Transcriptome Sequencing and Chemical Analysis Reveal the Formation Mechanism of White Florets in Carthamus tinctorius L.

Abstract
1. Introduction
2. Results
2.1. Contents of Safflower Yellow Pigments (SYP) and Safflower Red Pigments (SRP)
2.2. De Novo Transcriptome Assembly and RNA-Seq Analyses
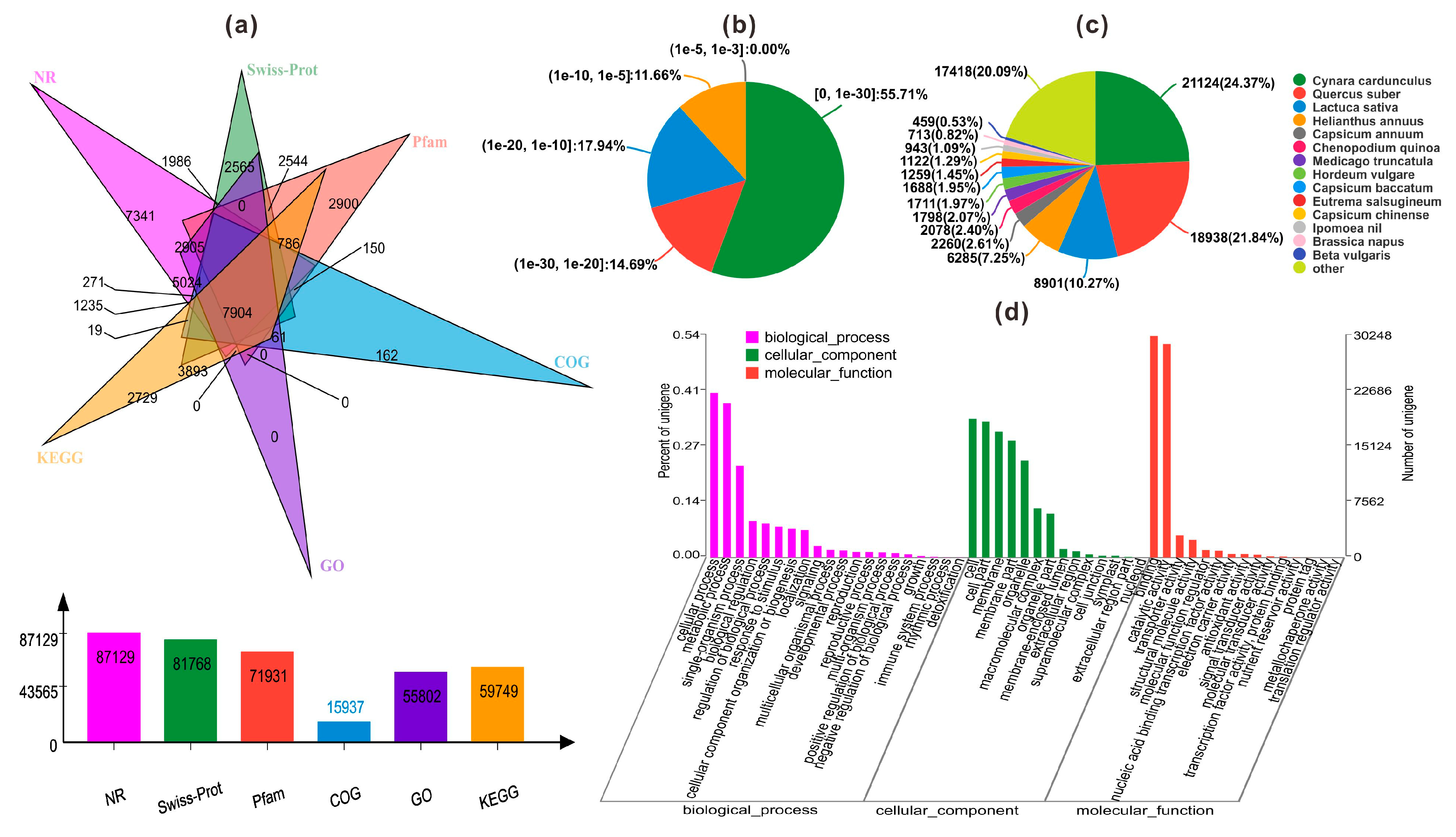
2.3. Functional Annotation and Classification
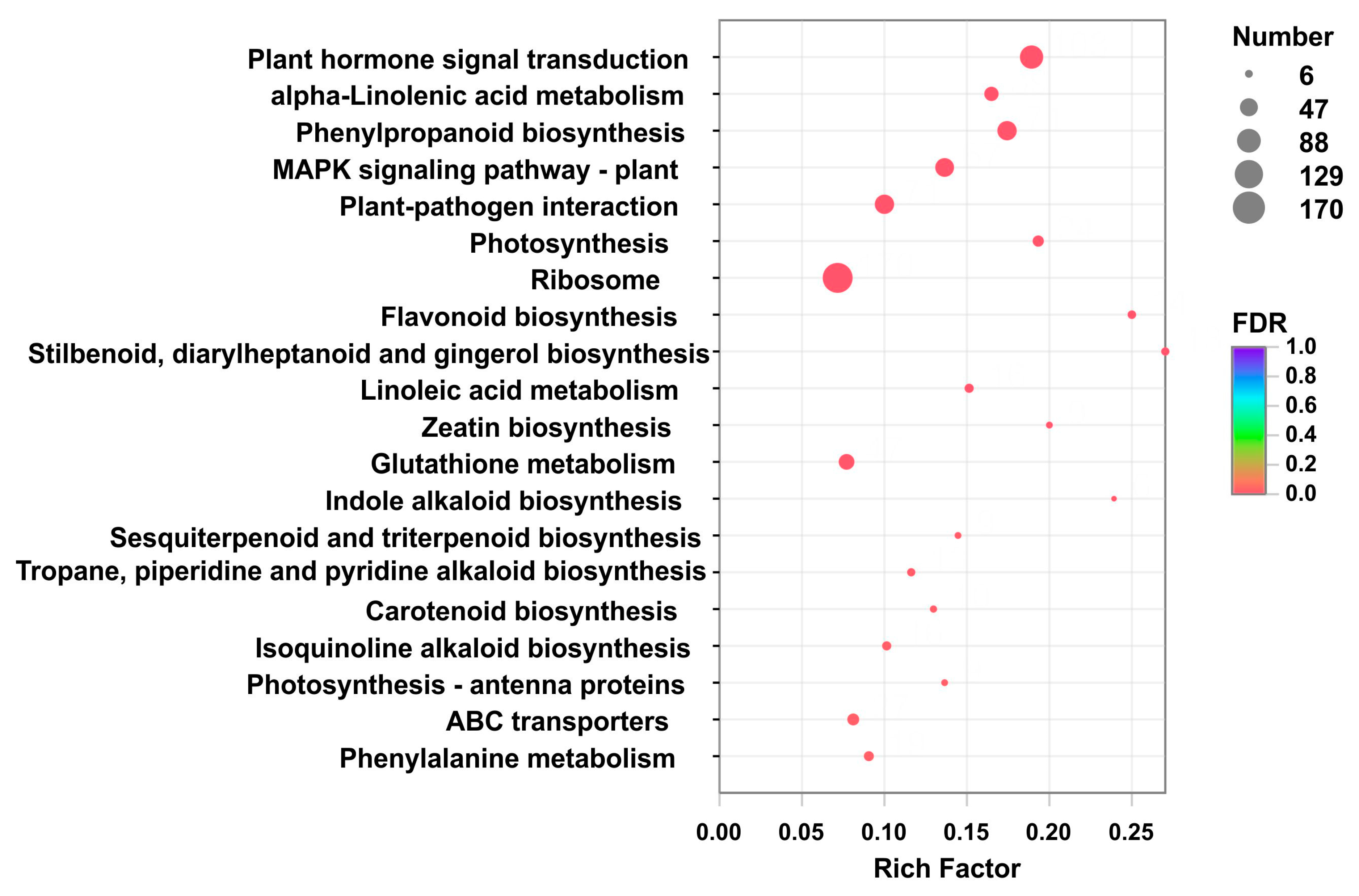
2.4. Identification and Functional Enrichment Analysis of Differentially Expressed Genes (DEGs)
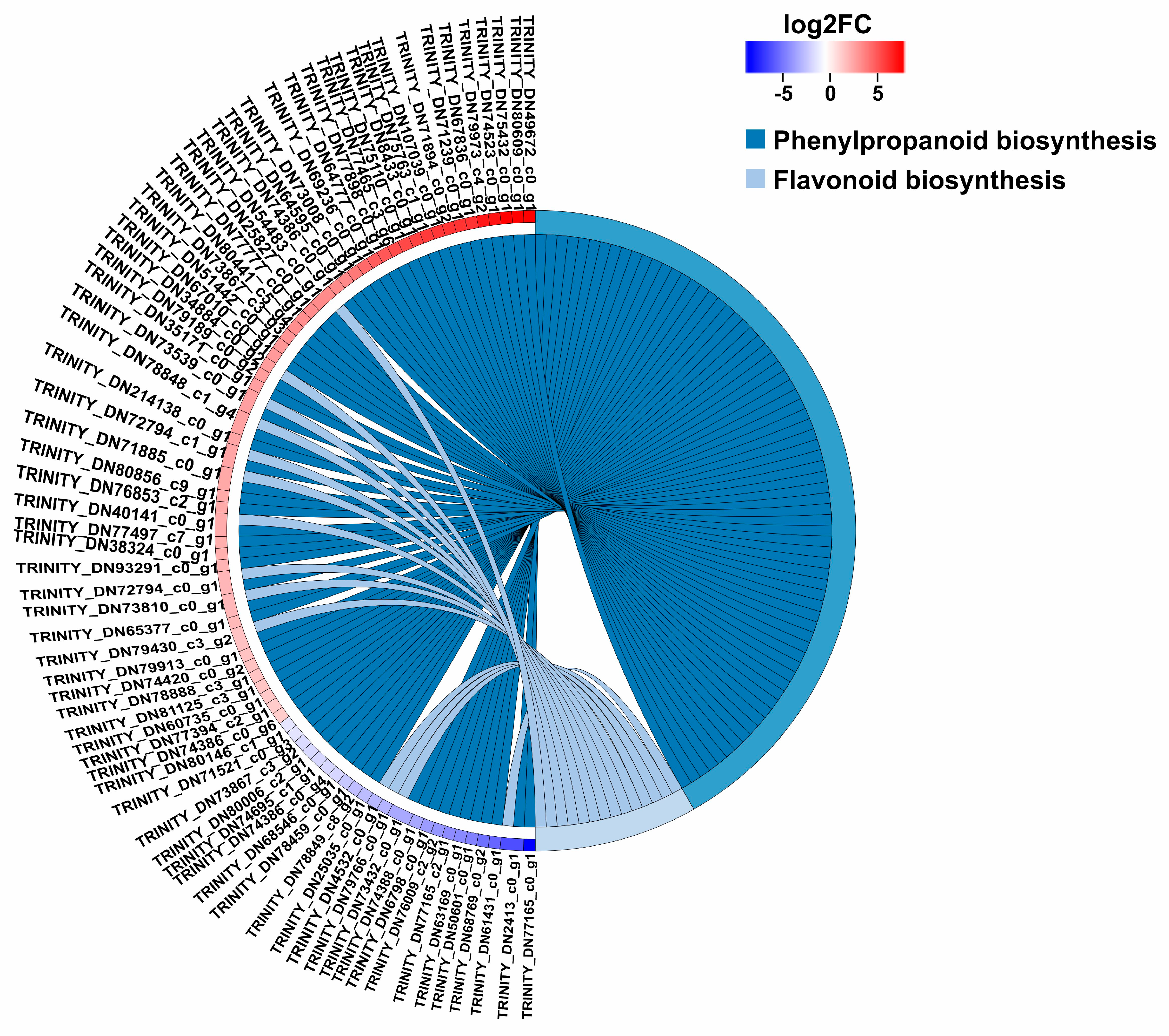
2.5. Genes Involved in the Safflower Pigment Biosynthesis
2.6. Characterization of 4 Chalcone Synthase (CHS) Unigenes
2.7. Candidate Genes Responsible for the Decrease of Safflower Pigments (SP) in Xinhonghua No. 7 (WXHH)
3. Discussion
4. Materials and Methods
4.1. Plants Materials
4.2. Preparation of Reference Solutions
4.3. Preparation of Samples and Content Analysis
4.4. RNA Extraction, Library Construction, and RNA Sequencing
4.5. De Novo Transcriptome Assembly and Annotation
4.6. Differential Expression and Functional Enrichment Analysis
4.7. Gene Expression Validation and Structure Analysis
4.8. Sequence Analysis of 4 CHS Unigenes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SP | safflower pigments |
| QCGs | quinochalcone C-glycosides |
| WXHH | Xinhonghua No. 7 |
| RYH | Yunhong No. 2 |
| PAL | phenylalanine ammonia-lyase |
| 4CL | 4-coumarate-CoA ligase |
| C4H | trans-cinnamate 4-monooxygenase |
| CHS | chalcone synthase |
| CHI | chalcone isomerase |
| F3H | flavanone 3-hydroxylase |
| DEGs | differentially expressed unigenes |
| SYP | safflower yellow pigments |
