Genetic Structure of Wild Germplasm of Macadamia: Species Assignment, Diversity and Phylogeographic Relationships




Abstract
1. Introduction
2. Results
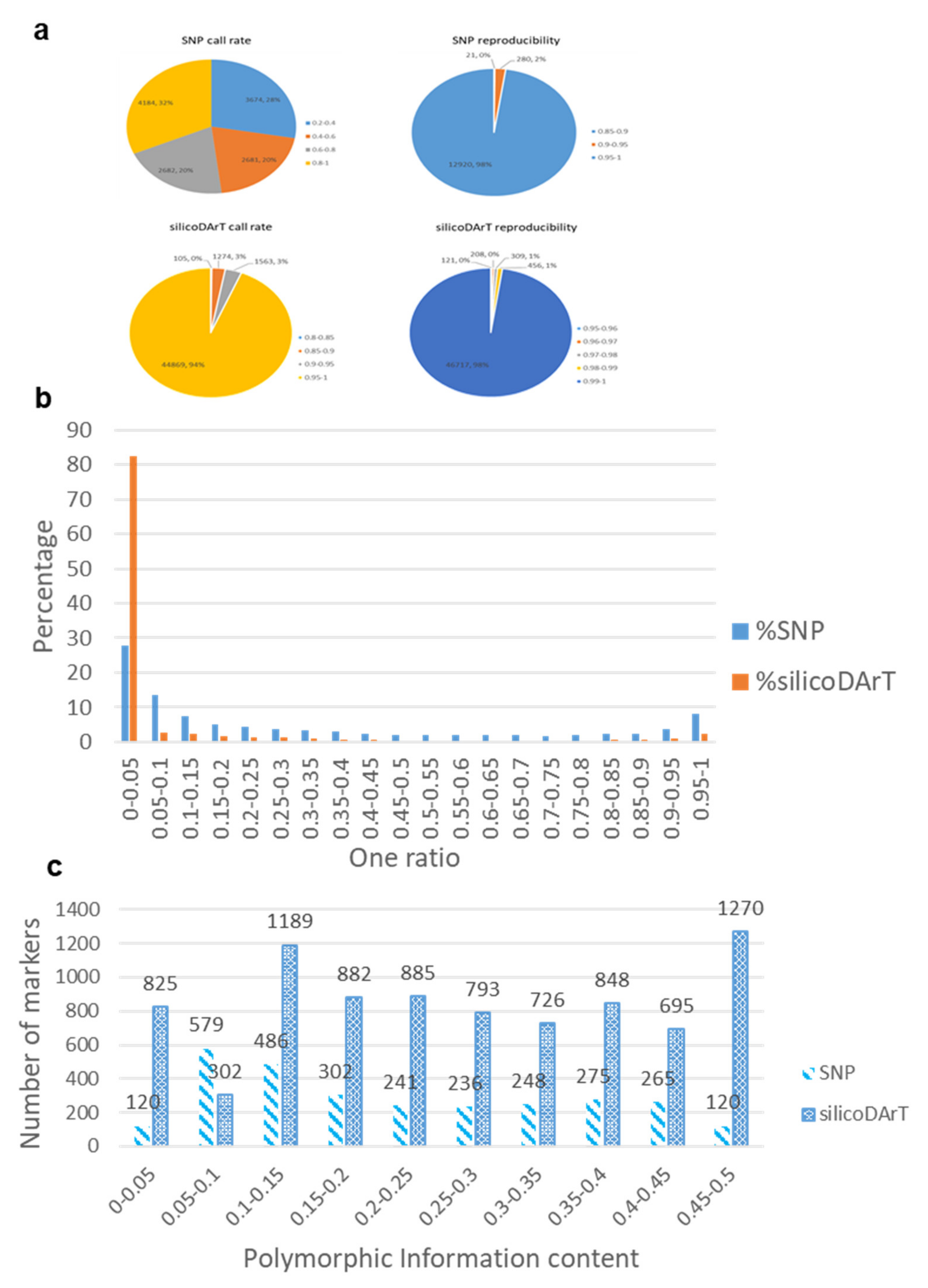
2.1. Quality of DArTseq Markers
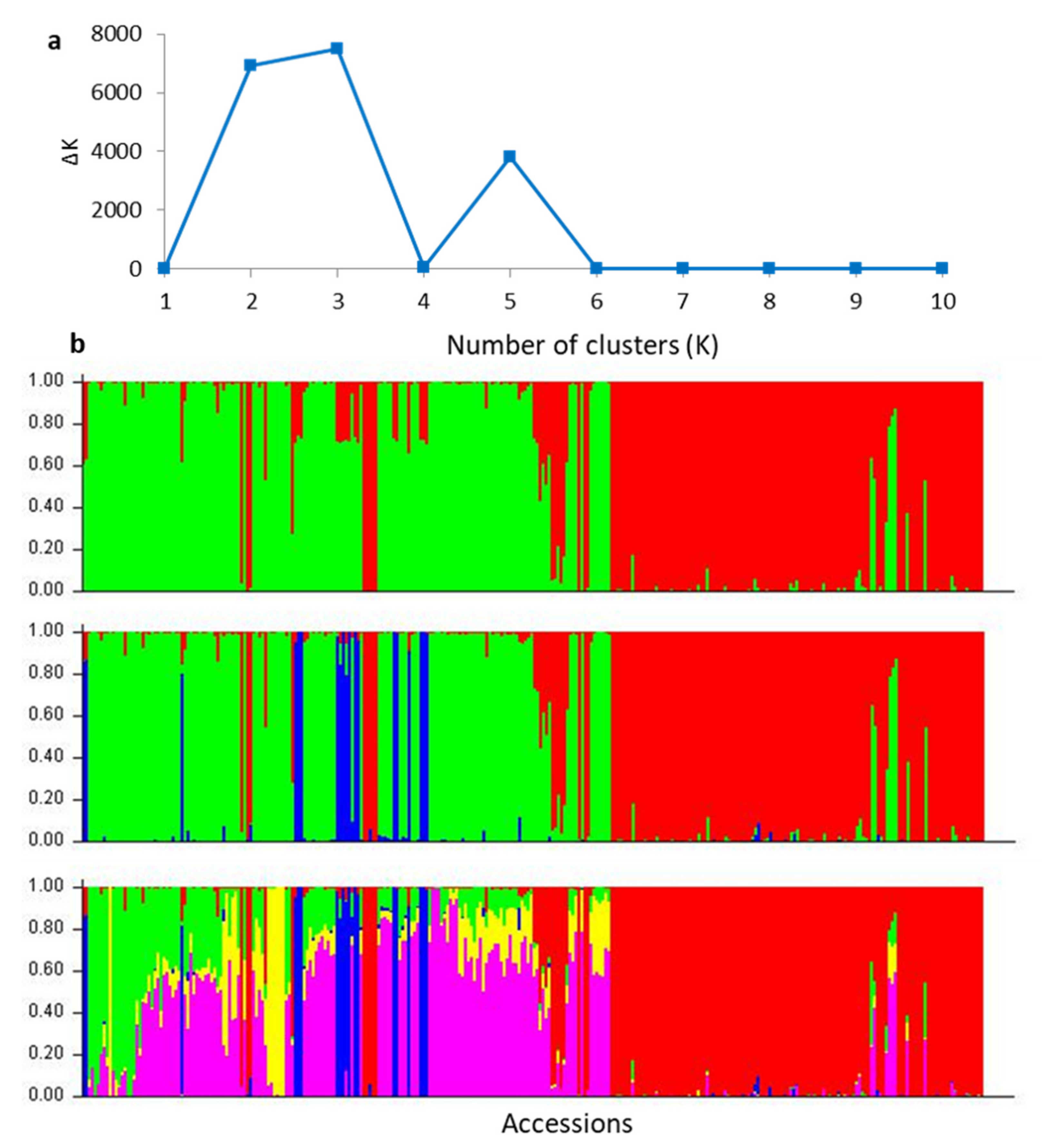
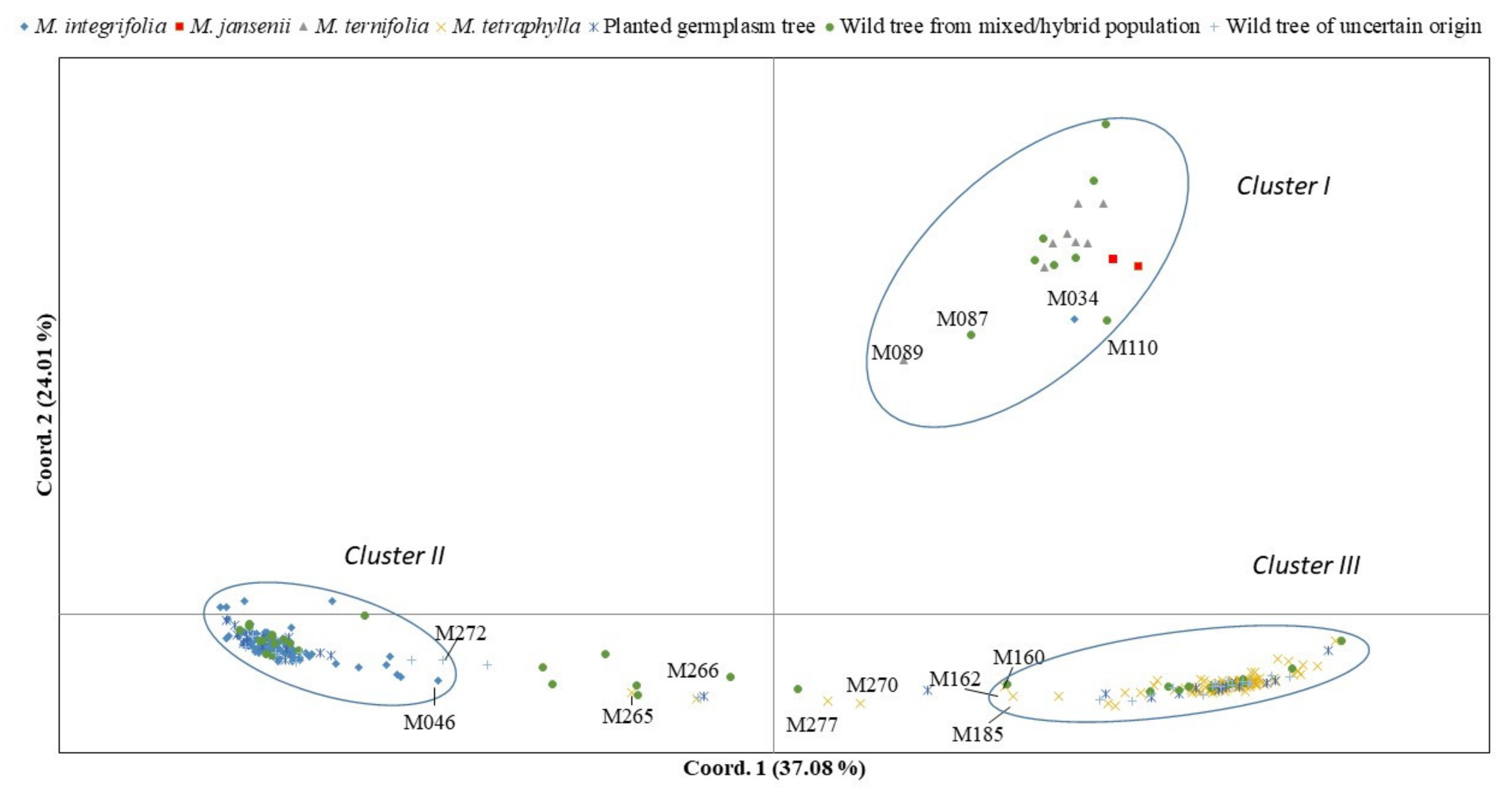
2.2. Population Assignment
2.3. Genetic Diversity
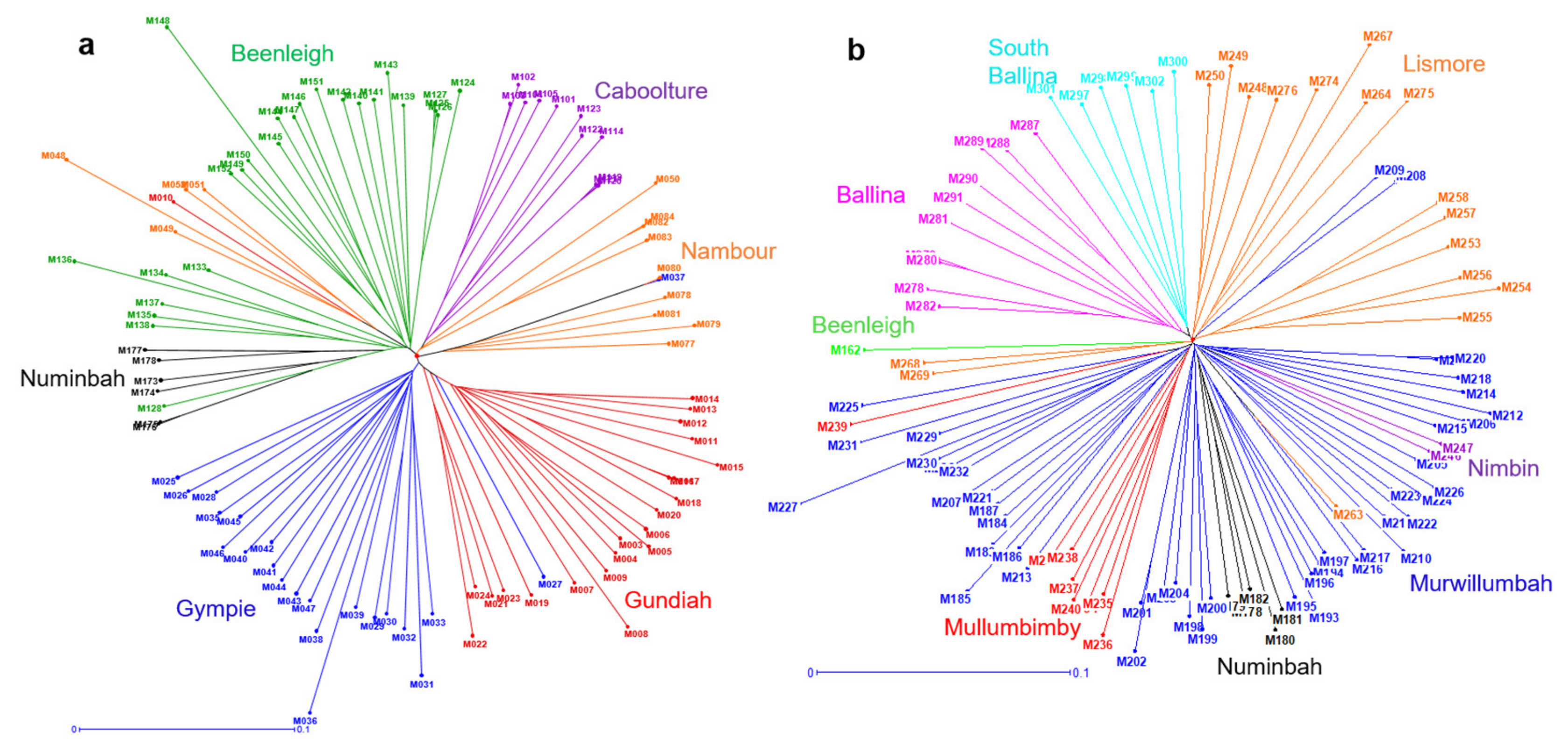
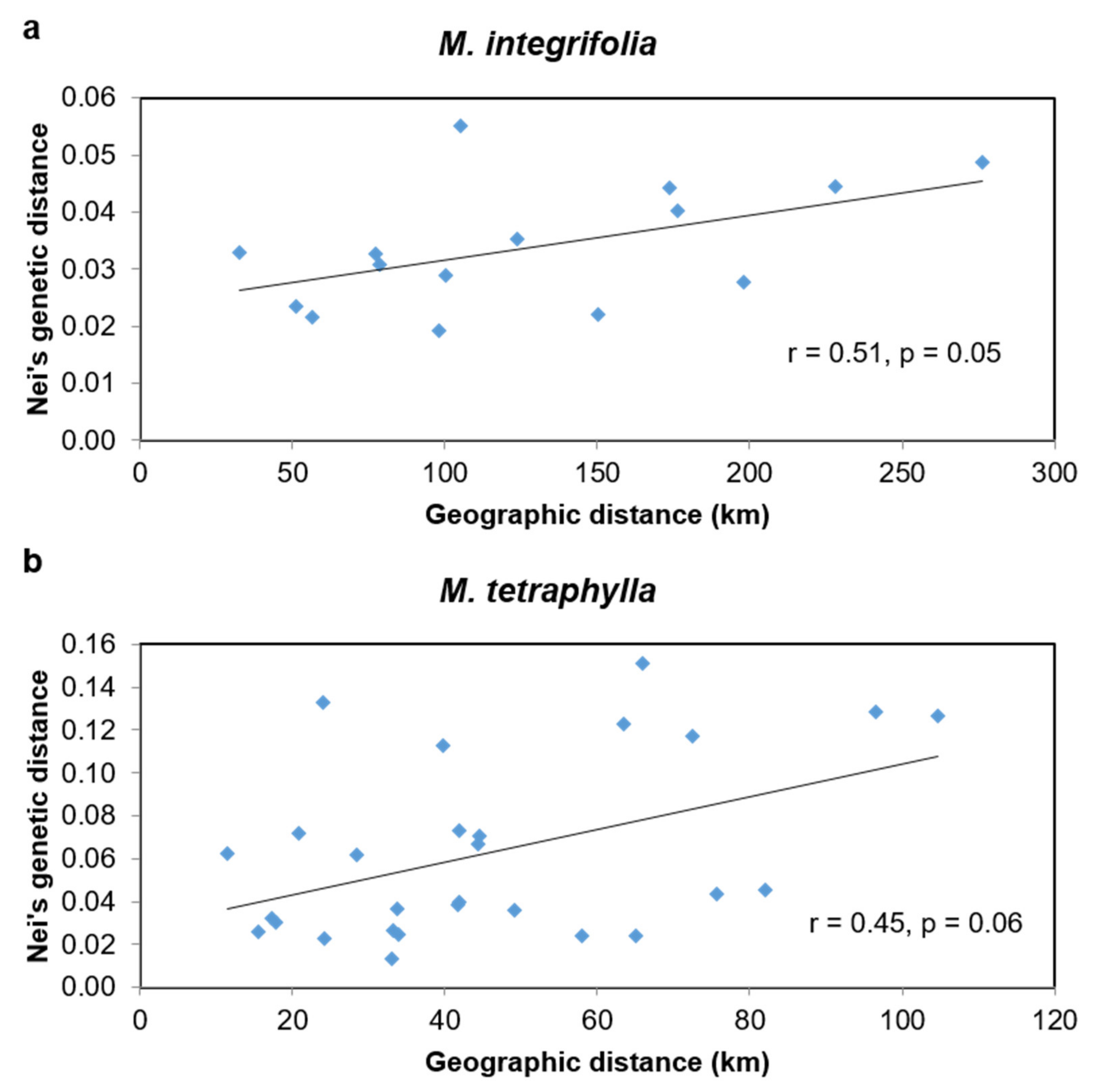
2.4. Phylogeographic Relationships among the Accessions of M. integrifolia and M. tetraphylla
3. Discussion
3.1. Species Assignment of Wild Macadamia Germplasm
3.2. Genetic Diversity of the Four Macadamia Species
3.3. Phylogeographic Relationship among the Accessions of M. integrifolia and M. tetraphylla
4. Materials and Methods
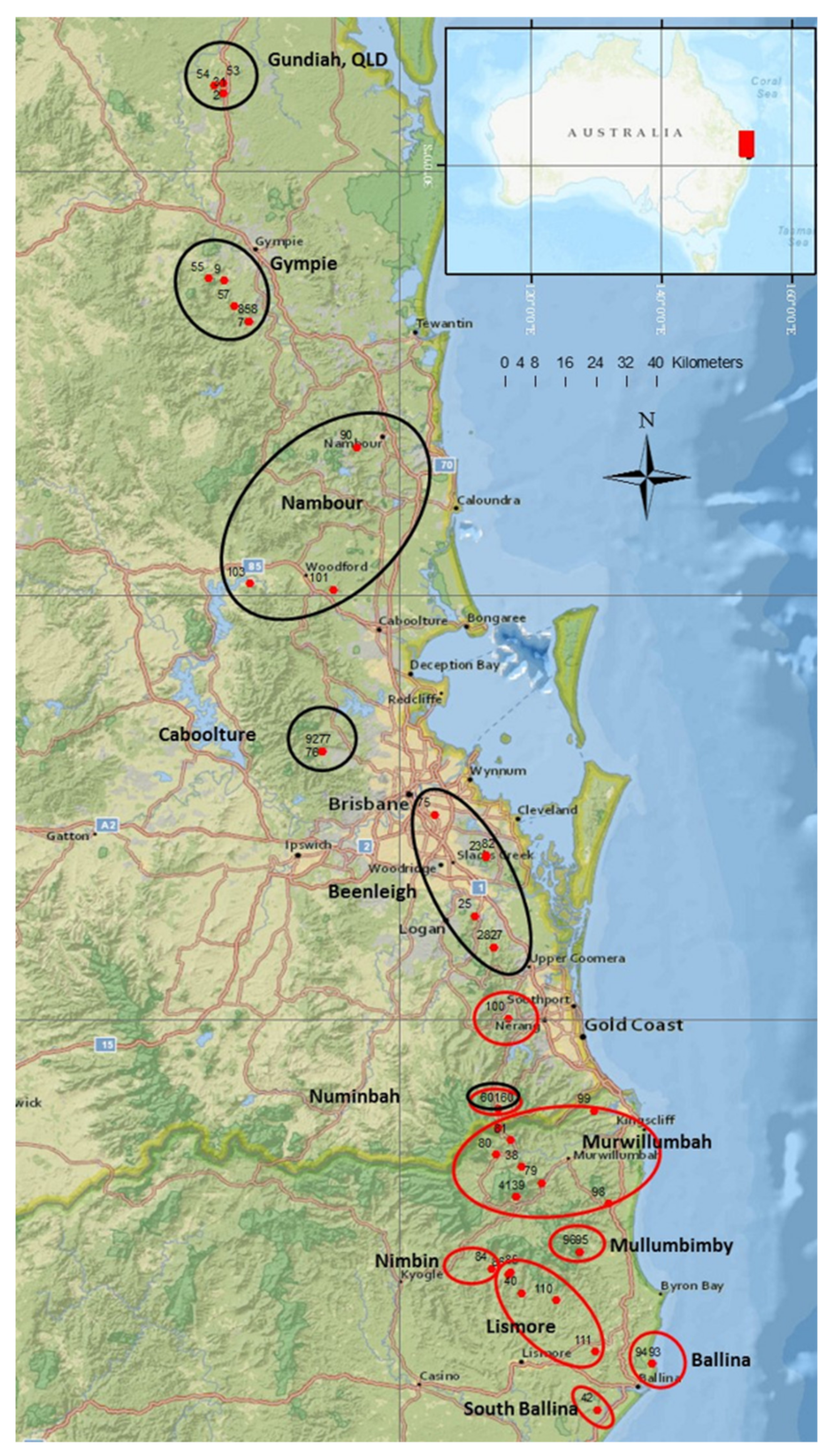
4.1. Collection of Plant Samples
4.2. DNA Extraction and Genotyping
4.3. Analysis of Processed Marker Data
4.4. Analysis of Population Structure and Genetic Diversity
4.5. Phylogeographic Relationships Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability Statement
References
- Hardner, C.M.; Peace, C.; Lowe, A.J.; Neal, J.; Pisanu, P.; Powell, M.; Schmidt, A.; Spain, C.; Williams, K. Genetic resources and domestication of macadamia. In Horticultural Reviews; Janick, J., Ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2009; Volume 35, pp. 1–125. [Google Scholar]
- Topp, B.L.; Nock, C.J.; Hardner, C.M.; Alam, M.; O’Connor, K.M. Macadamia (Macadamia spp.) Breeding. In Advances in Plant Breeding Strategies: Nut and Beverage Crops; Al-Khayri, J., Jain, S.D.J., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 221–251. [Google Scholar]
