High-Throughput Sequencing Facilitates Discovery of New Plant Viruses in Poland

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
2.1. Bioassay and Electron Microscopy
2.2. RT-PCR Detection
2.3. HTS for Identification of the Viruses in Selected Samples
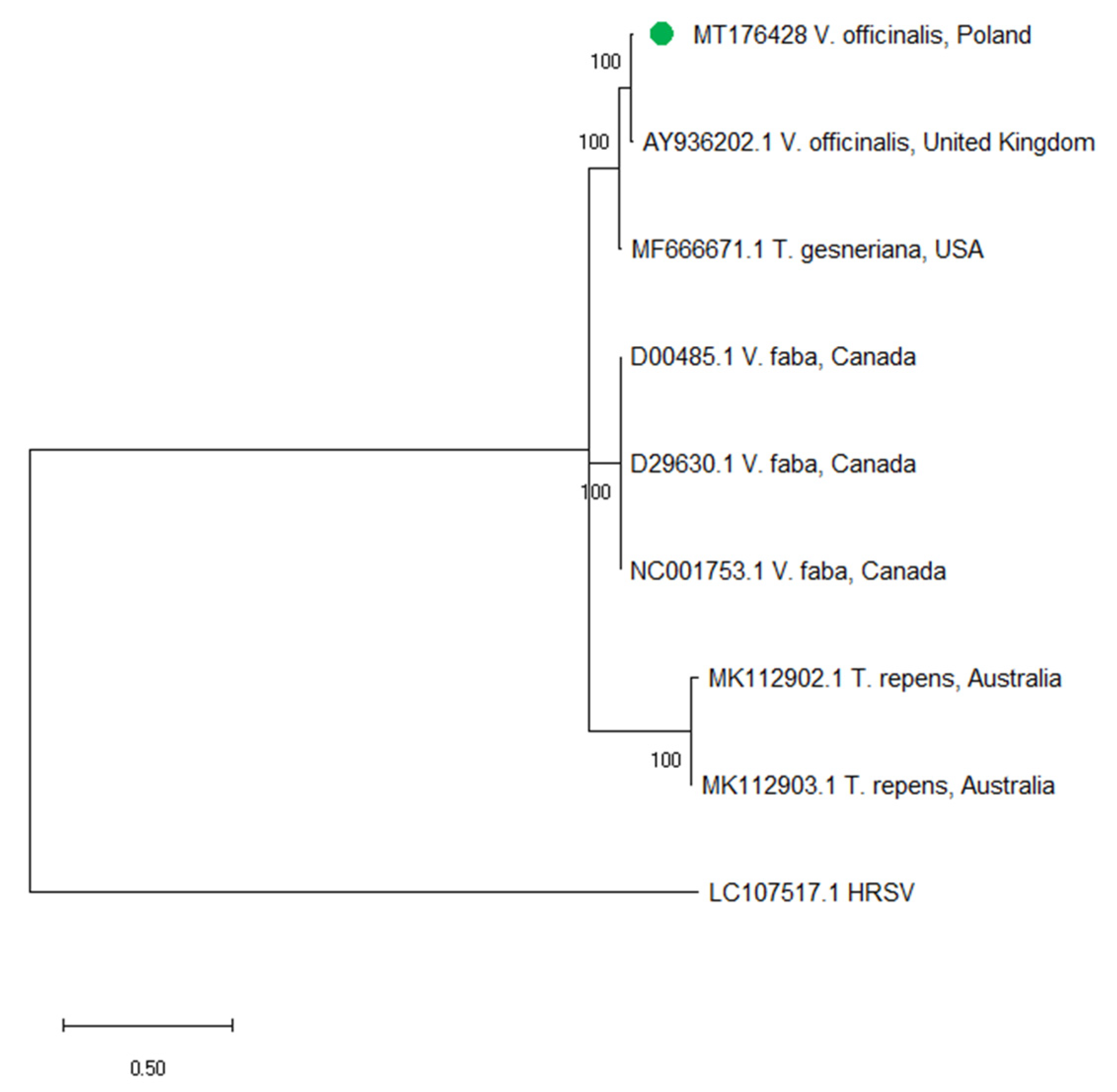
2.4. Sequence Analysis of ClYMV and MYFV
2.5. Phylogenetic Analysis of CABYV
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Bioassay and Electron Microscopy
4.3. RT-PCR Detection
4.4. Preparing the Samples for High-throughput Sequencing
4.5. Confirmation of Obtained Results by RT-PCR and Sanger Sequencing
4.6. Sequence Analysis of ClYMV and MYFV
4.7. Recombination and Phylogenetic Analyses of CABYV
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Oerke, E.C. Crop losses to pests. J. Agric. Sci. 2006, 144, 1–43. [Google Scholar] [CrossRef]
- Jones, R.A.C. Future scenarios for plant virus pathogens as climate change progresses. Adv. Virus Res. 2016, 95, 87–147. [Google Scholar] [PubMed]
- Rossinck, M. Mechanisms of plant virus evolution. Ann. Rev. Phytopathol. 1997, 35, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, M.; Koizumi, E.; Noguchi, M.; Sueda, K.; Shimura, H.; Ishikawa, N.; Matsuura, H.; Ohshima, K.; Natsuaki, T.; Kuwata, S.; et al. Infection dynamics in viral spread and interference under the synergism between Cucumber mosaic virus and Turnip mosaic virus. Mol. Plant Microbe Interact. 2012, 25, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, R.; García-Marcos, A.; Barajas, D.; Martiáñez, J.; Tenllado, F. PVX-potyvirus synergistic infections differentially alter microRNA accumulation in Nicotiana benthamiana. Virus Res. 2012, 165, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Hacker, D.L.; Fowler, B.C. Complementation of the Host Range Restriction of Southern Cowpea Mosaic Virus in Bean by Southern Bean Mosaic Virus. Virology 2000, 266, 140–149. [Google Scholar] [CrossRef]
- Wang, Y.; Lee, K.C.; Gaba, V.; Wong, S.M.; Palukaitis, P.; Gal-On, A. Breakage of resistance to Cucumber mosaic virus by co-infection with Zucchini yellow mosaic virus: Enhancement of CMV accumulation independent of symptom expression. Arch. Virol. 2004, 149, 379–396. [Google Scholar] [CrossRef]
- García-Cano, E.; Resende, R.O.; Fernández-Muñoz, R.; Moriones, E. Synergistic interaction between Tomato chlorosis virus and Tomato spotted wilt virus results in breakdown of resistance in tomato. Phytopathology 2006, 96, 1263–1269. [Google Scholar] [CrossRef]
- Wintermantel, W.M.; Cortez, A.A.; Anchieta, A.G.; Gulati-Sakhuja, A.; Hladky, L.L. Co-Infection by Two Criniviruses Alters Accumulation of Each Virus in a Host-Specific Manner and Influences Efficiency of Virus Transmission. Phytopathology 2008, 98, 1340–1345. [Google Scholar] [CrossRef]
- Vafaei, H.; Mahmoodi, M. Presence of Recombinant Strain of Cucurbit aphid borne yellows virus in Iran. Iran. J. Biotechnol. 2017, 15, e1541. [Google Scholar] [CrossRef]