| SRP | safflower red pigments |
| HSYA | hydroxysafflor yellow A |
| RT-qPCR | reverse transcription-quantitative real-time PCR |
| CDS | coding sequence |
| SNP | single nucleotide polymorphisms |
| SSR | simple sequence repeats |
| KO | KEGG Orthology |
| TCM | Traditional Chinese Medicine |
References
- Huang, X.; Yan, Z.; Liu, S.; Wang, X.; Song, S. Investigation of chemical constituents of safflower and their tyrosinase inhibitory activity. J. Asian Nat. Prod. Res. 2019, 31, 248–256. [Google Scholar] [CrossRef]
- Costantini, R.; Berghe, I.V.; Izzo, F.C. New insights into the fading problems of safflower red dyed textiles through a HPLC-PDA and colorimetric study. J. Cult. Herit. 2019, 38, 37–45. [Google Scholar] [CrossRef]
- Commission, C.P. Pharmacopoeia of the People’s Republic of China; China Medical Science Press: Beijing, China, 2015. [Google Scholar]
- Zhou, F.; Zhao, M.; Tu, P. Stimultaneous determination of four nucleosides in Carthamus tinctorius L. and Safllower injection using high performance liquid chromatography. J. Chin. Pharm. Sci. 2009, 18, 326–330. [Google Scholar]
- Luo, Z.; Zeng, H.; Ye, Y.; Liu, L.; Li, S.; Zhang, J.; Luo, R. Safflower polysaccharide inhibits the proliferation and metastasis of MCF-7 breast cancer cells. Mol. Med. Rep. 2015, 11, 4611–4616. [Google Scholar] [CrossRef]
- Luo, L.; Liu, S.; Yan, Z.; Lin, B.; Wang, X.; Huang, X.; Song, S. Tetrahydro-β-Carboline alkaloids from Carthamus tinctorius L. with tyrosinase inhibitory activity. Phytochem. Lett. 2017, 22, 107–112. [Google Scholar]