- Powell, M.; Accad, A.; Shapcott, A. Where they are, why they are there, and where they are going: Using niche models to assess impacts of disturbance on the distribution of three endemic rare subtropical rainforest trees of Macadamia (Proteaceae) species. Aust. J. Bot. 2014, 62, 322–334. [Google Scholar] [CrossRef]
- Peace, C.P. Genetic Characterisation of Macadamia with DNA Markers. Ph.D. Thesis, University of Queensland, Brisbane, Australia, 2004. [Google Scholar]
- Shapcott, A.; Powell, M. Demographic structure, genetic diversity and habitat distribution of the endangered, Australian rainforest tree help facilitate an introduction program. Aust. J. Bot. 2011, 59, 215–225. [Google Scholar] [CrossRef]
- Hardner, C.; Topp, B. Macadamia integrifolia and M. tetraphylla. In Trees for Life in Oceania: Conservation and Utilisation of Genetic Diversity; Lee, T., John, D., Clarke, B., Eds.; Australian Centre for International Agricultural Research: Canberra, Australia, 2018; pp. 147–151. [Google Scholar]
- Dahler, J.M.; McConchie, C.; Turnbull, C.G.N. Quantification of cyanogenic glycosides in seedlings of three Macadamia (Proteaceae) species. Aust. J. Bot. 1995, 43, 619–628. [Google Scholar] [CrossRef]
- Vivian-Smith, A. Cyanogenesis and the Fruit Growth of Three Macadamia Species. Bachelor’s Thesis, University of Queensland, Brisbane, Australia, 1995. [Google Scholar]
- Gross, C.L.; Weston, P.H. Macadamia jansenii (Proteaceae), a new species from central Queensland. Aust. Syst. Bot. 1992, 5, 725–728. [Google Scholar] [CrossRef]
- Shigeura, G.T.; Ooka, H. Macadamia nuts in Hawaii, history and production. In Research Series, Hawaii Institute of Tropical Agriculture and Human Resources; University of Hawaii: Honolulu, HI, USA, 1984; pp. 6–22. [Google Scholar]
- Hamilton, R.A.; Fukunaga, E.T. Growing Macadamia Nuts in Hawaii; Hawaii Agricultural Experiment Station: Honolulu, HI, USA, 1959; Volume 121, p. 59.
