
NCC—An Efficient Deep Learning Architecture for Non-Coding RNA Classification



Abstract
1. Introduction
1.1. RNA
- From DNA, messenger RNA (mRNA) is created. It transfers information to the ribosomes found in the cytoplasm from the nucleus. It is translated at the ribosomes, where protein synthesis occurs.
- Transfer RNA (tRNA) is a molecule that carries amino acids to the ribosomes during protein synthesis. Using mRNA codons, which are three nucleotide sequences that encode different amino acids, to match acids, this operation is accomplished. Then, these acids are incorporated into the expanding chain.
- A component of ribosomes, which are cellular structures in charge of protein synthesis, is ribosomal RNA (rRNA). In addition to supporting ribosomes, rRNA aids in accelerating chemical events that result in the synthesis of proteins.
- A class of RNA molecules that support cellular functions includes small nuclear RNA (snRNA). Participating in RNA splicing, which entails cutting out coding sections from mRNA called introns and putting together coding sequences called exons, is one of its roles.
1.2. Non-Coding RNA
2. Related Work
3. Implementing the Prediction Mechanism
- 1.
- The model’s input is the primary structure of RNA.
- 2.
- The proposed model should be simple, like ncRFP [22], for short training periods.
- 3.
- High level of accuracy for all prediction-related metrics.
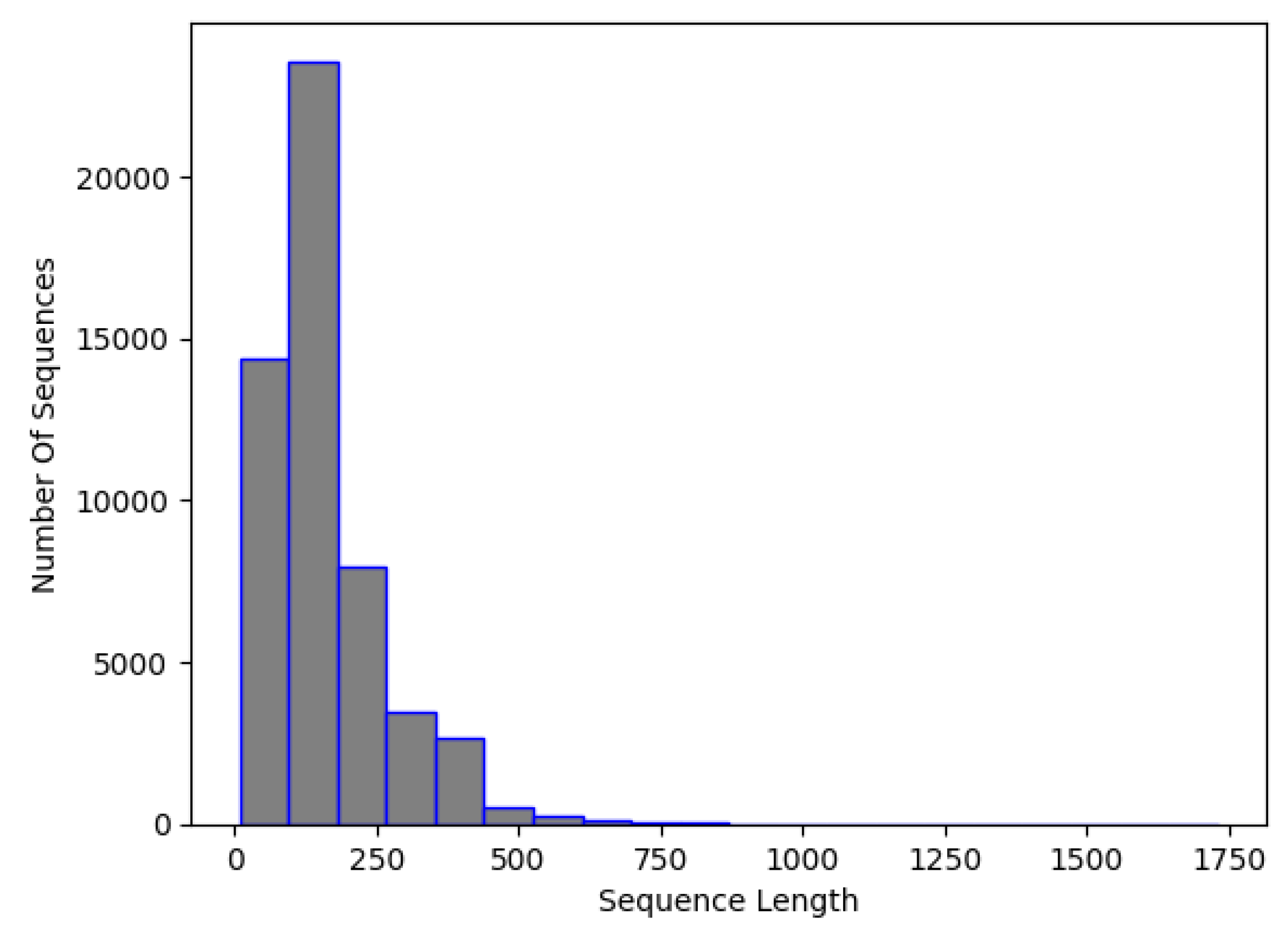
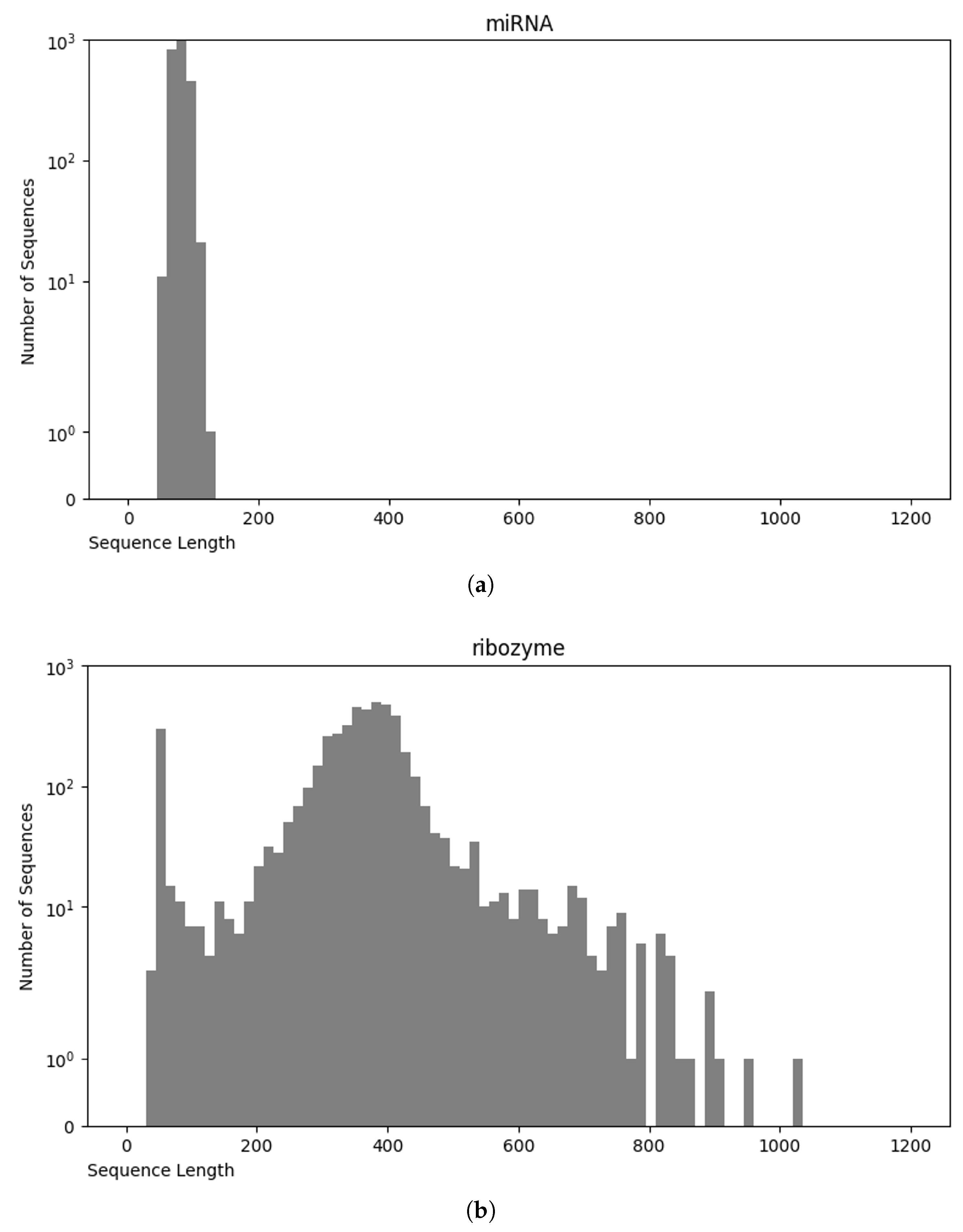
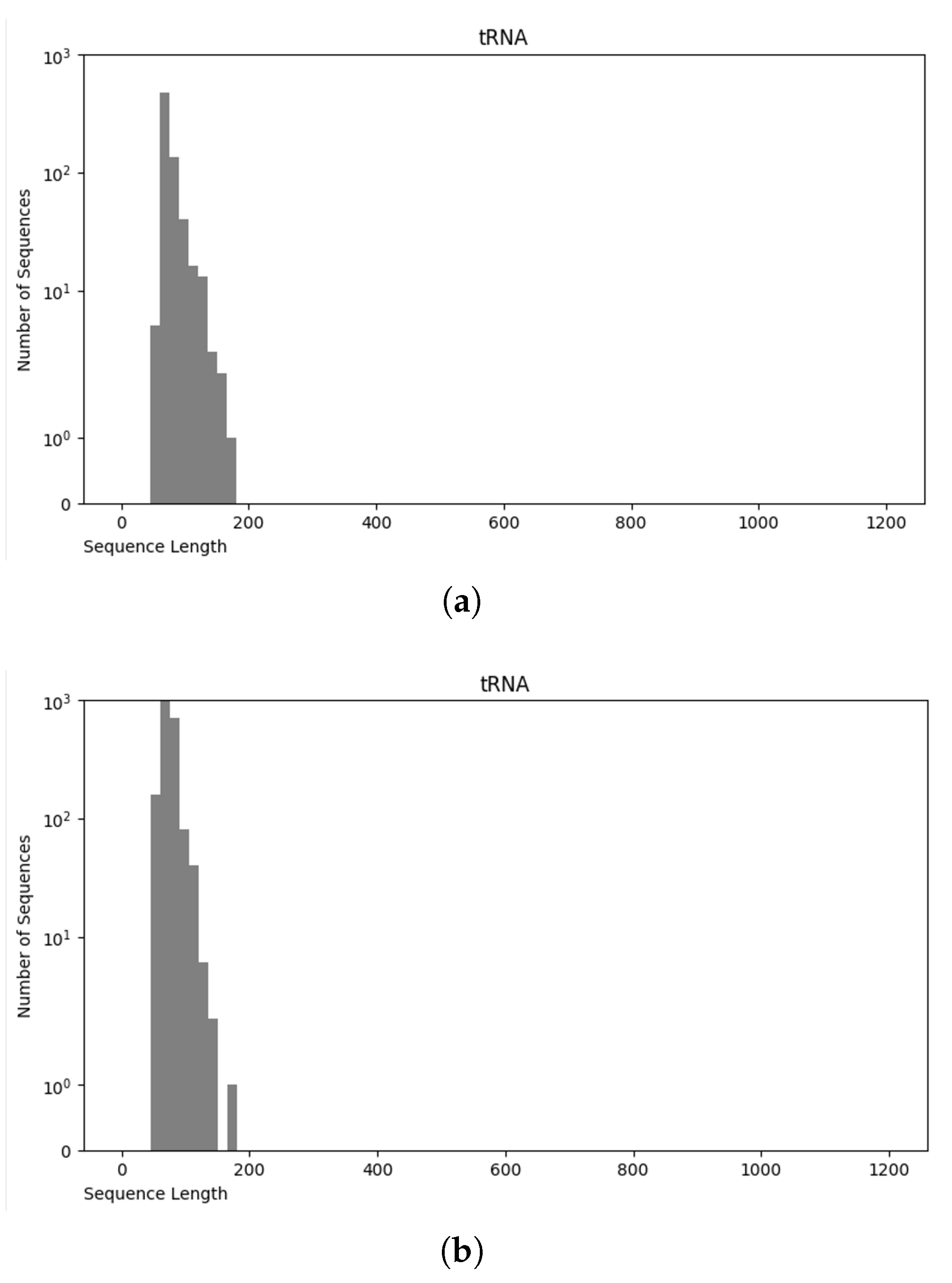
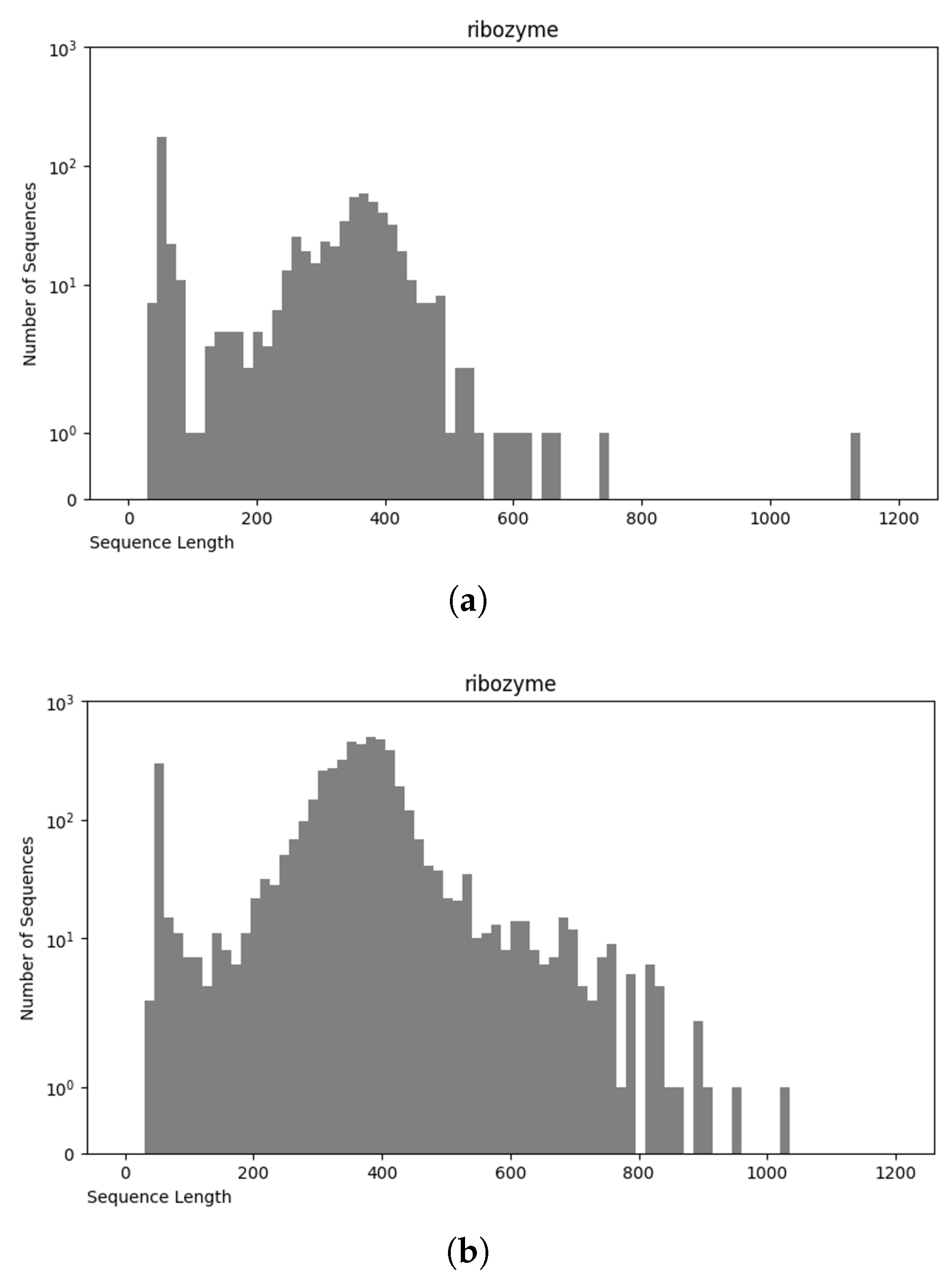
3.1. Data Preparation
3.1.1. Data Preprocessing
3.1.2. One-Hot Encoding
3.2. NCC Model Architecture
3.3. Data Collection