- Si, W.; Yang, W.; Guo, D.; Wu, J.; Zhang, J.; Qiu, S.; Yao, C.; Cui, Y.; Wu, W. Selective ion monitoring of quinochalcone C-glycoside markers for the simultaneous identification of Carthamus tinctorius L. in eleven Chinese patent medicines by UHPLC/QTOF MS. J. Pharm. Biomed. Anal. 2016, 117, 510–521. [Google Scholar] [CrossRef]
- Asgarpanah, J.; Kazemivash, N. Phytochemistry, pharmacology and medicinal properties of Carthamus tinctorius L. Chin. J. Integr. Med. 2013, 19, 153–159. [Google Scholar] [CrossRef]
- Tu, Y.; Xue, Y.; Guo, D.; Sun, L.; Guo, M. Carthami flos: A review of its ethnopharmacology, pharmacology and clinical applications. Rev. Bras. Farmacogn. 2015, 25, 553–566. [Google Scholar] [CrossRef]
- Emongor, V. Safflower (Carthamus tinctorius L.) the Underutilized and Neglected Crop: A Review. Asian J. Plant Sci. 2010, 9, 299–306. [Google Scholar] [CrossRef]
- Liu, J.-X.; Zhang, C.-X.; Hu, Y.; Zhang, M.-H.; Wang, Y.-N.; Qian, Y.-X.; Yang, J.; Yang, W.-Z.; Jiang, M.-M.; Guo, D.-A. Application of multiple chemical and biological approaches for quality assessment of Carthamus tinctorius L. (safflower) by determining both the primary and secondary metabolites. Phytomedicine 2019, 58, 152826. [Google Scholar]
- Pahlavani, M.H.; Mirlohi, A.F.; Saeidi, G. Inheritance of Flower Color and Spininess in Safflower (Carthamus tinctorius L.). J. Hered. 2004, 95, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, E.; Keleshteri, R.H.; Shahbazi, M.; Moradi, F.; Sadri, M. Optimization of extraction yield of carthamine and safflower yellow pigments from safflower (Carthamus tinctorious L.) under different treatments and solvent systems. Res. J. Pharmacogn. 2015, 2, 17–23. [Google Scholar]
- Ekin, Z. Resurgence of safflower (Carthamus tinctorius L.) utilization: A global view. J. Agron. 2005, 4, 83–87. [Google Scholar]