- Roosinck, M.J.; García-Arenal, F. Ecosystem simplification, biodiversity loss and plant virus emergence. Curr. Opin. Virol. 2015, 10, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Polischuk, V.; Budzanivska, I.; Shevchenko, T.; Oliynik, S. Evidence for plant viruses in the region of Argentina Islands, Antarctica. FEMS Microbiol. Ecol. 2007, 59, 409–417. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Rambaut, A.; Pybus, O.G.; Holmes, E.C. Rates of molecular evolution in RNA viruses: A quantitative phylogenetic analysis. J. Mol. Evol. 2002, 54, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, D.A.; Domingo, E.; Holland, J.J. Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 1992, 122, 281–288. [Google Scholar] [CrossRef]
- Hitchborn, J.H.; Hills, G.J. The use of negative staining in the electron microscopic examination of plant viruses in crude extract. Virology 1965, 27, 528–540. [Google Scholar] [CrossRef]
- Clark, M.F.; Adams, A.N. Characteristics of the microplate method of enzyme-linked immunosorbent assay for the detection of plant viruses. J. Gen. Virol. 1977, 34, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Mullis, K.B. The unusual origin of the polymerase chain reaction. Sci. Am. 1990, 262, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Siebert, P. RT-PCR: Methods and Applications; Book 1; Clontech Laboratories Inc.: San Diego, CA, USA, 1991. [Google Scholar]
- Ohan, N.W.; Heikkila, J.J. Reverse transcription-polymerase chain reaction: An overview of the technique and its applications. Biotechnol. Adv. 1993, 11, 13–29. [Google Scholar] [CrossRef]
- Domingo, E. Mechanisms of viral emergence. Vet. Res. 2010, 41, 38. [Google Scholar] [CrossRef]
- Martín, S.; Elena, S.F. Application of game theory to the interaction between 20 plant viruses during mixed infections. J. Gen. Virol. 2009, 90, 2815–2820. [Google Scholar] [CrossRef] [PubMed]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, T.M.; Martin, R.R.; Tzanetakis, I.E. High Throughput Sequencing for Plant Virus Detection and Discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Olmos, A.; Jijakli, H.; Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014, 188, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant virus metagenomics: Advances in virus discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, F.; Bennett, S.; Modha, S.; Murdoch, D.; Gunson, R.; Murcia, P.R. The Use of Next Generation Sequencing in the Diagnosis and Typing of Respiratory Infections. J. Clin. Virol. 2015, 69, 96–100. [Google Scholar] [CrossRef]
- Bačnik, K.; Kutnjak, D.; Pecman, A.; Mehle, N.; Tušek-Žnidarič, M.; Gutiérrez-Aguirre, I.; Ravnikar, M. Viromics and infectivity analysis reveal the release of infective plant viruses from wastewater into the environment. Water Res. 2020, 117, 115628. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Rowhani, A. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef]
- Donaire, L.; Wang, Y.; González-Ibeas, D.; Mayer, K.F.; Aranda, M.A.; Llave, C. Deep sequencing of plant viral small RNAs reveals effective and widespread targeting of viral genomes. Virology 2009, 392, 203–214. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Islas, C.; Rowhani, A. Comparison of next-generation sequencing versus biological indexing for the optimal detection of viral pathogens of grapevines. Phytopathology 2015, 105, 758–763. [Google Scholar] [CrossRef]