| >IRES ATACCTTTCTCGGCCTTTTGGCTAAGATCAAGTGTAGTATCTGTTCTTAT… >tRNA GCACCACTCTGGCCTTTTGGCTTAGATCAAGTGTAGTATCTGTTCTTATT… >tRNA ATACCTTTCTCGGCCTTTTGGCTAAGATCAAGTGTAGTATCTGTTTTTAT… >riboswitch ATTACTTCTCAGCCTTTTGGCTAAGATCAAGTGTAATAAATCTCATTGTG… >HACA-box CCAGCTCTCTTTGCCTTTTGGCTTAGATCAAGTGTAGTATCTGTTCTTTT… >tRNA ACAGCTGATGCCGCAGCTACACTATGTATTAATCGGATTTTTGAACTTGG… |
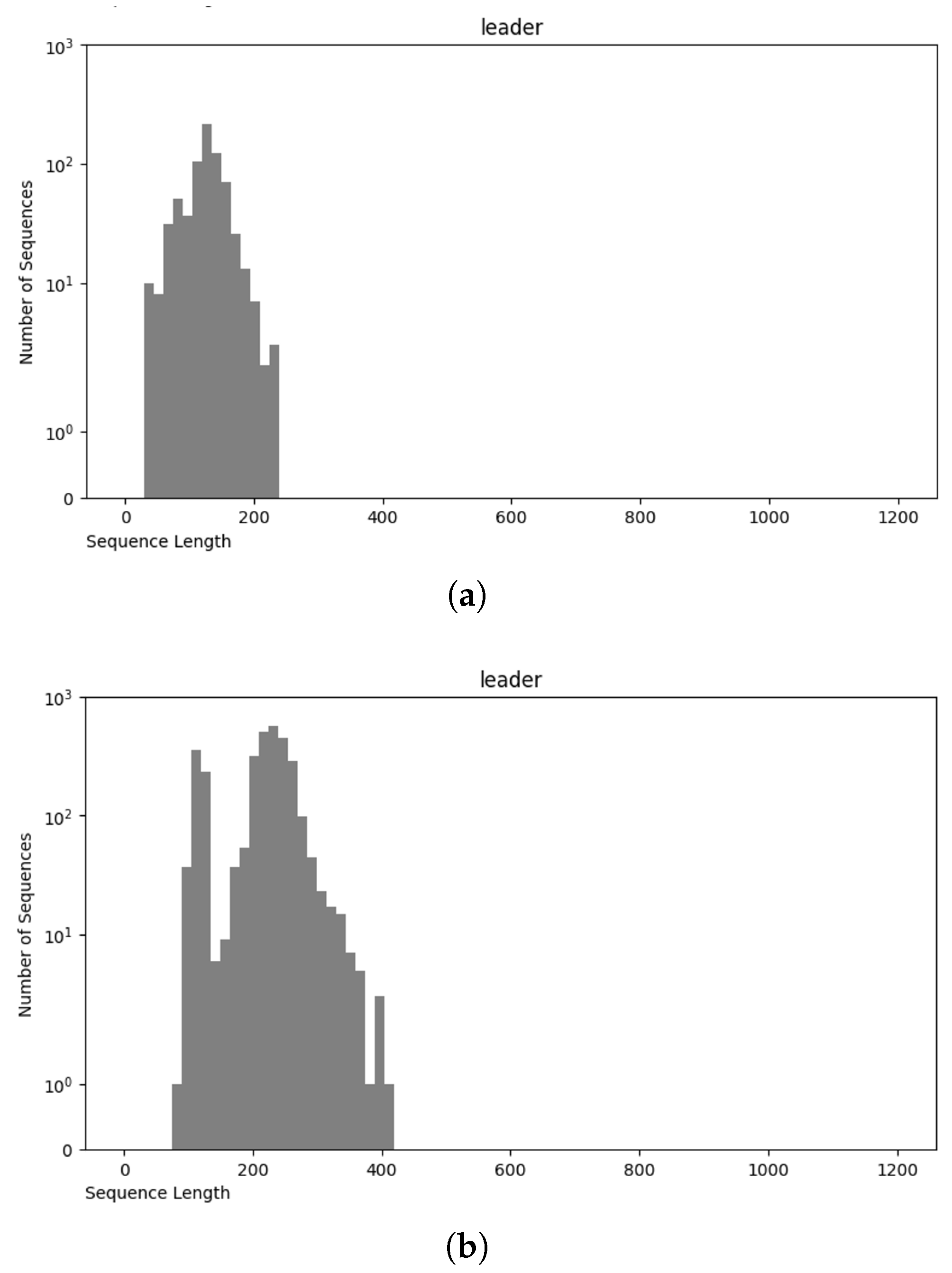
3.4. Final Dataset (NCC Dataset)
3.5. nRC Dataset
Comparative Analysis of NCC and nRC
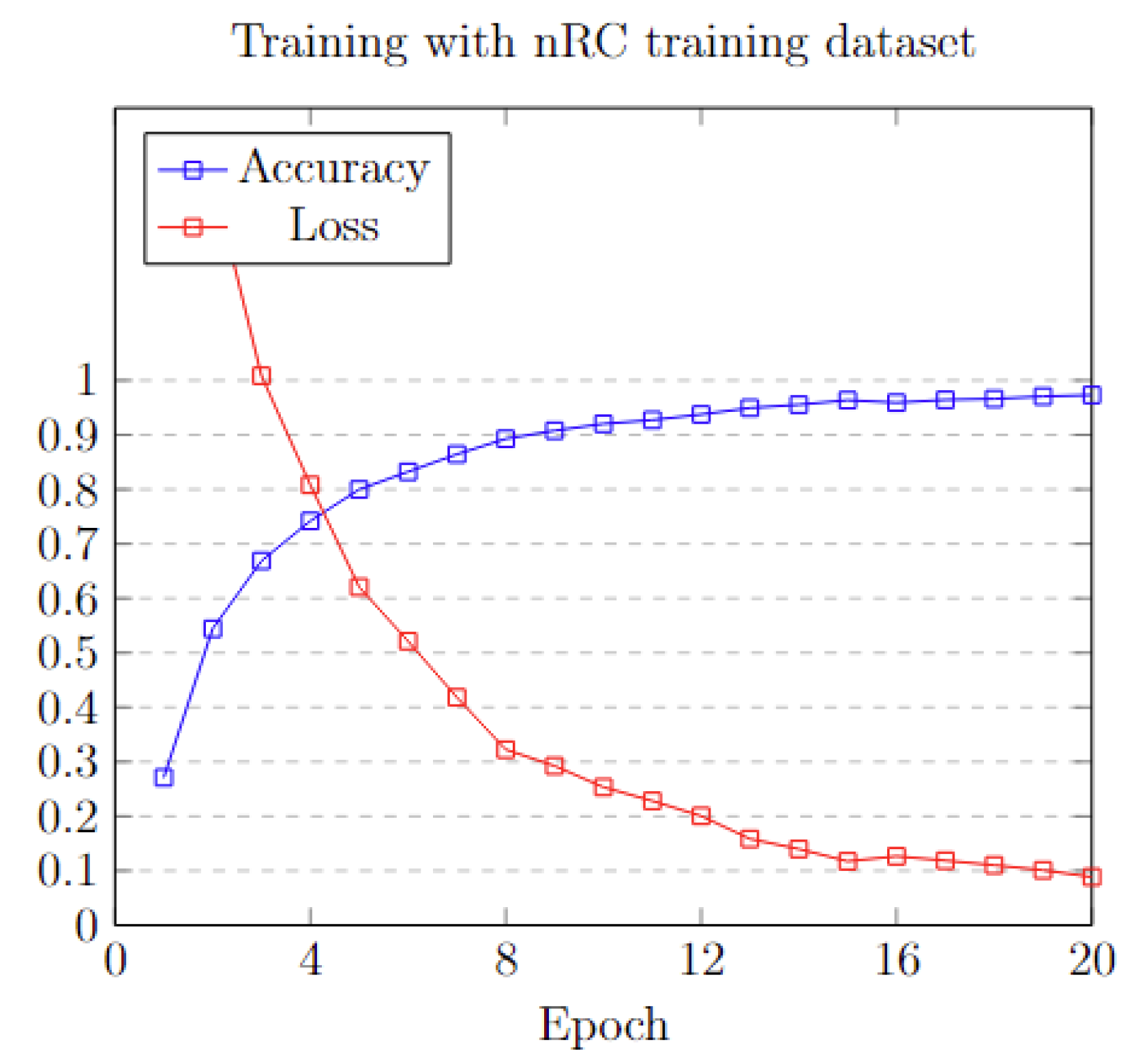
3.6. Preparing and Benchmarking the Model
- L is the loss.
