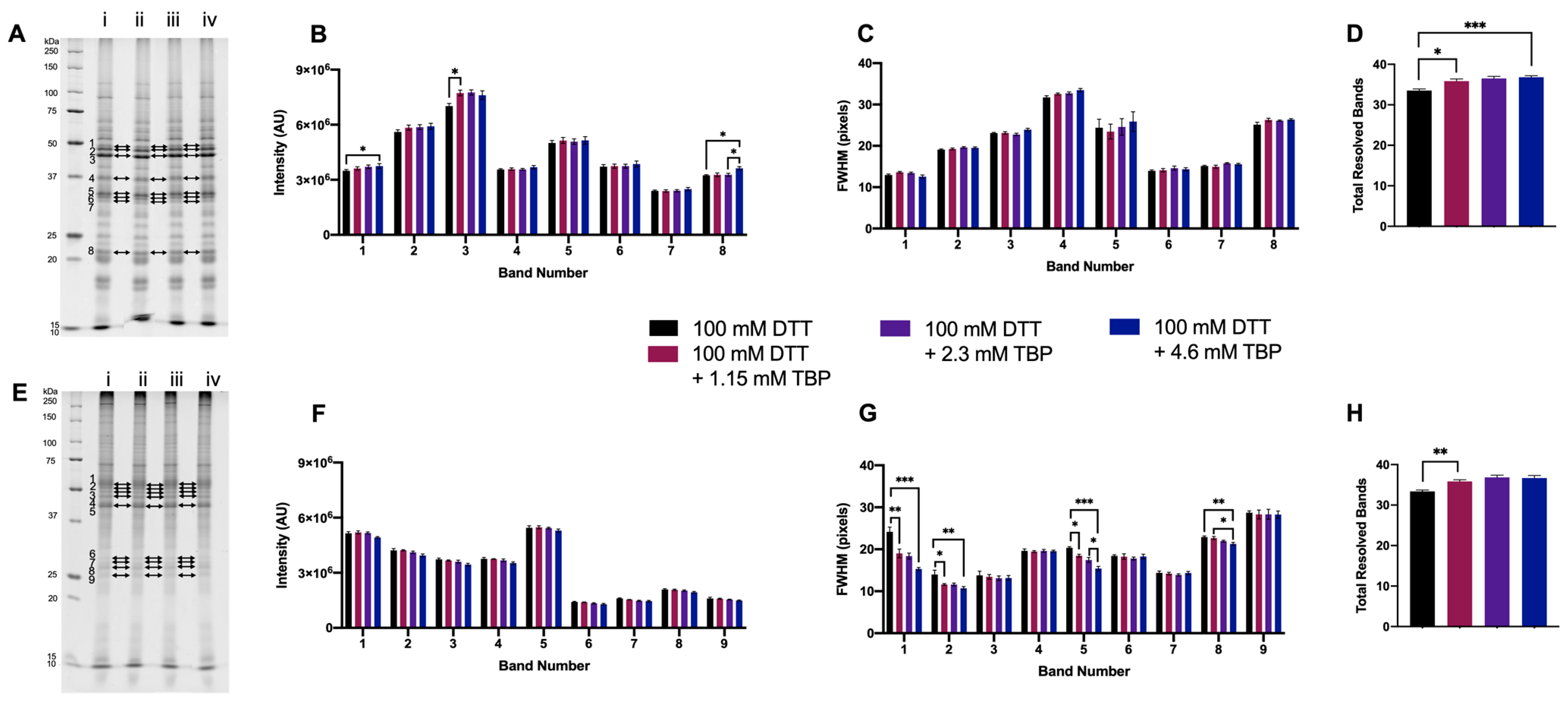
3.1.3. Supplementing DTT with TBP
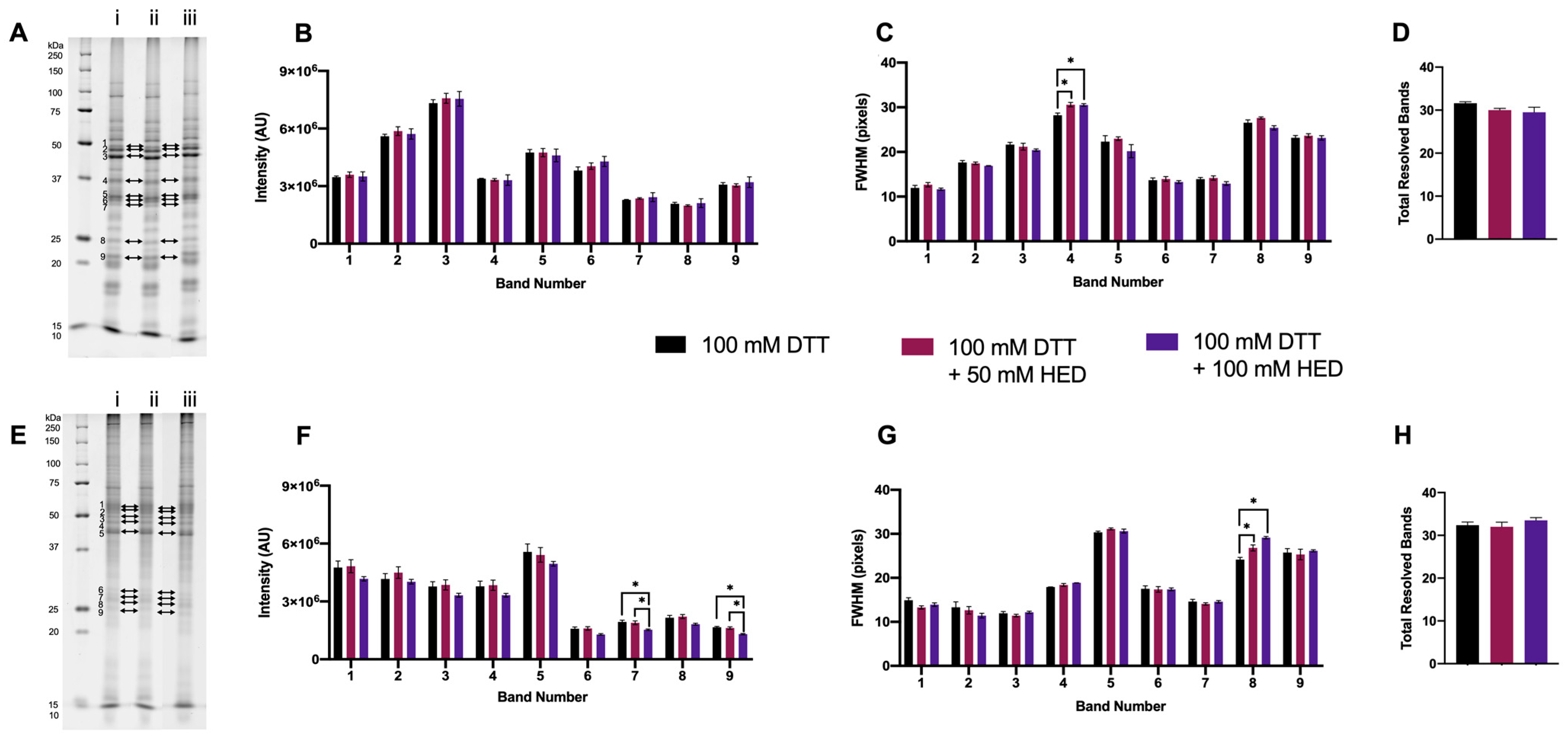
3.1.4. Supplementing DTT with HED
3.1.5. Final Testing of Reagents to Optimize for 2DE
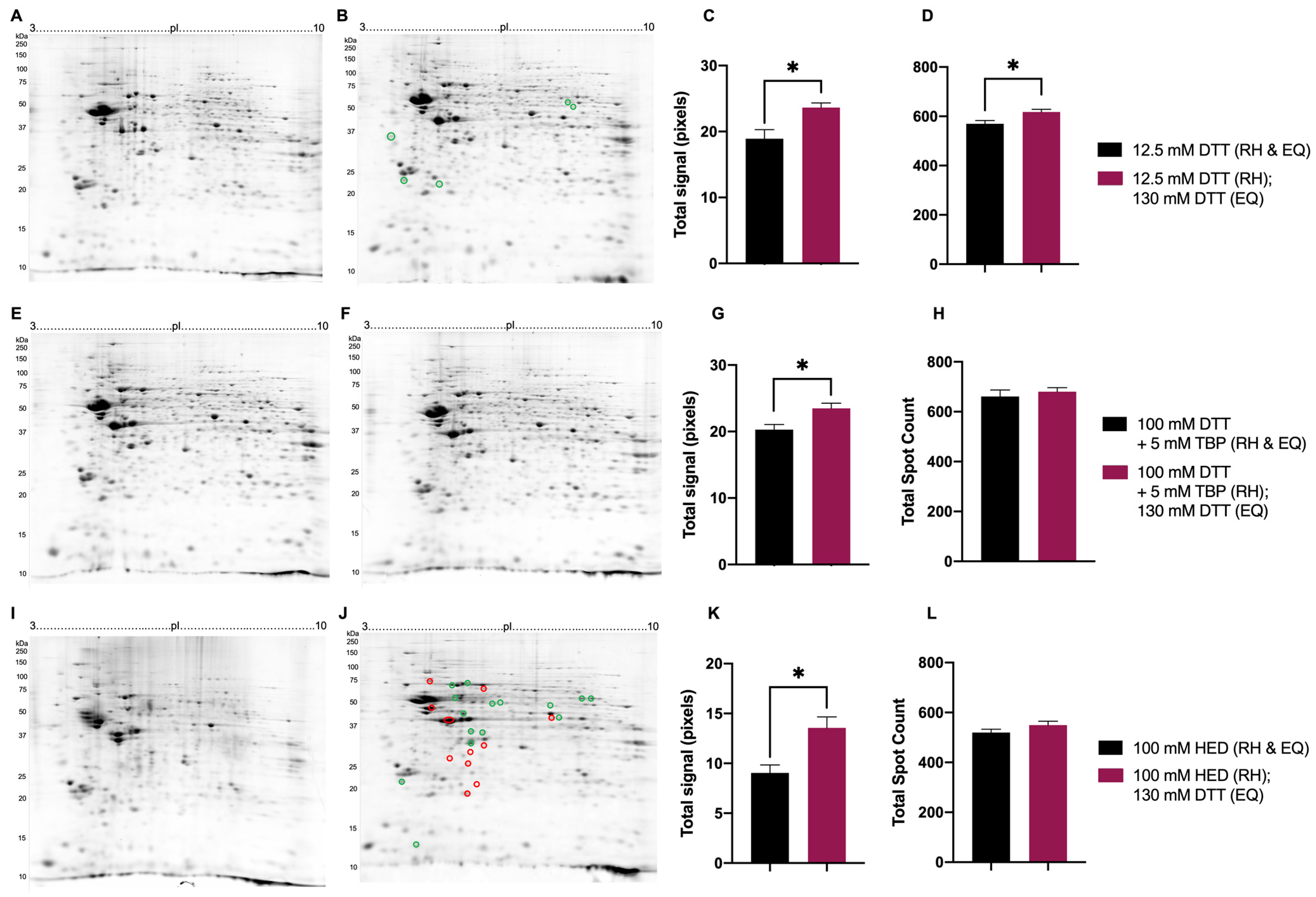
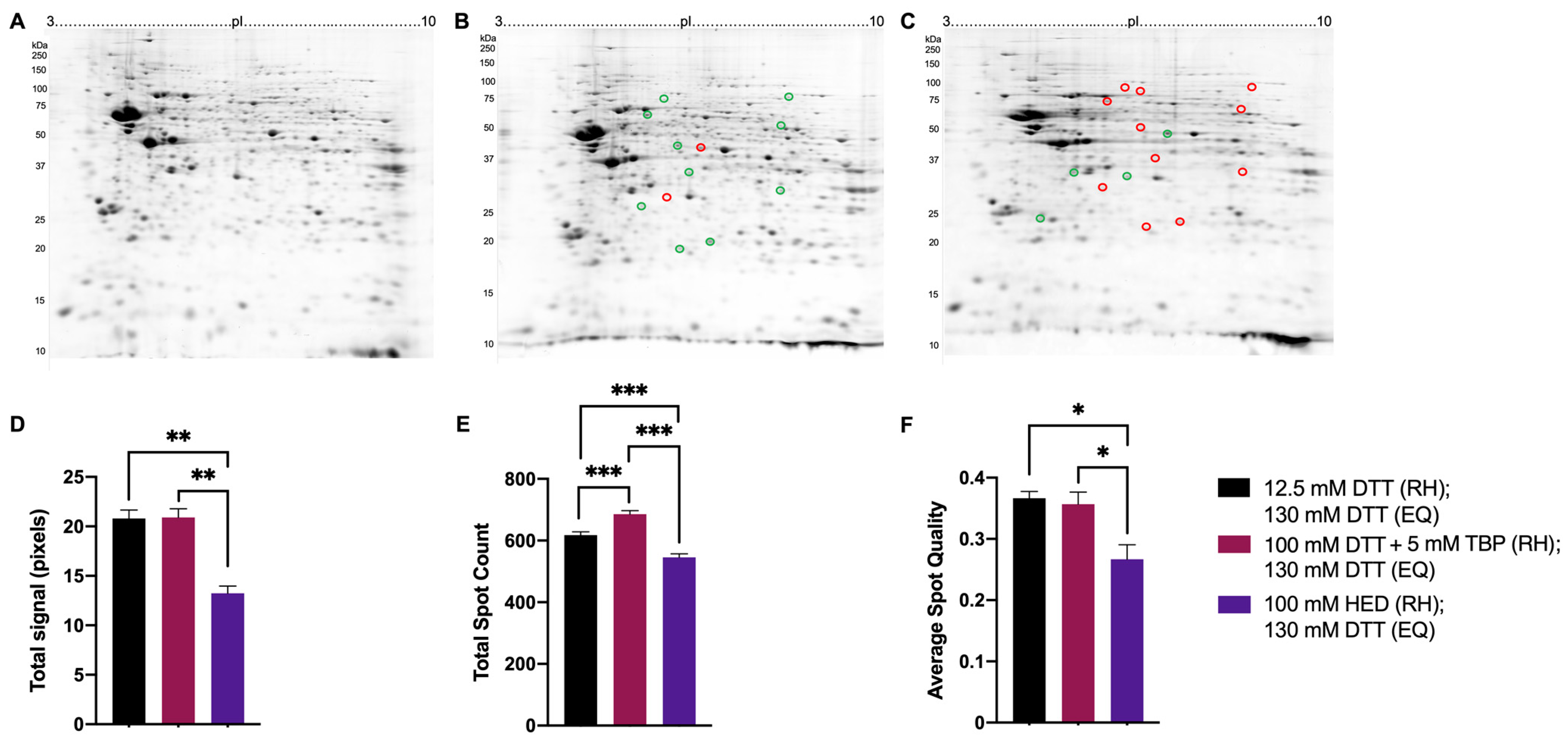
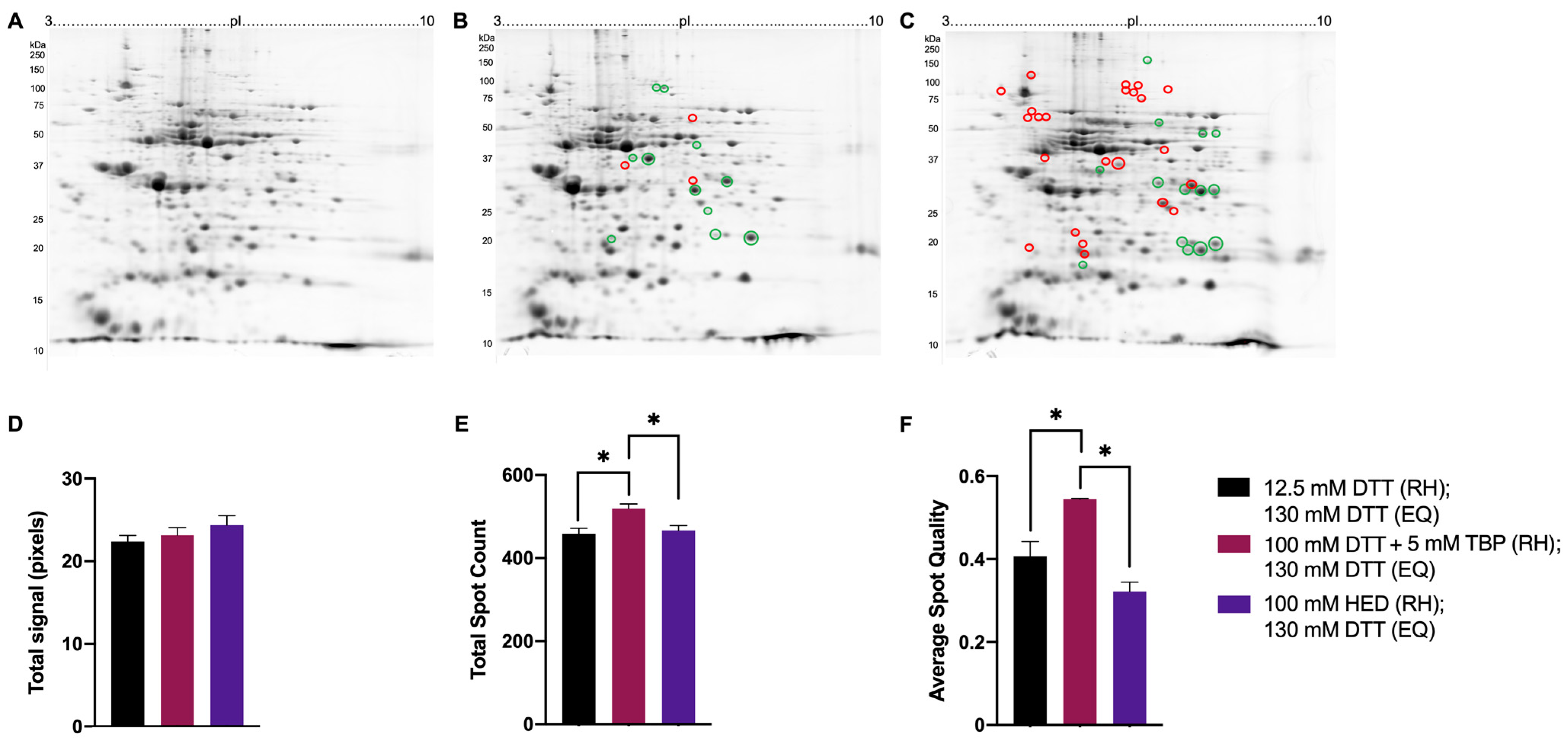
3.2. Testing in the Full 2DE Protocol
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coorssen, J.R.; Yergey, A.L. Proteomics Is Analytical Chemistry: Fitness-for-Purpose in the Application of Top-down and Bottom-up Analyses. Proteomes 2015, 3, 440–453. [Google Scholar] [CrossRef]
- Carbonara, K.; Andonovski, M.; Coorssen, J.R. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes 2021, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Agar, J.N.; Amster, J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E. How Many Human Proteoforms Are There? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef]
- Oliveira, B.M.; Coorssen, J.R.; Martins-de-Souza, D. 2DE: The Phoenix of Proteomics. J. Proteom. 2014, 104, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Li, B.; Zhan, X.; Schlüter, H.; Jungblut, P.R.; Coorssen, J.R. Innovating the Concept and Practice of Two-Dimensional Gel Electrophoresis in the Analysis of Proteomes at the Proteoform Level. Proteomes 2019, 7, 36. [Google Scholar] [CrossRef]