- Machewad, G.M.; Ghatge, P.U.; Chappalwar; Jadhav, B.T.; Chappalwar, A. Studies on Extraction of Safflower Pigments and its Utilization in Ice Cream. J. Food Process. Technol. 2012, 3, 1000172. [Google Scholar]
- Kouhi, F.; Sorkheh, K.; Ercisli, S. MicroRNA expression patterns unveil differential expression of conserved miRNAs and target genes against abiotic stress in safflower. PLoS ONE 2020, 15, e0228850. [Google Scholar] [CrossRef]
- Lin, H.; Wu, Z.; Zhang, L.; Liu, H.; Chen, Y.; Fang, Y.; Qin, R. Clustering and Correlation Analysis of Traits and Molecular Markers of 11 Cultivars of Safflower. Hans J. Agric. Sci. 2019, 9, 612–629. [Google Scholar]
- Grotewold, E. The genetics and biochemistry of floral pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Amaya, R.; Delia, B. Update on natural food pigments-A mini-review on carotenoids, anthocyanins, and betalains. Food Res. Int. 2019, 124, 200–205. [Google Scholar] [CrossRef]
- Watanabe, T.; Hasegawa, N.; Yamamoto, A.; Nagai, S.; Terabe, S. Separation and Determination of Yellow and Red Safflower Pigments in Food by Capillary Electrophoresis. Biosci. Biotechnol. Biochem. 1997, 61, 1179–1183. [Google Scholar] [CrossRef]
- Salem, N.; Msaada, K.; Elkahoui, S.; Mangano, G.; Azaeiz, S.; Slimen, I.B.; Kefi, S.; Pintore, G.; Limam, F.; Marzouk, B. Evaluation of Antibacterial, Antifungal, and Antioxidant Activities of Safflower Natural Dyes during Flowering. Biomed. Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Hu, Z.; Hu, X.; Yang, J.; Guo, L.; Liu, X. Cluster Analysis of Color Value and main Yield-related Traits of Germplasm Resources in safflower (Carthamus tinctorious L.). J. Plant Genet. Resour. 2018, 19, 857–864. [Google Scholar]