- M is the number of classes.
- is a binary indicator (0 or 1) of whether class label i is the correct classification for the observation.
- is the predicted probability of the observation belonging to class i.
- is a binary indicator (0 or 1) of whether class i is the correct classification for observation o.
- is the predicted probability of observation o being of class i.
3.6.1. Performance Indicators
- Accuracy is the percentage of correctly classified cases.
- Sensitivity, also known as Recall, is the ratio of accurately anticipated positive cases to all actual positive cases.
- Precision is the ratio of correctly predicted positive cases to the total predicted positive cases.
- The F1-score is the harmonic mean of a model’s precision and recall.
- The Matthews correlation coefficient (MCC) is an effective metric for unbalanced classes, well-known for evaluating this task.
- : the number of times class k truly occurred
- : the number of times class k was predicted
- c: the total number of samples correctly predicted
- s: the total number of samples
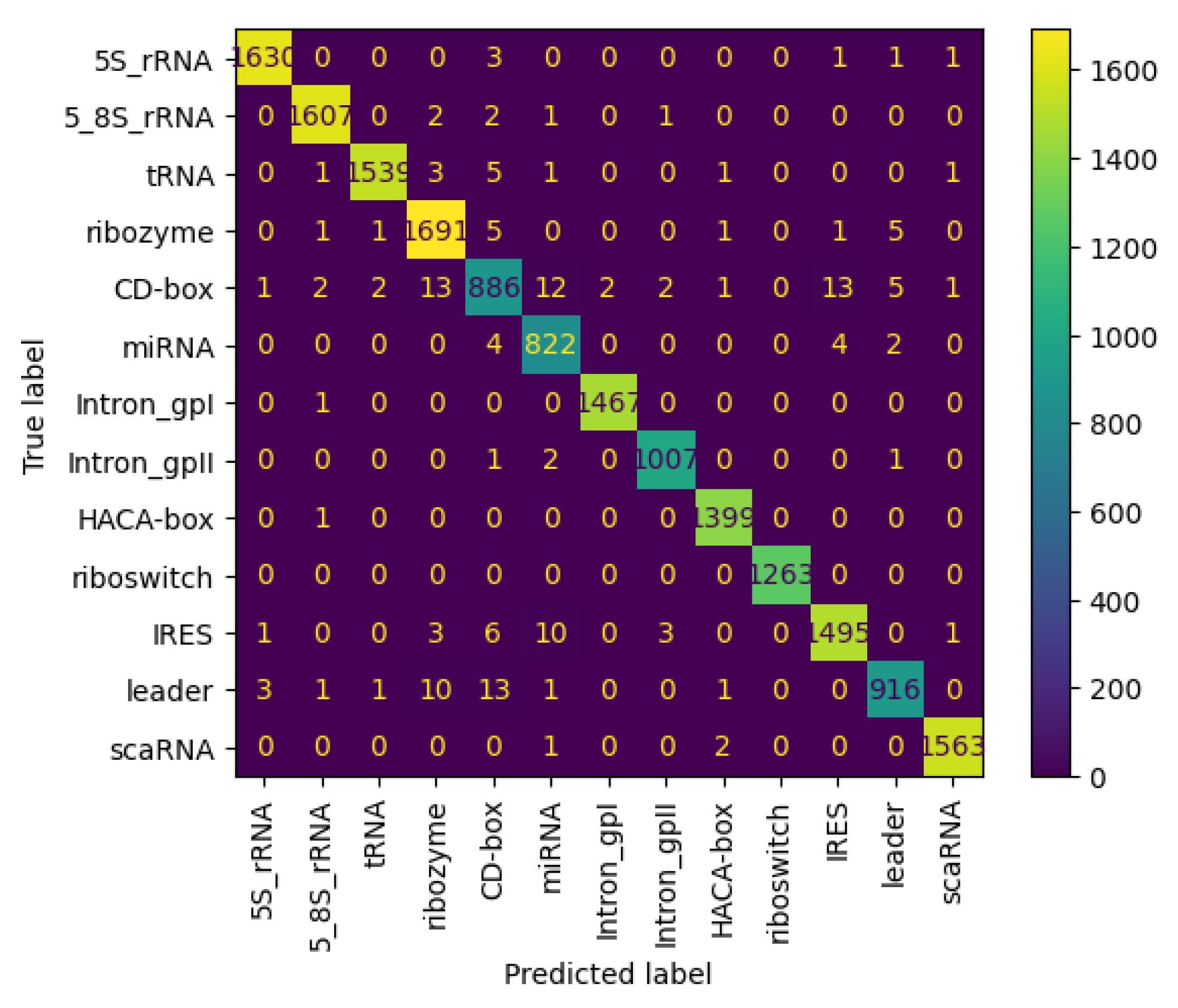
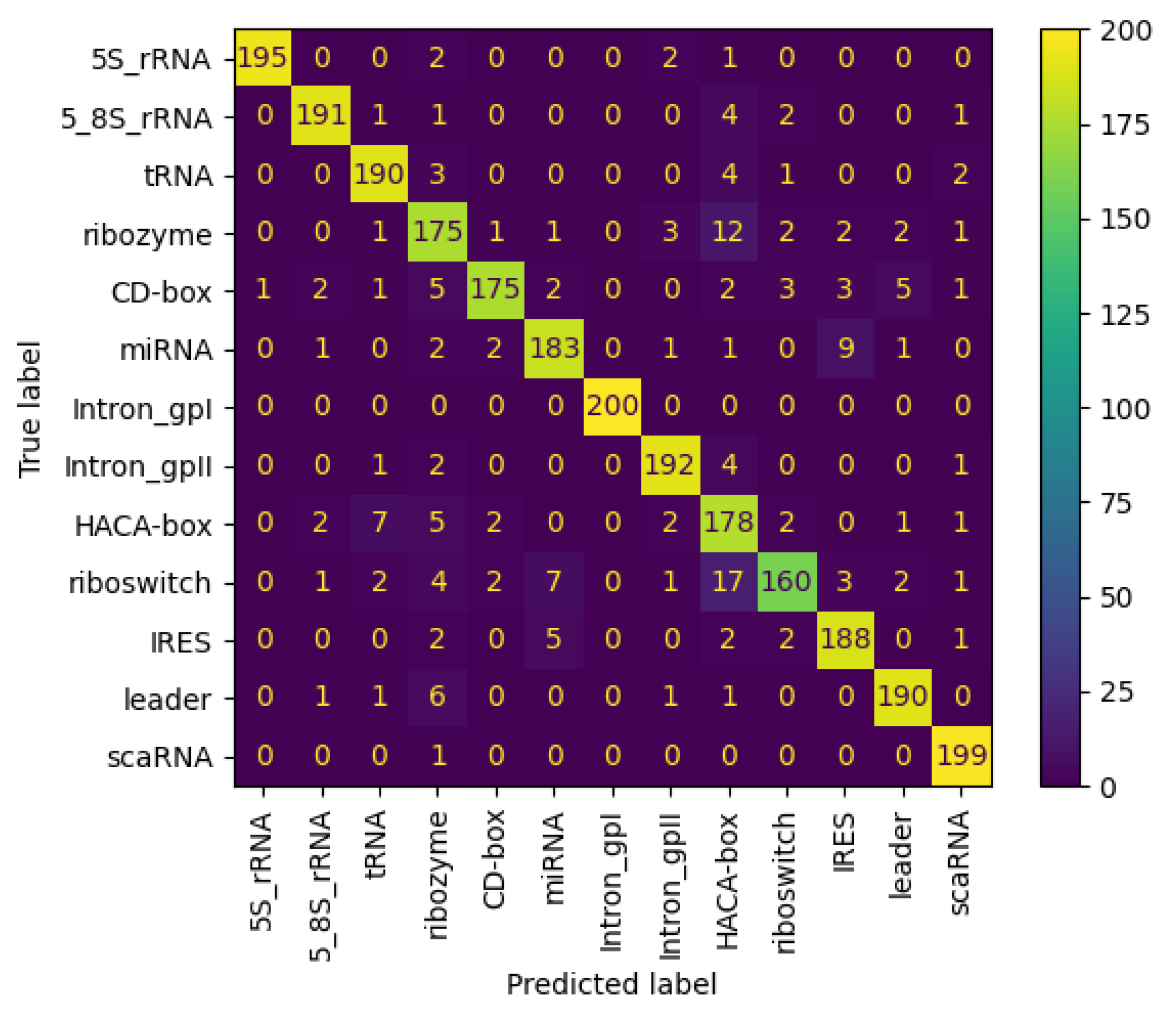
3.6.2. Results on NCC Dataset
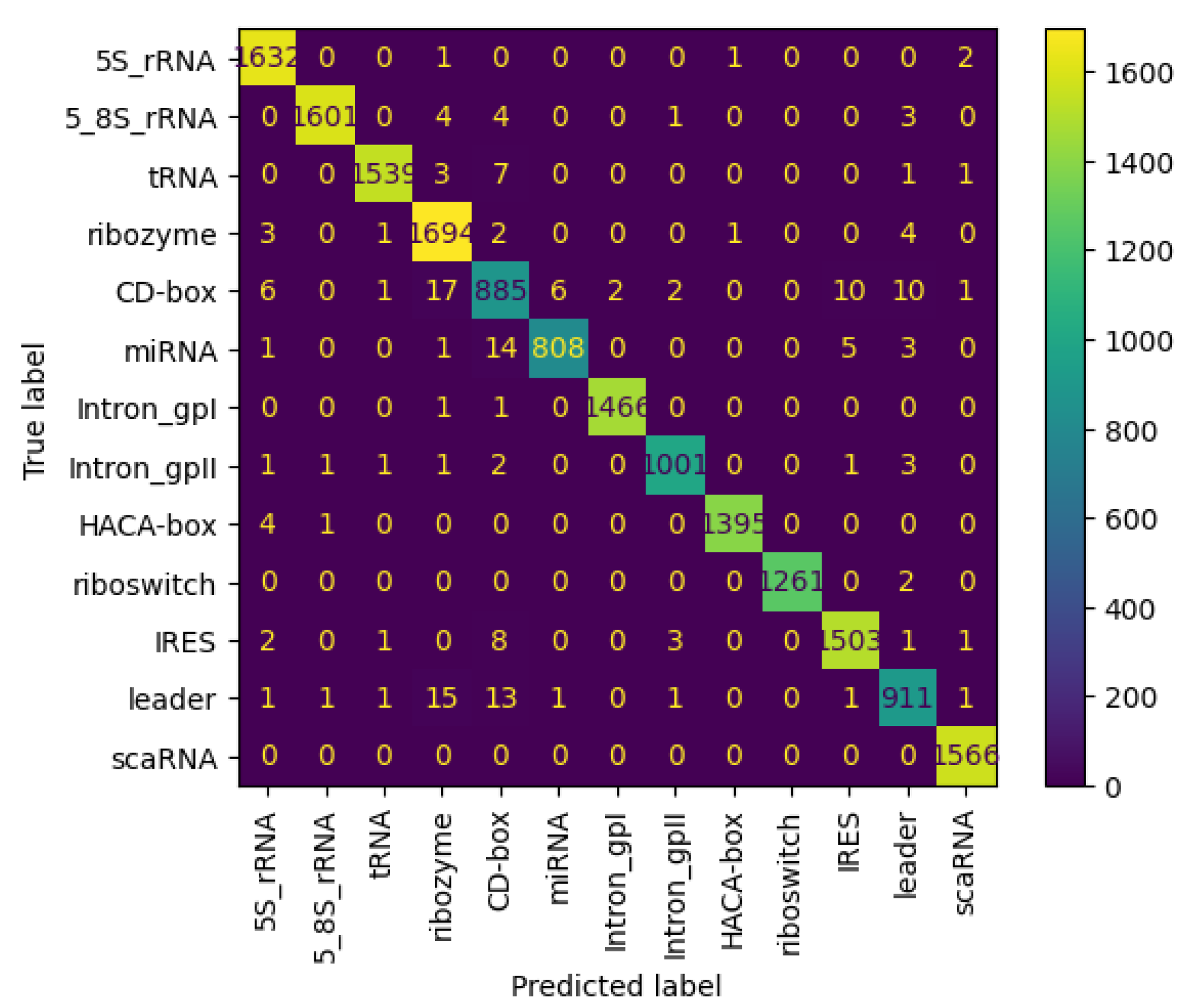
3.6.3. Results on nRC Dataset
4. Conclusions and Future Work
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hüttenhofer, A.; Vogel, J. Experimental approaches to identify non-coding RNAs. Nucleic Acids Res. 2006, 34, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.F.; Doolittle, R.F. Progressive sequence alignment as a prerequisitetto correct phylogenetic trees. J. Mol. Evol. 1987, 25, 351–360. [Google Scholar] [CrossRef] [PubMed]
- NCC: Non-Coding RNA Classifier. Available online: https://github.com/vassilas/NCC (accessed on 24 April 2025).