- Carbonara, K.; Padula, M.P.; Coorssen, J.R. Quantitative Assessment Confirms Deep Proteome Analysis by Integrative Top–down Proteomics. Electrophoresis 2022, 44, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Yang, H.; Peng, F.; Li, J.; Mu, Y.; Long, Y.; Cheng, T.; Huang, Y.; Li, Z.; Lu, M.; et al. How Many Proteins Can Be Identified in a 2DE Gel Spot within an Analysis of a Complex Human Cancer Tissue Proteome? Electrophoresis 2018, 39, 965–980. [Google Scholar] [CrossRef]
- Naryzhny, S.; Ronzhina, N.; Zorina, E.; Kabachenko, F.; Klopov, N.; Zgoda, V. Construction of 2DE Patterns of Plasma Proteins: Aspect of Potential Tumor Markers. Int. J. Mol. Sci. 2022, 23, 11113. [Google Scholar] [CrossRef]
- Naryzhny, S. Inventory of Proteoforms as a Current Challenge of Proteomics: Some Technical Aspects. J. Proteom. 2019, 191, 22–28. [Google Scholar] [CrossRef]
- Jungblut, P.R.; Holzhütter, H.G.; Apweiler, R.; Schlüter, H. The Speciation of the Proteome. Chem. Cent. J. 2008, 2, 16. [Google Scholar] [CrossRef]
- Butt, R.H.; Coorssen, J.R. Postfractionation for Enhanced Proteomic Analyses: Routine Electrophoretic Methods Increase the Resolution of Standard 2D-PAGE. J. Proteome Res. 2005, 4, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Churchward, M.A.; Butt, R.H.; Lang, J.C.; Hsu, K.K.; Coorssen, J.R. Enhanced Detergent Extraction for Analysis of Membrane Proteomes by Two-Dimensional Gel Eletrophoresis. Proteome Sci. 2005, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Coorssen, J.R. Pre-Extraction Sample Handling by Automated Frozen Disruption Significantly Improves Subsequent Proteomic Analyses. J. Proteome Res. 2006, 5, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.R.; Churchward, M.A.; Butt, R.H.; Coorssen, J.R. Assessing Detection Methods for Gel-Based Proteomic Analyses. J. Proteome Res. 2007, 6, 1418–1425. [Google Scholar] [CrossRef]