- Topp, B.; Hardner, C.M.; Neal, J.; Kelly, A.; Russell, D.; McConchie, C.; O’hare, P. Overview of the Australian macadamia industry breeding program. Acta Horticult. 2016, 1127, 45–50. [Google Scholar] [CrossRef]
- Yang, S.; Xue, S.; Kang, W.; Qian, Z.; Yi, Z. Genetic diversity and population structure of Miscanthus lutarioriparius, an endemic plant of China. PLoS ONE 2019, 14, e0211471. [Google Scholar] [CrossRef]
- Singh, B.P.; Singh, B.; Mishra, S.; Kumar, V.; Singh, N.K. Genetic diversity and population structure in Indian wild rice accessions. Aust. J. Crop Sci. 2016, 10, 144–151. [Google Scholar]
- Steiger, D.L.; Moore, P.H.; Zee, F.; Liu, Z.; Ming, R. Genetic relationships of macadamia cultivars and species revealed by AFLP markers. Euphytica 2003, 132, 269–277. [Google Scholar] [CrossRef]
- Nock, C.J.; Hardner, C.M.; Montenegro, J.D.; Ahmad Termizi, A.A.; Hayashi, S.; Playford, J.; Edwards, D.; Batley, J. Wild origins of macadamia domestication identified through intraspecific chloroplast genome sequencing. Front. Plant Sci. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Saeed, S.; Barozai, M.Y. A review on genetic diversity on wild plants by using different genetic markers. Pure Appl. Biol. 2012, 1, 68–71. [Google Scholar] [CrossRef]
- Aradhya, M.; Yee, L.; Zee, F.; Manshardt, R. Genetic variability in Macadamia. Genet. Resour. Crop Evol. 1998, 45, 19–32. [Google Scholar] [CrossRef]
- Mast, A.R.; Willis, C.L.; Jones, E.H.; Downs, K.M.; Weston, P.H. A smaller Macadamia from a more vagile tribe: Inference of phylogenetic relationships, divergence times, and diaspore evolution in Macadamia and relatives (tribe Macadamieae; Proteaceae). Am. J. Bot. 2008, 95, 843–870. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.M. The Impact of Habitat Fragmentation on Wild Macadamia Integrifolia Maiden and Betche (Proteaceae) Population Viability; University of New England: Armidale, Australia, 2007. [Google Scholar]