- Mattick, J.; Makunin, I. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S. Rfam: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef]
- RNAcentral Consortium. RNAcentral 2021: Secondary structure integration, improved sequence search and new member databases. Nucleic Acids Res. 2020, 49, D212–D220. [Google Scholar] [CrossRef]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.; Rivas, E.; Eddy, S.; Bateman, A.; Finn, R.; Petrov, A. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2017, 46, D335–D342. [Google Scholar] [CrossRef] [PubMed]
- Andrikos, C.; Makris, E.; Kolaitis, A.; Rassias, G.; Pavlatos, C.; Tsanakas, P. Knotify: An Efficient Parallel Platform for RNA Pseudoknot Prediction Using Syntactic Pattern Recognition. Methods Protoc. 2022, 5, 14. [Google Scholar] [CrossRef]
- Makris, E.; Kolaitis, A.; Andrikos, C.; Moulos, V.; Tsanakas, P.; Pavlatos, C. Knotify+: Toward the Prediction of RNA H-Type Pseudoknots, Including Bulges and Internal Loops. Biomolecules 2023, 13, 308. [Google Scholar] [CrossRef]
- Koroulis, C.; Makris, E.; Kolaitis, A.; Tsanakas, P.; Pavlatos, C. Syntactic Pattern Recognition for the Prediction of L-Type Pseudoknots in RNA. Appl. Sci. 2023, 13, 5168. [Google Scholar] [CrossRef]
- Makris, E.; Kolaitis, A.; Andrikos, C.; Moulos, V.; Tsanakas, P.; Pavlatos, C. An Intelligent Grammar-Based Platform for RNA H-type Pseudoknot Prediction. In Artificial Intelligence Applications And Innovations. AIAI 2022 IFIP WG 12.5 International Workshops; Springer: Cham, Switzerland, 2022; pp. 174–186. [Google Scholar]
- Jabbari, H.; Wark, I.; Montemagno, C.; Will, S. Knotty: Efficient and accurate prediction of complex RNA pseudoknot structures. Bioinformatics 2018, 34, 3849–3856. [Google Scholar] [CrossRef]
- Sato, K.; Kato, Y.; Hamada, M.; Akutsu, T.; Asai, K. IPknot: Fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics 2011, 27, i85–i93. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Rastegari, B.; Condon, A.; Hoos, H.H. HotKnots: Heuristic prediction of RNA secondary structures including pseudoknots. RNA 2005, 11, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Bellaousov, S.; Mathews, D.H. ProbKnot: Fast prediction of RNA secondary structure including pseudoknots. RNA 2010, 16, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Panwar, B.; Arora, A.; Raghava, G. Prediction and classification of ncRNAs using structural information. BMC Genom. 2014, 15, 127. [Google Scholar] [CrossRef]
- Li, G.; Zhao, B.; Su, X.; Yang, Y.; Hu, P.; Zhou, X.; Hu, L. Discovering consensus regions for interpretable identification of RNA N6-methyladenosine modification sites via graph contrastive clustering. IEEE J. Biomed. Health Inform. 2024, 28, 2362–2372. [Google Scholar] [CrossRef]
- Qiu, S.; Liu, R.; Liang, Y. GR-m6A: Prediction of N6-methyladenosine sites in mammals with molecular graph and residual network. Comput. Biol. Med. 2023, 163, 107202. [Google Scholar] [CrossRef]
- Childs, L.; Nikoloski, Z.; May, P.; Walther, D. Identification and classification of ncRNA molecules using graph properties. Nucleic Acids Res. 2009, 37, e66. [Google Scholar] [CrossRef]
- Nithin, C.; Mukherjee, S.; Basak, J.; Bahadur, R. NCodR: A multi-class support vector machine classification to distinguish non-coding RNAs in Viridiplantae. Quant. Plant Biol. 2022, 3, e23. [Google Scholar] [CrossRef]
- Dunkel, H.; Wehrmann, H.; Jensen, L.R.; Kuss, A.W.; Simm, S. MncR: Late Integration Machine Learning Model for Classification of ncRNA Classes Using Sequence and Structural Encoding. Int. J. Mol. Sci. 2023, 24, 8884. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, S.; Zhang, H.; Qiu, Z.; Zhong, X.; Liu, H.; Liu, Y. ncRFP: A Novel end-to-end Method for Non-Coding RNAs Family Prediction Based on Deep Learning. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 784–789. [Google Scholar] [CrossRef]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Briefings Bioinform. 2016, 18, 851–869. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Zhou, Z.; Liu, H.; Davuluri, R. DNABERT: Pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics 2021, 37, 2112–2120. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.; Cornman, A.; Kellogg, E.; Ovchinnikov, S.; Girguis, P. Genomic language model predicts protein co-regulation and function. Nat. Commun. 2024, 15, 2880. [Google Scholar] [CrossRef] [PubMed]
- Fiannaca, A.; La Rosa, M.; La Paglia, L.; Rizzo, R.; Urso, A. nRC: Non-coding RNA Classifier based on structural features. BioData Min. 2017, 10, 27. [Google Scholar] [CrossRef]
- Wang, L.; Zhong, X.; Wang, S.; Liu, Y. ncDLRES: A novel method for non-coding RNAs family prediction based on dynamic LSTM and ResNet. BMC Bioinform. 2021, 22, 447. [Google Scholar] [CrossRef]
- Chen, K.; Zhu, X.; Hao, L.; Wang, J.; Liu, Z.; Liu, Y. ncDENSE: A novel computational method based on a deep learning framework for non-coding RNAs family prediction. BMC Genom. 2022, 24, 68. [Google Scholar] [CrossRef]
- Avila Santos, A.P.; de Almeida, B.L.S.; Bonidia, R.P.; Stadler, P.F.; Stefanic, P.; Mandic-Mulec, I.; Rocha, U.; Sanches, D.S.; de Carvalho, A.C.P.L.F. BioDeepfuse: A hybrid deep learning approach with integrated feature extraction techniques for enhanced non-coding RNA classification. RNA Biol. 2024, 21, 1–12. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhao, Z.; Fan, X.; Yuan, Z.; Mao, Q.; Yao, Y. Review of machine learning methods for RNA secondary structure prediction. PLoS Comput. Biol. 2021, 17, e1009291. [Google Scholar] [CrossRef]
- Schuster, M.; Paliwal, K. Bidirectional recurrent neural networks. IEEE Trans. Signal Process. 1997, 45, 2673–2681. [Google Scholar] [CrossRef]
- Chollet, F. Others Keras. 2015. Available online: https://keras.io (accessed on 4 April 2025).