- Butt, R.H.; Coorssen, J.R. Coomassie Blue as a Near-Infrared Fluorescent Stain: A Systematic Comparison with Sypro Ruby for in-Gel Protein Detection. Mol. Cell. Proteom. 2013, 12, 3834–3850. [Google Scholar] [CrossRef]
- Gauci, V.J.; Padula, M.P.; Coorssen, J.R. Coomassie Blue Staining for High Sensitivity Gel-Based Proteomics. J. Proteom. 2013, 90, 96–106. [Google Scholar] [CrossRef]
- Wright, E.P.; Prasad, K.A.G.; Padula, M.P.; Coorssen, J.R. Deep Imaging: How Much of the Proteome Does Current Top-down Technology Already Resolve? PLoS ONE 2014, 9, e86058. [Google Scholar] [CrossRef]
- Wright, E.P.; Partridge, M.A.; Padula, M.P.; Gauci, V.J.; Malladi, C.S.; Coorssen, J.R. Top-down Proteomics: Enhancing 2D Gel Electrophoresis from Tissue Processing to High-Sensitivity Protein Detection. Proteomics 2014, 14, 872–889. [Google Scholar] [CrossRef]
- Noaman, N.; Abbineni, P.S.; Withers, M.; Coorssen, J.R. Coomassie Staining Provides Routine (Sub)Femtomole in-Gel Detection of Intact Proteoforms: Expanding Opportunities for Genuine Top-down Proteomics. Electrophoresis 2017, 38, 3086–3099. [Google Scholar] [CrossRef] [PubMed]
- Noaman, N.; Coorssen, J.R. Coomassie Does It (Better): A Robin Hood Approach to Total Protein Quantification. Anal. Biochem. 2018, 556, 53–56. [Google Scholar] [CrossRef]
- Carbonara, K.; Coorssen, J.R. A ‘Green’ Approach to Fixing Polyacrylamide Gels. Anal. Biochem. 2020, 605, 113853. [Google Scholar] [CrossRef]