- Bag, S.; Al Rwahnih, M.; Li, A.; Gonzalez, A.; Rowhani, A.; Uyemoto, J.K.; Sudarshana, M.R. Detection of a new luteovirus in imported nectarine trees: A case study to propose adoption of metagenomics in post-entry quarantine. Phytopathology 2015, 105, 840–846. [Google Scholar] [CrossRef]
- Zarzyńska-Nowak, A.; Hasiów-Jaroszewska, B.; Budzyńska, D.; Borodynko-Filas, N. First report of cucurbit aphid-borne yellows virus infecting zucchini plants (Cucurbita pepo convar. giromontiina) in Poland. Plant Dis. 2019, 103, 1047. [Google Scholar]
- Poudel, B.; Sabanadzovic, S.; Bujarski, J.; Tzanetakis, I.E. Population structure of Blackberry yellow vein associated virus, an emerging crinivirus. Virus Res. 2012, 169, 272–275. [Google Scholar] [CrossRef]
- Poudel, B.; Tzanetakis, I.E. Population structure of Blackberry chorotic ringspot virus. Arch. Virol. 2013, 158, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Antoniw, J.F. DPVweb: An open access internet resource on plant viruses and virus diseases. Outlooks Pest Manag. 2005, 16, 268–270. [Google Scholar] [CrossRef]
- Mumford, R.A.; Jarvis, B.; Harju, V.; Elmore, J.; Skelton, A. The first identification of two viruses infecting trailing verbena in the UK. N. Dis. Rep. 2005, 11, 7. [Google Scholar] [CrossRef]
- Veerakone, S.; Tang, J.; Zheng, A.; Ward, L.I.; Mason, C. First Report of Clover yellow mosaic virus in Postentry Quarantine Tulipa gesneriana Plants Imported from the United States. Plant Dis. Notes 2017, 102, 2. [Google Scholar] [CrossRef]
- Fránová, J.; Jakešová, H. Susceptibility of ten red clover (Trifolium pratense) cultivars to six viruses after artificial inoculation. Plant Prot. Sci. 2014, 50, 113–118. [Google Scholar] [CrossRef]
- Hollings, M.; Horváth, J. Melandrium yellow fleck virus, C.M.I./A.A.B. Descr. Plant Viruses 1981, 236, 39. [Google Scholar]
- Lecoq, H.; Desbiez, C. Cucurbit viruses in the Mediterranean region: An ever-changing picture. Adv. Virus Res. 2012, 84, 67–126. [Google Scholar]
- Dixon, G.R. Climate change—impact on crop growth and food production, and plant pathogens. Can. J. Plant Pathol. 2012, 34, 362–379. [Google Scholar] [CrossRef]
- Lecoq, H. Cucurbits. In Virus and Virus-Like Diseases of Major Crops in Developing Countries; Loebenstein, G., Thottapilly, G., Eds.; Kluwer: Dordrecht, The Netherlands, 2003; pp. 665–687. [Google Scholar]
- Pospieszny, H.; Cajza, M.; Plewa, R. First report of Cucumber leaf spot virus in Poland. Plant Dis. 2004, 88, 1381. [Google Scholar] [CrossRef]
- Segundo, E.; Janssen, D.; Velasco, L.; Ruiz, L.; Cuadrado, I.M. First Report of Cucumber leaf spot virus in Spain. Plant Dis. Notes 2007, 85, 10. [Google Scholar] [CrossRef] [PubMed]
- French, C.J.; Bouthillier, M.; Bernardy, M.; Ferguson, G.; Sabourin, M.; Johnson, R.C.; Masters, C.; Godkin, S.; Mumford, R. First report of Pepino mosaic virus in Canada and the United States. Plant Dis. 2001, 85, 1121. [Google Scholar] [CrossRef]
- Pospieszny, H.; Borodynko, N.; Palczewska, M. First record of Pepino mosaic virus in Poland. J. Plant Dis. Prot. 2003, 100, 97. [Google Scholar]