- O’Connor, K.; Powell, M.; Nock, C.; Shapcott, A. Crop to wild gene flow and genetic diversity in a vulnerable Macadamia (Proteaceae) species in New South Wales, Australia. Biol. Conserv. 2015, 191, 504–511. [Google Scholar] [CrossRef]
- Spain, C.S.; Lowe, A.J. Genetic consequences of subtropical rainforest fragmentation on Macadamia tetraphylla (Proteaceae). Silvae Genet. 2011, 60, 241–249. [Google Scholar] [CrossRef]
- Nock, C.J.; Elphinstone, M.S.; Ablett, G.; Kawamata, A.; Hancock, W.; Hardner, C.M.; King, G.J. Whole genome shotgun sequences for microsatellite discovery and application in cultivated and wild Macadamia (Proteaceae). Appl. Plant Sci. 2014, 2, apps.1300089. [Google Scholar] [CrossRef] [PubMed]
- Roorkiwal, M.; von Wettberg, E.J.; Upadhyaya, H.D.; Warschefsky, E.; Rathore, A.; Varshney, R.K. Exploring germplasm diversity to understand the domestication process in Cicer spp. using SNP and DArT markers. PLoS ONE 2014, 9, e102016. [Google Scholar] [CrossRef] [PubMed]
- Baloch, F.S.; Alsaleh, A.; Shahid, M.Q.; Çiftçi, V.; de Miera, L.E.S.; Aasim, M.; Nadeem, M.A.; Aktaş, H.; Özkan, H.; Hatipoğlu, R. A whole genome DArTseq and SNP analysis for genetic diversity assessment in durum wheat from Central Fertile Crescent. PLoS ONE 2017, 12, e0167821. [Google Scholar] [CrossRef] [PubMed]
- Grzebelus, D.; Iorizzo, M.; Senalik, D.; Ellison, S.; Cavagnaro, P.; Macko-Podgorni, A.; Heller-Uszynska, K.; Kilian, A.; Nothnagel, T.; Allender, C.; et al. Diversity, genetic mapping, and signatures of domestication in the carrot (Daucus carota L.) genome, as revealed by Diversity Arrays Technology (DArT) markers. Mol. Breed. 2014, 33, 625–637. [Google Scholar] [CrossRef]