- Carbonara, K.; Coorssen, J.R. Sometimes Faster Can Be Better: Microneedling IPG Strips Enables Higher Throughput for Integrative Top-down Proteomics. Proteomics 2022, 23, 2200307. [Google Scholar] [CrossRef]
- Henningsen, R.; Gale, B.L.; Straub, K.M.; DeNagel, D.C. Application of Zwitterionic Detergents to the Solubilization of Integral Membrane Proteins for Two-Dimensional Gel Electrophoresis and Mass Spectrometry. Proteomics 2002, 2, 1479–1488. [Google Scholar] [CrossRef]
- Görg, A.; Boguth, G.; Obermaier, C.; Posch, A.; Weiss, W. Two-Dimensional Polyacrylamide Gel Electrophoresis with Immobilized PH Gradients in the First Dimension (IPG-Dalt): The State of the Art and the Controversy of Vertical versus Horizontal Systems. Electrophoresis 1995, 16, 1079–1086. [Google Scholar] [CrossRef]
- Herbert, B.; Galvani, M.; Hamdan, M.; Olivieri, E.; MacCarthy, J.; Pedersen, S.; Righetti, P.G. Reduction of Alkylation of Proteins in Preparation of Two-Dimensional Map Analysis: Why, When, and How? Electrophoresis 2001, 22, 2046–2057. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T. Solubilization of Proteins for Electrophoretic Analyses. Electrophoresis 1996, 17, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Herbert, B.R.; Molloy, M.P.; Gooley, A.A.; Walsh, B.J.; Bryson, W.G.; Williams, K.L. Improved Protein Solubility in Two-Dimensional Electrophoresis Using Tributyl Phosphine as Reducing Agent. Electrophoresis 1998, 19, 845–851. [Google Scholar] [CrossRef]
- Cleland, W.W. Dithiothreitol, a New Protective Reagent for SH Groups. Biochemistry 1964, 3, 480–482. [Google Scholar] [CrossRef] [PubMed]