- Maroon-Lango, C.J.; Guaragna, M.A.; Jordan, R.L.; Hammond, J.; Bandla, M.; Marquardt, S.K. Two unique US isolates of Pepino mosaic virus from a limited source of pooled tomato tissue are distinct from a third (European-like) US isolate. Arch. Virol. 2005, 150, 1187–1201. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Pérez, M.G.; Pagán, I.; Aragón-Caballero, L.; Cáceres, F.; Fraile, A.; García-Arenal, F. Ecological and genetic determinants of Pepino mosaic virus emergence. J. Virol. 2014, 88, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.S. Molecular characterization of two Pepino mosaic virus variants from imported tomato seed reveals high levels of sequence identity between Chilean and US isolates. Virus Genes 2007, 34, 1–8. [Google Scholar] [CrossRef]
- Van der Vlugt, R.A.; Stijger, C.M.; Verhoeven, J.T.J.; Lesemann, D.E. First report of Pepino mosaic virus on tomato. Plant Dis. 2000, 84, 103–108. [Google Scholar] [CrossRef]
- Roggero, P.; Masenga, V.; Lenzi, R.; Coghe, F.; Ena, S.; Winter, S. First report of Pepino mosaic virus in tomato in Italy. Plant Pathol. 2001, 50, 798. [Google Scholar] [CrossRef]
- Spence, N.J.; Basham, J.; Mumford, R.A.; Hayman, G.; Edmondson, R.; Jones, D.R. Effect of Pepino mosaic virus on the yield and quality of glasshouse-grown tomatoes in the UK. Plant Pathol. 2006, 55, 595–606. [Google Scholar] [CrossRef]
- Hasiów, B.; Borodynko, N.; Pospieszny, H. Complete genomic RNA sequence of the Polish Pepino mosaic virus isolate belonging to the US2 strain. Virus Genes 2008, 36, 1–8. [Google Scholar] [CrossRef]
- Hasiów-Jaroszewska, B.; Pospieszny, H.; Borodynko, N. New necrotic isolates of Pepino mosaic virus representing the CH2 genotype. J. Phytopathol. 2009, 157, 494–496. [Google Scholar] [CrossRef]
- Hasiów-Jaroszewska, B.; Paeleman, A.; Ortega-Parra, N.; Borodynko, N.; Minicka, J.; Czerwoniec, A.; Thomma, B.P.H.J.; Hanssen, I.M. Ratio of mutated versus wild-type coat protein sequences in Pepino mosaic virus determines the nature and severity of yellowing symptoms on tomato plants. Mol. Plant Pathol. 2013, 14, 923–933. [Google Scholar] [CrossRef]
- Hanssen, I.M.; Thomma, B.P.H.J. Pepino mosaic virus: A successful pathogen that rapidly evolved from emerging to endemic in tomato crops. Mol. Plant Pathol. 2010, 11, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Blystad, D.R.; van der Vlugt, R.; Alfaro-Fernandez, A.; Cordoba, M.D.; Bese, G.; Hristova, D.; Pospieszny, H.; Mehle, N.; Ravnikar, M.; Tomassoli, L.; et al. Host range and symptomatology of Pepino mosaic virus strains occurring in Europe. Eur. J. Plant Pathol. 2015, 143, 43–56. [Google Scholar] [CrossRef]
- Zarzyńska-Nowak, A.; Rymelska, N.; Borodynko, N.; Hasiów-Jaroszewska, B. The occurrence of Tomato yellow ring virus on tomato in Poland. Plant Dis. 2016, 100, 234. [Google Scholar] [CrossRef]
- Syller, J. Facilitative and antagonistic interactions between plant viruses in mixed infections. Mol. Plant Pathol. 2012, 13, 204–216. [Google Scholar] [CrossRef]
- Borodynko, N.; Hasiów, B.; Figlerowicz, M.; Pospieszny, H. Identification of the New Strain of Strawberry latent ringspot virus Isolated from Black Locust (Robinia pseudoacacia L.). J. Phytopathol. 2007, 155, 738–742. [Google Scholar] [CrossRef]