- Fan, S.; Lin, N.; Shan, G.; Zuo, P.; Cui, L. Safflower yellow for acute ischemic stroke: A systematic review of randomized controlled trials. Complement. Ther. Med. 2014, 22, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Shimokoriyama, M.; Hattori, S. On the formation of carthamin in the flowers of Carthamus tinctorius. Arch. Biochem. Biophys. 1955, 54, 93–101. [Google Scholar] [CrossRef]
- Zhou, Y. Study on the Extraction of Red Safflower Pigment and Hyperlipidemia. Master’s Thesis, Tianjin University of Sciences & Technology, Tianjin, China, 2009. [Google Scholar]
- Dai, Y.; Verpoorte, R.; Choi, Y. Natural deep eutectic solvents providing enhanced stability of natural colorants from safflower (Carthamus tinctorius). Food Chem. 2014, 159, 116–121. [Google Scholar] [CrossRef]
- Kazuma, K.; Takahashi, T.; Sato, K.; Takeuchi, H.; Matsumoto, T.; Okuno, T. Quinochalcones and Flavonoids from Fresh Florets in Different Cultivars of Carthamus tinctorius L. Biosci. Biotechnol. Biochem. 2000, 64, 1588–1599. [Google Scholar] [CrossRef]
- Fatahi, N.; Carapetian, J.; Heidari, R. Comparing Stability of Carthamin and Safflower Yellow Pigments at PH, Temperture and Light, from Safflower (Carthamus tinctorius L.) Florets. Res. J. Biol. Sci. 2009, 4, 250–253. [Google Scholar]
- Zhang, L.-L.; Tian, K.; Tang, Z.-H.; Chen, X.-J.; Bian, Z.-X.; Wang, Y.-T.; Lu, J.-J. Phytochemistry and Pharmacology of Carthamus tinctorius L. Am. J. Chin. Med. 2016, 44, 197–226. [Google Scholar] [CrossRef]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 20 June 2020).