- Righetti, P.G.; Tudor, G.; Gianazza, E. Effect of 2-Mercaptoethanol on PH Gradients in Isoelectric Focusing. J. Biochem. Biophys. Methods 1982, 6, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Cannon-Carlson, S.; Tang, J. Modification of the Laemmli Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis Procedure to Eliminate Artifacts on Reducing and Nonreducing Gel. Anal. Biochem. 1997, 246, 146–148. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nat. Publ. Group 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. High Resolution Two-Dimensional Electrophoresis of Proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xu, C.; Wang, W. Reduction and Alkylation of Proteins in 2D Gel Electrophoresis: Before or after Isoelectric Focusing? Front. Chem. 2017, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Han, J.C.; Han, G.Y. A Procedure for Quantitative Determination of Tris(2-Carboxyethyl)Phosphine, an Odorless Reducing Agent More Stable and Effective than Dithiothreitol. Anal. Biochem. 1994, 220, 5–10. [Google Scholar] [CrossRef]
- Vâlcu, C.M.; Schlink, K. Reduction of Proteins during Sample Preparation and Two-Dimensional Gel Electrophoresis of Woody Plant Samples. Proteomics 2006, 6, 1599–1605. [Google Scholar] [CrossRef] [PubMed]
- Cline, D.J.; Redding, S.E.; Brohawn, S.G.; Psathas, J.N.; Schneider, J.P.; Thorpe, C. New Water-Soluble Phosphines as Reductants of Peptide and Protein Disulfide Bonds: Reactivity and Membrane Permeability. Biochemistry 2004, 43, 15195–15203. [Google Scholar] [CrossRef]