- Alam, M.; Neal, J.; O’Connor, K.; Kilian, A.; Topp, B. Ultra-high-throughput DArTseq-based silicoDArT and SNP markers for genomic studies in macadamia. PLoS ONE 2018, 13, e0203465. [Google Scholar] [CrossRef]
- O’Connor, K.; Kilian, A.; Hayes, B.; Hardner, C.; Nock, C.; Baten, A.; Alam, M.; Topp, B. Population structure, genetic diversity and linkage disequilibrium in a macadamia breeding population using SNP and silicoDArT markers. Tree Genet. Genomes 2019, 15, 24. [Google Scholar] [CrossRef]
- Sharp, D.; Playford, J. Native macadamia germplasm resources. In Australian Macadamia Society Annual Conference Report; Australian Macadamia Society: Lismore, Australia, 1997; p. 20. [Google Scholar]
- Peace, C.; Vithanage, V.; Turnbull, C.; Carroll, B.J. Characterising macadamia germplasm with codominant radiolabelled DNA fingerprinting (RAF) markers. Acta Horticult. 2002, 575, 371–380. [Google Scholar] [CrossRef]
- Storey, W.B. The ternifolia group of Macadamia species. Pac. Sci. 1965, 19, 507–514. [Google Scholar]
- Peace, C.; Vithanage, V.; Neal, J.; Turnbull, C.; Carroll, B. A comparison of molecular markers for genetic analysis of macadamia. J. Horticult. Sci. Biotechnol. 2004, 79, 965–970. [Google Scholar] [CrossRef]
- Sun, M. Effects of population size, mating system, and evolutionary origin on genetic diversity in Spiranthes sinensis and S. hongkongensis. Conserv. Biol. 1996, 10, 785–795. [Google Scholar] [CrossRef]
- Shapcott, A.; Lamont, R.W.; O’Connor, K.M.; James, H.; Conroy, G.C. Population genetics of Philotheca sporadica (Rutaceae) to advise an offset translocation program. Conserv. Genet. 2015, 16, 687. [Google Scholar] [CrossRef]