- Zhao, J.; Li, Y.; Wang, C.; Zhang, H.; Zhang, H.; Jiang, B.; Guo, X.; Song, X. IRESbase: A Comprehensive Database of Experimentally Validated Internal Ribosome Entry Sites. Genom. Proteom. Bioinform. 2020, 18, 129–139. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Kingma, D.; Ba, J. Adam: A Method for Stochastic Optimization. In Proceedings of the International Conference on Learning Representations, Banff, AB, Canada, 14–16 April 2014. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IUPAC Code | Meaning | Complement | Encoding |
|---|---|---|---|
| A | A | T | 1000 0000 0000 0000 |
| C | C | G | 0100 0000 0000 0000 |
| G | G | C | 0010 0000 0000 0000 |
| T/U | T | A | 0001 0000 0000 0000 |
| M | A or C | K | 0000 1000 0000 0000 |
| R | A or G | Y | 0000 0100 0000 0000 |
| W | A or T | W | 0000 0010 0000 0000 |
| S | C or G | S | 0000 0001 0000 0000 |
| Y | C or T | R | 0000 0000 1000 0000 |
| K | G or T | M | 0000 0000 0100 0000 |
| V | A or C or G | B | 0000 0000 0010 0000 |
| H | A or C or T | D | 0000 0000 0001 0000 |
| D | A or G or T | H | 0000 0000 0000 1000 |
| B | C or G or T | V | 0000 0000 0000 0100 |
| N | G or A or T or C | N | 0000 0000 0000 0010 |
| X | None | - | 0000 0000 0000 0000 |
| Base | 4 Digits | 8 Digits |
|---|---|---|
| A | 1000 | 1000 0010 |
| T/U | 0100 | 0100 0001 |
| G | 0010 | 0010 1000 |
| C | 0001 | 0001 0100 |
| X | 0000 | 0000 0000 |
| RNA Family | Source | # of Sequences |
|---|---|---|
| IRES | Rfam | 1472 |
| IRES | IRESbase | 1328 |
| leader | Rfam | 31,662 |
| riboswitch | Rfam | 69,465 |
| miRNA | Rfam | 387,173 |
| ribozyme | Rfam | 220,007 |
| CD-box | Rfam | 132,915 |
| HACA-box | Rfam | 36,938 |
| scaRNA | Rfam | 2962 |
| tRNA | Rfam | 1,432,442 |
| 5S_rRNA | Rfam | 140,644 |
| 5_8S_rRNA | Rfam | 4940 |
| Intron_gpI | Rfam | 2611 |
| Intron_gpII | Rfam | 15,729 |
| RNA Family | Number of Sequences |
|---|---|
| IRES | 2800 |
| leader | 3061 |
| riboswitch | 3791 |
| miRNA | 4317 |
| ribozyme | 4630 |
| CD-box | 4661 |
| HACA-box | 4931 |
| scaRNA | 2962 |
| tRNA | 4882 |
| 5S_rRNA | 4882 |
| 5_8S_rRNA | 4940 |
| Intron_gpI | 2611 |
| Intron_gpII | 4409 |
| Number of epochs | 20/50/100 |
| Batch size | 128 |
| Steps per epoch | Default = 277 |
| Optimizer | Adam |
| Loss function | Categorical Cross Entropy |
| Shuffle train data | True |
| One-Hot Enc | Accuracy | Sensitivity | Precision | F-Score | MCC |
|---|---|---|---|---|---|
| 8 Digits | 0.98922 | 0.98651 | 0.98789 | 0.98718 | 0.98828 |
| 4 Digits | 0.99054 | 0.98864 | 0.98886 | 0.98873 | 0.98971 |
| Model/Method | Accuracy | Sensitivity | Precision | F-Score | MCC |
|---|---|---|---|---|---|
| RNAcon | 0.3737 | 0.3787 | 0.4500 | 0.3605 | 0.3341 |
| GraPPLE | 0.6487 | 0.6684 | 0.7325 | 0.7050 | 0.6857 |
| nRC | 0.6960 | 0.6889 | 0.6878 | 0.6878 | 0.6627 |
| ncRFP | 0.7972 | 0.7878 | 0.7904 | 0.7883 | 0.7714 |
| ncDLRES | 0.8430 | 0.8344 | 0.8419 | 0.8407 | 0.8335 |
| ncDENSE | 0.8687 | 0.8677 | 0.8703 | 0.8667 | 0.8574 |
| NCC 4d | 0.9269 | 0.9269 | 0.9286 | 0.9268 | 0.9210 |
| NCC 8d | 0.9292 | 0.9292 | 0.9311 | 0.9293 | 0.9234 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilas, K.; Makris, E.; Pavlatos, C.; Maglogiannis, I. NCC—An Efficient Deep Learning Architecture for Non-Coding RNA Classification. Technologies 2025, 13, 196. https://doi.org/10.3390/technologies13050196
Vasilas K, Makris E, Pavlatos C, Maglogiannis I. NCC—An Efficient Deep Learning Architecture for Non-Coding RNA Classification. Technologies. 2025; 13(5):196. https://doi.org/10.3390/technologies13050196
Chicago/Turabian StyleVasilas, Konstantinos, Evangelos Makris, Christos Pavlatos, and Ilias Maglogiannis. 2025. "NCC—An Efficient Deep Learning Architecture for Non-Coding RNA Classification" Technologies 13, no. 5: 196. https://doi.org/10.3390/technologies13050196
APA StyleVasilas, K., Makris, E., Pavlatos, C., & Maglogiannis, I. (2025). NCC—An Efficient Deep Learning Architecture for Non-Coding RNA Classification. Technologies, 13(5), 196. https://doi.org/10.3390/technologies13050196