- Bashir, N.S.; Kalhor, M.R.; Zarghani, S.N. Detection, differentiation and phylogenetic analysis of Cucumber mosaic virus isolates from cucurbits in the northwest region of Iran. Virus Genes 2006, 32, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-K.; Yoon, J.-Y.; Choi, G.-S. Biological and Molecular Characterization of a Korean Isolate of Cucurbit aphidborne yellows virus Infecting Cucumis Species in Korea. Plant Pathol. J. 2015, 31, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Posis, K.; Maroon-Lango, C.J.; Mavrodieva, V.; Haymes, S.; Pitman, T.L.; Falk, W.B. First Report of Cucumber green mottle mosaic virus on Melon in the United States. Plant Dis. Notes 2014, 98, 1163. [Google Scholar] [CrossRef]
- Chin, M.; Rojas, Y.; Moret, J.; Fermin, G.; Tennant, P.; Gonsalves, D. Varying genetic diversity of Papaya ringspot virus isolates from two time-separated outbreaks in Jamaica and Venezuela. Arch. Virol. 2007, 152, 2101–2106. [Google Scholar] [CrossRef]
- Hasiów-Jaroszewska, B.; Budzyńska, D.; Borodynko, N.; Pospieszny, H. Rapid detection of genetically diverse tomato black ring virus isolates using reverse transcription loop-mediated isothermal amplification. Arch. Virol. 2015, 160, 3075–3078. [Google Scholar] [CrossRef]
- Sharifi, M.; Massumi, H.; Heydarnejad, J.; Pour, A.H.; Shaabanian, M.; Rahimian, H. Analysis of the biological and molecular variability of Watermelon mosaic virus isolates from Iran. Virus Genes 2008, 37, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Thomson, K.G.; Dietzgen, R.G.; Gibbs, A.J.; Tang, Y.C.; Liesack, W.; Teakle, D.S.; Stackebrandt, E. Identification of Zucchini yellow mosaic potyvirus by RT-PCR and analysis of sequence variability. J. Virol. Methods 1995, 55, 83–96. [Google Scholar] [CrossRef]
- Hasiów-Jaroszewska, B.; Borodynko, N. Detection of Pepino mosaic virus isolates from tomato by one-step reverse transcription loop-mediated isothermal amplification. Arch. Virol. 2013, 158, 2153–2156. [Google Scholar] [CrossRef] [PubMed]
- Rigotti, S.; Gugerli, P. Rapid identification of potato virus Y strains by one-step triplex RT-PCR. J. Virol. Methods 2007, 140, 90–94. [Google Scholar] [CrossRef]
- Petrzik, K. Complete genome sequence of broad bean true mosaic virus. Arch. Virol. 2010, 155, 1179–1181. [Google Scholar] [CrossRef]
- Digiaro, M.; Elbeaino, T.; Martelli, G.P. Development of degenerate and species-specific primers for the differential and simultaneous RT-PCR detection of grapevine-infecting nepoviruses of subgroups A, B and C. J. Virol. Methods 2007, 141, 34–40. [Google Scholar] [CrossRef]
- Zarzyńska-Nowak, A.; Hasiów-Jaroszewska, B.; Korbecka-Glinka, G.; Przybyś, M.; Borodynko-Filas, N. A multiplex RT-PCR assay for simultaneous detection of Tomato spotted wilt virus and Tomato yellow ring virus in tomato plants. Can. J. Plant Pathol. 2018, 40, 580–586. [Google Scholar] [CrossRef]
- Song, A.; You, Y.; Chen, F.; Li, P.; Jiang, J.; Chen, S. A multiplex RT-PCR for rapid and simultaneous detection of viruses and viroids in chrysanthemum. Lett. Appl. Microbiol. 2012, 56, 8–13. [Google Scholar] [CrossRef]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 3 August 2016).