- Luo, Q.; Liu, Y.; Qi, Y.; Jiao, S.; Tian, F.; Jiang, L.; Wang, Y. Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower colour in grape hyacinth. J. Exp. Bot. 2014, 65, 3157–3164. [Google Scholar]
- Wu, Q.; Wu, J.; Li, S.-S.; Zhang, H.-J.; Feng, C.-Y.; Yin, D.-D.; Wu, R.-Y.; Wang, L.-S. Transcriptome sequencing and metabolite analysis for revealing the blue flower formation in waterlily. BMC Genom. 2016, 17, 897. [Google Scholar] [CrossRef]
- Cho, K.; Cho, K.S.; Sohn, H.B.; Ha, I.J.; Hong, S.Y.; Lee, H.; Kim, Y.M.; Nam, M.H. Network analysis of the metabolome and transcriptome reveals novel regulation of potato pigmentation. J. Exp. Bot. 2016, 67, 1519–1533. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.-D.; Liu, F.; Tu, Y.-H.; He, B.-X.; Gao, Y.; Guo, M.-L. Expression Patterns of Three UGT Genes in Different Chemotype Safflower Lines and under MeJA Stimulus Revealed Their Potential Role in Flavonoid Biosynthesis. PLoS ONE 2016, 11, e0158159. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Liu, F.; Guo, D.; Fan, L.; Zhu, Z.; Xue, Y.; Gao, Y.; Guo, M. Molecular characterization of flavanone 3-hydroxylase gene and flavonoid accumulation in two chemotyped safflower lines in response to methyl jasmonate stimulation. BMC Plant Biol. 2016, 16, 132. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Xue, Y.; Li, D.; He, B.; Jia, X.; Dong, X.; Guo, M. Overexpression of CtCHS1 Increases Accumulation of Quinochalcone in Safflower. Front. Plant Sci. 2017, 8, 1409. [Google Scholar] [CrossRef]
- Ren, C.; Tang, X.; Chen, C.; Chen, J.; Pei, J.; Wu, Y.; Wu, Q. Cloning and expression analysis of a new chalcone isomerase gene duringflowering in safflower. Turk. J. Bot. 2019, 43, 143–150. [Google Scholar] [CrossRef]
- Liu, X.; Dong, Y.; Yao, N.; Zhang, Y.; Wang, N.; Cui, X.; Li, X.; Wang, Y.; Wang, F.; Yang, J.; et al. De Novo Sequencing and Analysis of the Safflower Transcriptome to Discover Putative Genes Associated with Safflor Yellow in Carthamus tinctorius L. Int. J. Mol. Sci. 2015, 16, 25657–25677. [Google Scholar] [CrossRef]
- Schijlen, E.G.W.M.; Vos, C.H.R.d.; Martens, S.; Jonker, H.H.; Rosin, F.M.; Molthoff, J.W.; Tikunov, Y.M.; Angenent, G.C.; Tunen, A.J.v.; Bovy, A.G. RNA Interference Silencing of Chalcone Synthase, the First Step in the Flavonoid Biosynthesis Pathway, Leads to Parthenocarpic Tomato Fruits. Plant Physiol. 2007, 144, 1520–1530. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Flavonoid Biosynthesis. A Colorful Model for Genetics, Biochemistry, Cell Biology, and Biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB–bHLH–WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef]
- Petrussa, E.; Braidot, E.; Zancani, M.; Peresson, C.; Bertolini, A.; Patui, S.; Vianello, A. Plant Flavonoids—Biosynthesis, Transport and Involvement in Stress Responses. Int. J. Mol. Sci. 2013, 14, 14950–14973. [Google Scholar] [CrossRef]