- Olsson, I.; Larsson, K.; Palmgren, R.; Bjellqvist, B. Organic Disulfides as a Means to Generate Streak-Free Two-Dimensional Maps with Narrow Range Basic Immobilized Ph Gradient Strips as First Dimension. Proteomics 2002, 2, 1630–1632. [Google Scholar] [CrossRef]
- Acín, P.; Rayó, J.; Guerrero, A.; Quero, C. Improved Resolution in the Acidic and Basic Region of 2-DE of Insect Antennae Proteins Using Hydroxyethyl Disulfide. Electrophoresis 2009, 30, 2613–2616. [Google Scholar] [CrossRef]
- Piersma, S.R.; Warmoes, M.O. Whole Gel Processing Procedure for GeLC-MS/MS Based Proteomics. Proteome Sci. 2013, 11, 17. [Google Scholar] [CrossRef]
- Grimsley, G.R.; Pace, C.N. Spectrophotometric Determination of Protein Concentration. Curr. Protoc. Protein Sci. 2003, 33, 3.1.1–3.1.9. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Albertorio, F.; Bezrukov, L.; Kolosova, I.; Backlund, P.S.; Zimmerberg, J. Quantitative Femto- to Attomole Immunodetection of Regulated Secretory Vesicle Proteins Critical to Exocytosis. Anal. Biochem. 2002, 307, 54–62. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Sternberg, S.R. Biomedical Image Processing. IEEE Comput. 1983, 16, 22–32. [Google Scholar] [CrossRef]
- Holm, S. A Simple Sequentially Rejective Multiple Test Procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar] [CrossRef]
- Using DeStreakTM Rehydration Solution. Sigma Aldrich 2022. Available online: https://www.sigmaaldrich.com/CA/en/technical-documents/protocol/protein-biology/gel-electrophoresis/using-destreak-rehydration-solution (accessed on 11 January 2023).