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 34, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evolut. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evolut. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Host Plant | Plant Used for RNA Isolation | Number of Total Raw Reads | Percent of Reference Genome Covered by Reads | Number of Reads Mapped to Corresponding Reference Sequence from Viral RefSeq | Average Depth of Coverage for Corresponding Viral Species | Identified Viruses |
|---|---|---|---|---|---|---|---|
| 1 | R. pseudoacacia | N. benthamiana | 11,027,620 | 100% 83.05% 64.64% 100% | 41,097 67,428 16,277 71,540 | 3629.22 730.04 201.40 1139.87 | satRNA peanut stunt virus (NC_003855) * RNA1 peanut stunt virus (NC_002038) RNA2 peanut stunt virus (NC_002039) RNA3 peanut stunt virus (NC_002040) |
| 2 | V. officinalis | C. quinoa | 98,173,277 | 90.27% | 401,558 | 1945.10 | clover yellow mosaic virus (NC_001753) |
| 3 | D. belladonna | N. benthamiana | 46,911,057 | 91.73% 97.48% | 63,133 92,157 | 295.03 837.80 | RNA1 arabis mosaic virus (NC_006057) RNA2 arabis mosaic virus (NC_006056) |
| 4 | C. pepo | C. pepo | 18,034,634 | 99.46% 93.72% 97.49% | 41,742 21,359 3140 | 154.49 75.70 19.60 | zucchini yellow mosaic virus (NC_003224) watermelon mosaic virus (NC_006262) cucurbit aphid-borne yellows virus (NC_003688) |
| 5 | C. pepo | C. pepo | 600,806 | 99.94% 99.73% 99.86% 97.61% 67.7% 72.57% | 19,301 19,814 53,922 1238 1361 205 | 341.50 340.47 1293.96 7.47 2.19 1.91 | RNA1 cucumber mosaic virus (NC_002034) RNA2 cucumber mosaic virus (NC_002035) RNA3 cucumber mosaic virus (NC_001440) zucchini yellow mosaic virus (NC_003224) watermelon mosaic virus (NC_006262) cucurbit aphid-borne yellows virus (NC_003688) |
| 6 | C. pepo | C. pepo | 56,369,111 | 91.8% 67.29% 84/09% 97.15% | 59,518 206 198 488 | 206.72 2.1 2.26 7.84 | watermelon mosaic virus (NC_006262) RNA1 cucumber mosaic virus (NC_002034) RNA2 cucumber mosaic virus (NC_002035) RNA3 cucumber mosaic virus (NC_001440) |
| 7 | V. faba | N. benthamiana | 19,979,239 | 85.62% | 186,607 | 700.15 | bean yellow mosaic virus (NC_003492) |
| 8 | S. latifolia | N. benthamiana | 302,784 | 99.63% 98.25% 99.21% | 71,934 25,849 7627 | 1662.62 708.24 250.10 | RNA1 melandrium yellow fleck virus (NC_013266) RNA2 melandrium yellow fleck virus (NC_013267) RNA3 melandrium yellow fleck virus (NC_013268) |
| 9 | C. pepo | C. pepo | 35,797,184 | 96.79% 94.45% 95.43% | 41,613 12,029 3567 | 152.35 42.04 21.99 | zucchini yellow mosaic virus (NC_003224) watermelon mosaic virus (NC_006262) cucurbit aphid-borne yellows virus (NC_003688) |
| 10 | C. pepo | C. pepo | 26,025,392 | 97.09% | 74,858 | 261.66 | watermelon mosaic virus (NC_006262) |
| 11 | C. sativus | C. sativus | 773,010 | 99.91% 99.44% 99.77% | 10,882 20,570 40,511 | 157.57 321.49 880.88 | RNA1 cucumber mosaic virus (NC_002034) RNA2 cucumber mosaic virus (NC_002035) RNA3 cucumber mosaic virus (NC_001440) |