- Sniderman, J.K.; Jordan, G.J.; Cowling, R.M. Fossil evidence for a hyperdiverse sclerophyll flora under a non–Mediterranean-type climate. Proc. Natl. Acad. Sci. USA 2013, 110, 3423–3428. [Google Scholar] [CrossRef]
- Brunet, J.; Larson-Rabin, Z.; Stewart, C.M. The distribution of genetic diversity within and among populations of the rocky mountain Columbine: The impact of gene flow, pollinators, and mating system. Int. J. Plant Sci. 2012, 173, 484–494. [Google Scholar] [CrossRef]
- Pisanu, P.C. Survivorship of the Threatened Subtropical Rainforest Tree Macadamia tetraphylla L. Johnson (Proteaceae) in Small Habitat Fragments. Ph.D. Thesis, University of New England, Armidale, Australia, 2001. [Google Scholar]
- Halász, J.; Kodad, O.; Galiba, G.; Skola, I.; Ercisli, S.; Ledbetter, C.; Hegedűs, A. Genetic variability is preserved among strongly differentiated and geographically diverse almond germplasm: An assessment by simple sequence repeat markers. Tree Genet. Genomes 2019, 15, 1–13. [Google Scholar] [CrossRef]
- Nkatha, G.M.; Alice, M.; Anmin, Y.; Jianchu, X.; Aizhong, L. Genetic diversity analysis reveals genetic differentiation and strong population structure in Calotropis plants. Sci. Rep. 2018, 8, 1–10. [Google Scholar]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C. Diversity arrays technology: A generic genome profiling technology on open platforms. In Data Production and Analysis in Population Genomics: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2012; pp. 67–89. [Google Scholar]
- Pritchard, J.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef]