- Saraygord-Afshari, N.; Naderi-Manesh, H.; Naderi, M. Enhanced Reproducibility of the Human Gel-Based Tear Proteome Maps in the Presence of Di-(2-Hydroxyethyl) Disulfide. Biotechnol. Appl. Biochem. 2014, 61, 660–667. [Google Scholar] [CrossRef]
- Mi, W.; Liu, X.; Jia, W.; Li, L.; Cai, Y.; Ying, W.T.; Qian, X.H. Toward a High Resolution 2-DE Profile of the Normal Human Liver Proteome Using Ultra-Zoom Gels. Sci. China Life Sci. 2011, 54, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, C.; Pessione, E.; Giuffrida, M.G.; Mazzoli, R.; Barello, C.; Conti, A.; Giunta, C. Combined Cup Loading, Bis(2-Hydroxyethyl) Disulfide, and Protein Precipitation Protocols to Improve the Alkaline Proteome of Lactobacillus Hilgardii. Electrophoresis 2007, 28, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, J.J.; Bjerneld, E.J.; Cetinkaya, S.; Goscinski, J.; Grigorescu, I.; Haid, D.; Laurin, Y.; Bjellqvist, B. A Simplified 2-D Electrophoresis Protocol with the Aid of an Organic Disulfide. Proteomics 2005, 5, 3088–3096. [Google Scholar] [CrossRef]
- Cortón, M.; Villuendas, G.; Botella, J.I.; San Millán, J.L.; Escobar-Morreale, H.F.; Peral, B. Improved Resolution of the Human Adipose Tissue Proteome at Alkaline and Wide Range PH by the Addition of Hydroxyethyl Disulfide. Proteomics 2004, 4, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wang, J.R.; Wong, K.W.V.; Hsiao, W.L.; Zhou, H.; Jiang, Z.H.; Kam, K.T.R.; Liu, L. Optimization of 2-Dimensional Gel Electrophoresis for Proteomic Studies of Solid Tumor Tissue Samples. Mol. Med. Rep. 2014, 9, 626–632. [Google Scholar] [CrossRef]
- Khoudoli, G.A.; Porter, I.M.; Blow, J.J.; Swedlow, J.R. Optimisation of the Two-Dimensional Gel Electrophoresis Protocol Using the Taguchi Approach. Proteome Sci. 2004, 2, 6. [Google Scholar] [CrossRef]
- Stimpson, S.E.; Lauto, A.; Coorssen, J.R.; Myers, S.J. Isolation and Identification of ER Associated Proteins with Unique Expression Changes Specific to the V144D SPTLC1 Mutations in HSN-I. Biochem. Anal. Biochem. 2016, 5, 1. [Google Scholar] [CrossRef]
- Stroud, L.J.; Šlapeta, J.; Padula, M.P.; Druery, D.; Tsiotsioras, G.; Coorssen, J.R.; Stack, C.M. Comparative Proteomic Analysis of Two Pathogenic Tritrichomonas Foetus Genotypes: There Is More to the Proteome than Meets the Eye. Int. J. Parasitol. 2017, 47, 203–213. [Google Scholar] [CrossRef] [PubMed]
- D’Silva, A.M.; Hyett, J.A.; Coorssen, J.R. A Routine “top-down” Approach to Analysis of the Human Serum Proteome. Proteomes 2017, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- D’Silva, A.M.; Hyett, J.A.; Coorssen, J.R. Proteomic Analysis of First Trimester Maternal Serum to Identify Candidate Biomarkers Potentially Predictive of Spontaneous Preterm Birth. J. Proteom. 2018, 178, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Kurgan, N.; Noaman, N.; Pergande, M.R.; Cologna, S.M.; Coorssen, J.R.; Klentrou, P. Changes to the Human Serum Proteome in Response to High Intensity Interval Exercise: A Sequential Top-down Proteomic Analysis. Front. Physiol. 2019, 10, 362. [Google Scholar] [CrossRef]
- Mazinani, S.A.; Noaman, N.; Pergande, M.R.; Cologna, S.M.; Coorssen, J.; Yan, H. Exposure to Microwave Irradiation at Constant Culture Temperature Slows the Growth of: Escherichia Coli DE3 Cells, Leading to Modified Proteomic Profiles. RSC Adv. 2019, 9, 11810–11817. [Google Scholar] [CrossRef] [PubMed]