| 12 | C. pepo | C. pepo | 32,033,000 | 99.32% | 18,647,504 | 185,152.26 | watermelon mosaic virus (NC 006262) |
| 13 | C. pepo | C. pepo | 31,632,000 | 99.16% 99.58% 99.37% 99.09% | 4,166,267 33,803 42,961 58,722 | 41,361.90 1006.61 1387.22 2573.19 | watermelon mosaic virus (NC_006262) RNA1 cucumber mosaic virus (NC_002034) RNA2 cucumber mosaic virus (NC_002035) RNA3 cucumber mosaic virus (NC_001440) |
| 14 | C. sativus | C. sativus | 30,564,000 | 99.85% 100% 99.9% 98.07% 99.21% | 4,179,988 4,387,800 11,630,859 113,703 4396 | 123,854.64 144,669.22 527,062.54 1987.71 14.38 | RNA1 cucumber mosaic virus (NC_002034) RNA2 cucumber mosaic virus (NC_002035) RNA3 cucumber mosaic virus (NC_001440) cucurbit aphid-borne yellows virus (NC_003688) cucumber leaf spot virus (NC_007216) |
| 15 | C. pepo | C. pepo | 31,237,000 | 100% 100% 99.61% 98.55% 100% | 7,434,123 4,038,168 24,208 31,393 39,424 | 77,638.71 40,148.47 722.10 1017.05 1740.52 | zucchini yellow mosaic virus (NC_003224) watermelon mosaic virus (NC_006262) RNA1 cucumber mosaic virus (NC_002034) RNA2 cucumber mosaic virus (NC_002035) RNA3 cucumber mosaic virus (NC_001440) |
| 16 | R. × prostrata | N. benthamiana | 29,986,000 | 97.49% 89.90% 98.45% 96.93% | 5,884,878 311 5015 602 | 58,322.65 9.25 16.63 26.44 | turnip mosaic virus (NC_002509) RNA1 cucumber mosaic virus (NC_002034) RNA2 cucumber mosaic virus (NC_002035) RNA3 cucumber mosaic virus (NC_001440) |
| 17 | S. lycopersicum | S. lycopersicum | 38,560,000 | 98.48% 99.95% 97.88% 94.87% | 29,548,236 1,297,591 2,472,631 2,594,718 | 433,514.97 14,364.57 50,882.36 85,169.46 | pepino mosaic virus (NC_004067) Segment L tomato yellow ring virus (JN 560178) Segment M tomato yellow ring virus (JN 560177) Segment S tomato yellow ring virus (DQ 462163) |
| 18 | C. pepo | C. pepo | 36,021,000 | 100% 100% 100% | 8,440,062 12,232,893 35,606,110 | 250,241.34 403,564.26 1,612,531.81 | RNA1 cucumber mosaic virus (NC_002034) RNA2 cucumber mosaic virus (NC_002035) RNA3 cucumber mosaic virus (NC_001440) |
| 19 | S. lycopersicum | S. lycopersicum | 36,694,000 | 99.94% 99.73% 99.68% 99.98% | 1,565,570 1,386,012 6,284,859 24,007,721 | 499,564.13 820,381.91 1,764,993.94 116,343.05 | RNA1 cucumber mosaic virus (NC_002034) RNA2 cucumber mosaic virus (NC_002035) RNA3 cucumber mosaic virus (NC_001440) pepino mosaic virus (NC 004067) |
| 20 | S. lycopersicum | S. lycopersicum | 39,382,000 | 100% 100% 99.68% 99.81% | 13,891,631 21,749,691 34,738,425 6438 | 412,119.76 704,894.27 1,539,186.77 49.33 | RNA1 cucumber mosaic virus (NC_002034) RNA2 cucumber mosaic virus (NC_002035) RNA3 cucumber mosaic virus (NC_001440) pepino mosaic virus (NC_004067) |
| Virus | Primer | Sequence 5′-3′ | Reference |
|---|---|---|---|
| cucumber mosaic virus | CMV CPf CMV CPr | GCTTCTCCGCGAG GCCGTAAGCTGGATGGAC | [60] |
| cucurbit aphid-borne yellows virus | CABYVCPF CABYVCPRev | ATGAATACGGCCGCGGCTAGAAATC CTATTTCGGGTTCTGGACCTGGCA | [61] |