- Earl, D.; von Holdt, B. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Bernard, A.; Barreneche, T.; Lheureux, F.; Dirlewanger, E. Analysis of genetic diversity and structure in a worldwide walnut (Juglans regia L.) germplasm using SSR markers. PLoS ONE 2018, 13, e0208021. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Perrier, X.; Jacquemoud-Collet, J.P. DARwin Software. 2006. Available online: http://darwin.cirad.fr/darwin (accessed on 3 June 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | N | Na | Ne | Ho | He | I | %P | |
|---|---|---|---|---|---|---|---|---|
| SNPs | ||||||||
| M. ternifolia/M. jansenii (Cluster I) | Mean | 19 | 1.28 | 1.08 | 0.04 | 0.07 | 0.11 | 32.00 |
| SE | 0.01 | 0.01 | 0.00 | 0.00 | 0.00 | |||
| M. integrifolia (Cluster II) | Mean | 135 | 1.87 | 1.28 | 0.12 | 0.18 | 0.29 | 86.53 |
| SE | 0.01 | 0.01 | 0.00 | 0.00 | 0.00 | |||
| M. tetraphylla (Cluster III) | Mean | 133 | 1.72 | 1.20 | 0.08 | 0.12 | 0.20 | 72.32 |
| SE | 0.01 | 0.01 | 0.00 | 0.00 | 0.00 | |||
| Admixture | Mean | 15 | 1.75 | 1.34 | 0.18 | 0.21 | 0.33 | 74.93 |
| SE | 0.01 | 0.01 | 0.00 | 0.00 | 0.00 | |||
| Total | Mean | 302 | 1.66 | 1.22 | 0.11 | 0.15 | 0.23 | 47.20 |
| SE | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 | 19.30 | ||
| silicoDArTs | ||||||||
| M. ternifolia/M. jansenii (Cluster I) | Mean | 19 | 0.58 | 1.11 | - | 0.07 | 0.11 | 24.01 |
| SE | 0.01 | 0.00 | - | 0.00 | 0.00 | |||
| M. integrifolia (Cluster II) | Mean | 135 | 1.66 | 1.35 | - | 0.21 | 0.33 | 79.94 |
| SE | 0.01 | 0.00 | - | 0.00 | 0.00 | |||
| M. tetraphylla (Cluster III) | Mean | 133 | 1.47 | 1.26 | - | 0.16 | 0.24 | 70.86 |
| SE | 0.01 | 0.00 | - | 0.00 | 0.00 | |||
| Admixture | Mean | 15 | 1.53 | 1.36 | - | 0.22 | 0.34 | 70.18 |
| SE | 0.01 | 0.00 | - | 0.00 | 0.00 | |||
| Total | Mean | 302 | 1.31 | 1.27 | - | 0.16 | 0.26 | 61.25 |
| SE | 0.01 | 0.00 | - | 0.00 | 0.00 | 12.61 |
| Group | M. integrifolia | M. tetraphylla | M. ternifolia/M. jansenii |