- Ferreyra, M.L.F.; Rius, S.P.; Casati, P. Flavonoids: Biosynthesis, Biological functions and Biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar]
- Tohge, T.; Souza, L.P.d.; Fernie, A.R. Current understanding of the pathways of flavonoid biosynthesis in model and crop plants. J. Exp. Bot. 2017, 68, 4013–4028. [Google Scholar] [CrossRef] [PubMed]
- He, B.-X.; Xue, Y.-R.; Tu, Y.-H.; Gao, Y.; Guo, M.-L. CtCHS4 induces the accumulation of safflower quinone chalcones in response to methyl jasmonate induction. Acta Pharm. Sin. 2018, 53, 636–645. [Google Scholar]
- Fatahi, N.; Carapetian, J.; Heidari, R. Spectrophotometric Measurement of Valuble Pigments from Petals of Safflower (Carthamus tinctorius L.) and their Identification by TLC Method. Res. J. Biol. Sci. 2008, 3, 761–763. [Google Scholar]
- Sigurdson, G.T.; Tang, P.; Giusti, M.M. Natural Colorants: Food Colorants from Natural Sources. Annu. Rev. Food Sci. 2017, 8, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Du, C.; Zhang, N.; Shi, X.; Wu, Z.; Qiao, Y. Color spaces of safflower (Carthamus tinctorius L.) for quality assessment. J. Tradit. Chin. Med. Sci. 2016, 3, 168–175. [Google Scholar] [CrossRef]
- Patanè, C.; Cosentino, S.L.; Calcagno, S.; Pulvirenti, L.; Siracusa, L. How do sowing time and plant density affect the pigments safflomins and carthamin in florets of safflower? Ind. Crop. Prod. 2020, 148, 112313. [Google Scholar] [CrossRef]
- Steberl, K.; Hartung, J.; Munz, S.; Graeff-Hönninger, S. Effect of Row Spacing, Sowing Density, and Harvest Time on Floret Yield and Yield Components of Two Safflower Cultivars Grown in Southwestern Germany. Agronomy 2020, 10, 664. [Google Scholar] [CrossRef]
- Eryiğit, T.; Yıldırım, B.; Kumlay, A.M.; Sancaktaroğlu, S. The Effects of Different Row Distances and Nitrogen Fertilizer Rates on Yield and Yield Components of Safflower (Carthamus tinctorious) Under Micro-Climate Conditions of Iğdır Plain—Turkey. In Proceedings of the 3rd International Conference on Biological, Chemical & Environmental Sciences, Kuala Lumpur, Malaysia, 21–22 September 2015; pp. 21–22. [Google Scholar]
- Kumar, S.; Ambreen, H.; Variath, M.T.; Rao, A.R.; Agarwal, M.; Kumar, A.; Goel, S.; Jagannath, A. Utilization of Molecular, Phenotypic, and Geographical Diversity to Develop Compact Composite Core Collection in the Oilseed Crop, Safflower (Carthamus tinctorius L.) through Maximization Strategy. Front. Plant. Sci. 2016, 7, 1554. [Google Scholar] [CrossRef]
- Ma, G. Study on the Extraction and Purification of Safflower Yellow and Carthamin. Master’s Thesis, Jiangnan University, Wuxi, China, 2015. [Google Scholar]
- SeqPrep. Available online: http://github.com/jstjohn/SeqPrep (accessed on 1 February 2013).
- Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files. Available online: http://github.com/najoshi/sickle (accessed on 9 February 2011).