| cucumber green mottle mosaic virus | CGMMV-F5370 CGMMV-R6390 | CTAATTATTCTGTCGTGGCTGCGGATGC CTTGCAGAATTACTGCCCATA | [62] |
| papaya ringspot virus | 04-02 04-04 | TACTAGTGTACCATGAATC CTCTCATTCTAAGAGGCTC | [63] |
| tomato black ring virus | TBRV CPF TBRV CPR | GCCTGTCTCTCTCGCAATG AAGGAGCCAAACTGAAATG | [64] |
| watermelon mosaic virus | WMV F WMVR | GAA TCA GTG TCT CTG CAA TCA GG ATT CAC GTC CCT TGC AGT GTG | [65] |
| zucchini yellow mosaic virus | ZY-1, ZY-2 | CACAATTTTCCCATGAGAACCAGC GCTCCATACATAGCTGAGACAGC | [66] |
| pepino mosaic virus | TGB3F TGB3R | GGTGGACAATATCAAGACCGG CTGTATTGGGTTTGAGAAGTC | [67] |
| potato virus Y | PVYc3 PVYf PVY3+ PVY3− CP2+ CP1− | CAACGCAAAAACACTCA(CT)AAA(AC)GC TAAGTG(AG)ACAGACCCTCT(CT)TTCTC TGTAACGAAAGGGACTAGTGCAAAG CCGCTATGAGTAAGTCCTGCACA CCAGTCAAACCCGAACAAAGG GGCATAGCGTGCTAAACCCA | [68] |
| broad bean true mosaic virus | BBTMV-IGGf BBTMV-VQTr | CnAThGGnGGnGGnGCnGG CACyTGnGTnGACCAnGC | [69] |
| bean yellow mosaic virus | BYMV-CP-5 BYMV-CP-3 | GAACTGTTGGAACGTTTTCAATTCC TCTGTTCCAACATTGCCATCAAG | |
| Nepovirus genus | Nepo-AF Nepo-AR Nepo-BF Nepo-BR Nepo-CF Nepo-CR | GGHDTBCAKTMYSARRARTGG TGDCCASWVARYTCYCCATA ATGTGYGCHACYACWGGHATGCA TTCTCTDHAAGAAATGCCTAAGA TTRKDYTGGYKAAMYYCCA TMATCSWASCRHGTGSKKGCCA | [70] |
| tomato spotted wilt virus | TS1-F TS1-R | GCCTATGGATTACCTCTTG GTTTCACTGTAATGTTCCA | [71] |
| chrysanthemum virus B | CVB-F CVB-R | AGTCACAATGCCTCCCAAAC CATACCTTTCTTAGAGTGCTATGCT | [72] |
| Virus | Primer | Sequence 5′-3′ | Amplified Region of the Genome | Amplicon Size [bp] | Reference |
|---|---|---|---|---|---|
| TYRV | TYRVLF1473 TYRVLR2068 | GGAGAAATGAATTTTAA CTTTGTATCATTGAAT | RdRp | 595 | This study |
| CIMYV | ClMYVF1574 ClMYVR2620 | CAAGTCCTGAACAGAGT AGTTTCCAGGGTAGTTC | RdRp | 1046 | This study |
| TuMV | TuMVF1194 TuMVR2108 | TGAGCCATAAGATTGTGCAT AGTGGATCACCTGATTC | MP | 914 | This study |
| MYFV | MYFMV2F2577 MYFMV2R2840 | CTAAGTAAGTTGCTAATGC GGTCTCCTTTATGACCACTAATC | 2a/3’UTR | 263 | This study |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minicka, J.; Zarzyńska-Nowak, A.; Budzyńska, D.; Borodynko-Filas, N.; Hasiów-Jaroszewska, B. High-Throughput Sequencing Facilitates Discovery of New Plant Viruses in Poland. Plants 2020, 9, 820. https://doi.org/10.3390/plants9070820
Minicka J, Zarzyńska-Nowak A, Budzyńska D, Borodynko-Filas N, Hasiów-Jaroszewska B. High-Throughput Sequencing Facilitates Discovery of New Plant Viruses in Poland. Plants. 2020; 9(7):820. https://doi.org/10.3390/plants9070820
Chicago/Turabian StyleMinicka, Julia, Aleksandra Zarzyńska-Nowak, Daria Budzyńska, Natasza Borodynko-Filas, and Beata Hasiów-Jaroszewska. 2020. "High-Throughput Sequencing Facilitates Discovery of New Plant Viruses in Poland" Plants 9, no. 7: 820. https://doi.org/10.3390/plants9070820
APA StyleMinicka, J., Zarzyńska-Nowak, A., Budzyńska, D., Borodynko-Filas, N., & Hasiów-Jaroszewska, B. (2020). High-Throughput Sequencing Facilitates Discovery of New Plant Viruses in Poland. Plants, 9(7), 820. https://doi.org/10.3390/plants9070820