|---|---|---|---|
| SNPs | |||
| M. tetraphylla | 0.20 | ||
| M. ternifolia/M. jansenii | 0.27 | 0.23 | |
| Admixture | 0.06 | 0.07 | 0.22 |
| silicoDArTs | |||
| M. tetraphylla | 0.17 | ||
| M. ternifolia/M. jansenii | 0.17 | 0.16 | |
| Admixture | 0.08 | 0.05 | 0.16 |
| Source | df | SS | MS | Est. Var. | % Var. | PhiPT Statistic | p-Value |
|---|---|---|---|---|---|---|---|
| SNPs | |||||||
| Among species | 3 | 175,814 | 58,604 | 951 | 45% | ||
| Within species | 298 | 344,429 | 1155 | 1155 | 55% | ||
| Total | 301 | 520,243 | 2107 | 100% | 0.45 | 0.001 | |
| silicoDArTs | |||||||
| Among species | 3 | 74,996 | 24,999 | 401 | 34% | ||
| Within species | 298 | 235,037 | 789 | 789 | 66% | ||
| Total | 301 | 310,033 | 1190 | 100% | 0.34 | 0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mai, T.; Alam, M.; Hardner, C.; Henry, R.; Topp, B. Genetic Structure of Wild Germplasm of Macadamia: Species Assignment, Diversity and Phylogeographic Relationships. Plants 2020, 9, 714. https://doi.org/10.3390/plants9060714
Mai T, Alam M, Hardner C, Henry R, Topp B. Genetic Structure of Wild Germplasm of Macadamia: Species Assignment, Diversity and Phylogeographic Relationships. Plants. 2020; 9(6):714. https://doi.org/10.3390/plants9060714
Chicago/Turabian StyleMai, Thuy, Mobashwer Alam, Craig Hardner, Robert Henry, and Bruce Topp. 2020. "Genetic Structure of Wild Germplasm of Macadamia: Species Assignment, Diversity and Phylogeographic Relationships" Plants 9, no. 6: 714. https://doi.org/10.3390/plants9060714
APA StyleMai, T., Alam, M., Hardner, C., Henry, R., & Topp, B. (2020). Genetic Structure of Wild Germplasm of Macadamia: Species Assignment, Diversity and Phylogeographic Relationships. Plants, 9(6), 714. https://doi.org/10.3390/plants9060714