- Furber, K.L.; Backlund, P.S.; Yergey, A.L.; Coorssen, J.R. Unbiased Thiol-Labeling and Top-down Proteomic Analyses Implicate Multiple Proteins in the Late Steps of Regulated Secretion. Proteomes 2019, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.K.; Almuslehi, M.S.M.; Gyengesi, E.; Myers, S.J.; Shortland, P.J.; Mahns, D.A.; Coorssen, J.R. Suppression of the Peripheral Immune System Limits the Central Immune Response Following Cuprizone-Feeding: Relevance to Modelling Multiple Sclerosis. Cells 2019, 8, 1314. [Google Scholar] [CrossRef]
- Almuslehi, M.S.M.; Sen, M.K.; Shortland, P.J.; Mahns, D.A.; Coorssen, J.R. Histological and Top-Down Proteomic Analyses of the Visual Pathway in the Cuprizone Demyelination Model. J. Mol. Neurosci. 2022, 72, 1374–1401. [Google Scholar] [CrossRef]
- Görg, A.; Obermaier, C.; Boguth, G.; Csordas, A.; Diaz, J.-J.; Madjar, J.-J. Very Alkaline Immobilized PH Gradients for Two-Dimensional Electrophoresis of Ribosomal and Nuclear Proteins. Electrophoresis 1998, 18, 328–337. [Google Scholar] [CrossRef]
- Kusch, K.; Uecker, M.; Liepold, T.; Möbius, W.; Hoffmann, C.; Neumann, H.; Werner, H.B.; Jahn, O. Partial Immunoblotting of 2D-Gels: A Novel Method to Identify Post-Translationally Modified Proteins Exemplified for the Myelin Acetylome. Proteomes 2017, 5, 3. [Google Scholar] [CrossRef]
- Qian, S.; Yang, Y.; Li, N.; Cheng, T.; Wang, X.; Liu, J.; Li, X.; Desiderio, D.M.; Zhan, X. Prolactin Variants in Human Pituitaries and Pituitary Adenomas Identified with Two-Dimensional Gel Electrophoresis and Mass Spectrometry. Front. Endocrinol. 2018, 9, 468. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, X.; Yang, C.; Wen, S.; Li, J.; Li, N.; Long, Y.; Mu, Y.; Liu, J.; Liu, Q.; et al. Human Growth Hormone Proteoform Pattern Changes in Pituitary Adenomas: Potential Biomarkers for 3P Medical Approaches. EPMA J. 2021, 12, 67–89. [Google Scholar] [CrossRef]
- Braakman, I.; Lamriben, L.; van Zadelhoff, G.; Hebert, D.N. Analysis of Disulfide Bond Formation. Curr. Protoc. Protein Sci. 2017, 90, 14.1.1–14.1.21. [Google Scholar] [CrossRef] [PubMed]
- Boja, E.S.; Fales, H.M. Overalkylation of a Protein Digest with Iodoacetamide. Anal. Chem. 2001, 73, 3576–3582. [Google Scholar] [CrossRef]
- Müller, T.; Winter, D. Systematic Evaluation of Protein Reduction and Alkylation Reveals Massive Unspecific Side Effects by Iodine-Containing Reagents. Mol. Cell. Proteom. 2017, 16, 1173–1187. [Google Scholar] [CrossRef]
- Thiede, B.; Koehler, C.J.; Strozynski, M.; Treumann, A.; Stein, R.; Zimny-Arndt, U.; Schmid, M.; Jungblut, P.R. High Resolution Quantitative Proteomics of Hela Cells Protein Species Using Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC), Two-Dimensional Gel Electrophoresis (2DE) and Nano-Liquid Chromatograpohy Coupled to an LTQ-Orbitrap Mass Spectrometer. Mol. Cell. Proteom. 2013, 12, 529–538. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woodland, B.; Necakov, A.; Coorssen, J.R. Optimized Proteome Reduction for Integrative Top–Down Proteomics. Proteomes 2023, 11, 10. https://doi.org/10.3390/proteomes11010010
Woodland B, Necakov A, Coorssen JR. Optimized Proteome Reduction for Integrative Top–Down Proteomics. Proteomes. 2023; 11(1):10. https://doi.org/10.3390/proteomes11010010
Chicago/Turabian StyleWoodland, Breyer, Aleksandar Necakov, and Jens R. Coorssen. 2023. "Optimized Proteome Reduction for Integrative Top–Down Proteomics" Proteomes 11, no. 1: 10. https://doi.org/10.3390/proteomes11010010
APA StyleWoodland, B., Necakov, A., & Coorssen, J. R. (2023). Optimized Proteome Reduction for Integrative Top–Down Proteomics. Proteomes, 11(1), 10. https://doi.org/10.3390/proteomes11010010