- Grabherr, M.G.; Hass, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garciagomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: a universal tool for annotation visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Arndt, W.; Miller, B.L.; Wheeler, T.J.; Schreiber, F.; Bateman, A.; Eddy, S.R. HMMER web server: 2015 update. Nucleic Acids Res. 2015, 43, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Tang, S.; Lomsadze, A.; Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 2015, 43, e78. [Google Scholar] [CrossRef]
- Narasimhan, V.; Danecek, P.; Scally, A.; Xue, Y.; Chris, T.; Durbin, R. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 2016, 32, 1749–1751. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics rescourse portal. Nucleis Acids Res. 2012, 40, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Sudhir, K.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample No. | Variety | Color | AY | CY (g/g) | Average (CY) (g/g) | AR | Average (AR) |
|---|---|---|---|---|---|---|---|
| W1 | WXHH | White | 0.125 | 0.0080 | 0.0079 | 0.108 | 0.110 |
| W2 | WXHH | White | 0.121 | 0.0078 | 0.111 | ||
| W3 | WXHH | White | 0.124 | 0.0079 | 0.112 | ||
| R1 | RYH | Red | 0.876 | 0.0613 | 0.0588 | 4.000 | 3.337 |
| R2 | RYH | Red | 0.889 | 0.0622 | 2.910 | ||
| R3 | RYH | Red | 0.753 | 0.0529 | 3.101 |
| WXHH | RYH | |
|---|---|---|
| Average Clean Reads | 45,345,156 | 47,156,100 |
| Average Clean Bases | 6,793,392,182 | 7,063,377,560 |
| Average Error Rate (%) | 0.0229 | 0.0229 |
| Average Clean Reads Q20 (%) | 98.95 | 98.97 |
| Average Clean Reads Q30 (%) | 96.15 | 96.21 |
| Average GC Content (%) | 46.07 | 46.31 |
| Total transcripts | 304,392 | |
| Total unigenes | 225,008 | |
| Average length (bp) | 697.36 | |
| N50 (bp) | 1080 | |
| GC percent (%) | 42.32 | |
| Function | Gene | Enzyme | KO Id (EC No.) | No. |
|---|---|---|---|---|
| Phenylpropanoid biosynthesis | PAL | Phenylalanine ammonia-lyase | K10775 (4.3.1.24) | 7 |
| 4CL | 4-coumarate-CoA ligase | K01904 (6.2.1.12) | 20 | |
| C4H | Trans-cinnamate 4-monooxygenase | K00487 (1.14.14.91) | 1 | |
| Flavonoid biosynthesis | CHS | Chalcone synthase | K00660 (2.3.1.74) | 7 |
| CHI | Chalcone isomerase | K01859 (5.5.1.6) | 4 | |
| F3H | Flavanone 3-hydroxylase | K00475 (1.14.11.9) | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiang, T.; Liu, J.; Dong, Y.; Ma, Y.; Zhang, B.; Wei, X.; Liu, H.; Xiao, P. Transcriptome Sequencing and Chemical Analysis Reveal the Formation Mechanism of White Florets in Carthamus tinctorius L. Plants 2020, 9, 847. https://doi.org/10.3390/plants9070847
Qiang T, Liu J, Dong Y, Ma Y, Zhang B, Wei X, Liu H, Xiao P. Transcriptome Sequencing and Chemical Analysis Reveal the Formation Mechanism of White Florets in Carthamus tinctorius L. Plants. 2020; 9(7):847. https://doi.org/10.3390/plants9070847
Chicago/Turabian StyleQiang, Tingyan, Jiushi Liu, Yuqing Dong, Yinbo Ma, Bengang Zhang, Xueping Wei, Haitao Liu, and Peigen Xiao. 2020. "Transcriptome Sequencing and Chemical Analysis Reveal the Formation Mechanism of White Florets in Carthamus tinctorius L." Plants 9, no. 7: 847. https://doi.org/10.3390/plants9070847
APA StyleQiang, T., Liu, J., Dong, Y., Ma, Y., Zhang, B., Wei, X., Liu, H., & Xiao, P. (2020). Transcriptome Sequencing and Chemical Analysis Reveal the Formation Mechanism of White Florets in Carthamus tinctorius L. Plants, 9(7), 847. https://doi.org/10.3390/plants9070847