Transcriptome Analysis Identifies Candidate Target Genes Involved in Glyphosate-Resistance Mechanism in Lolium multiflorum

 , ,
, ,
Abstract
1. Introduction
2. Results
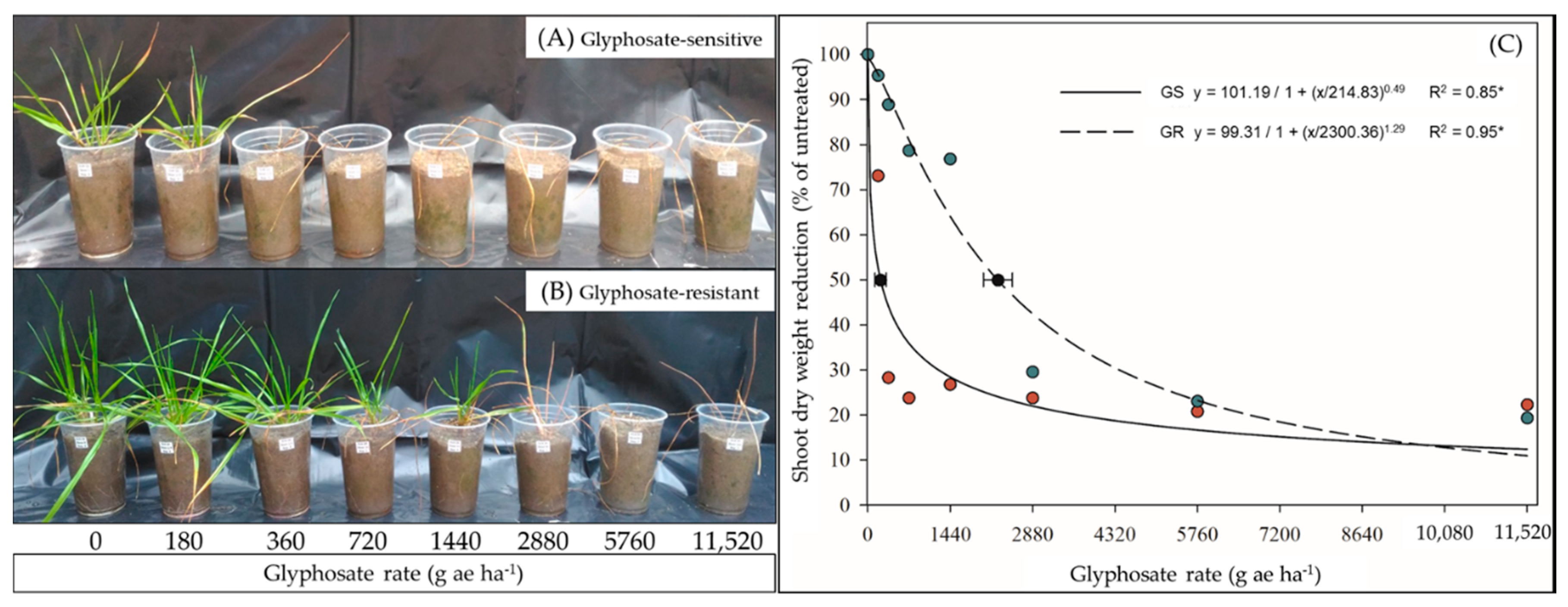
2.1. Glyphosate Dose–Response and Shikimic Acid Content
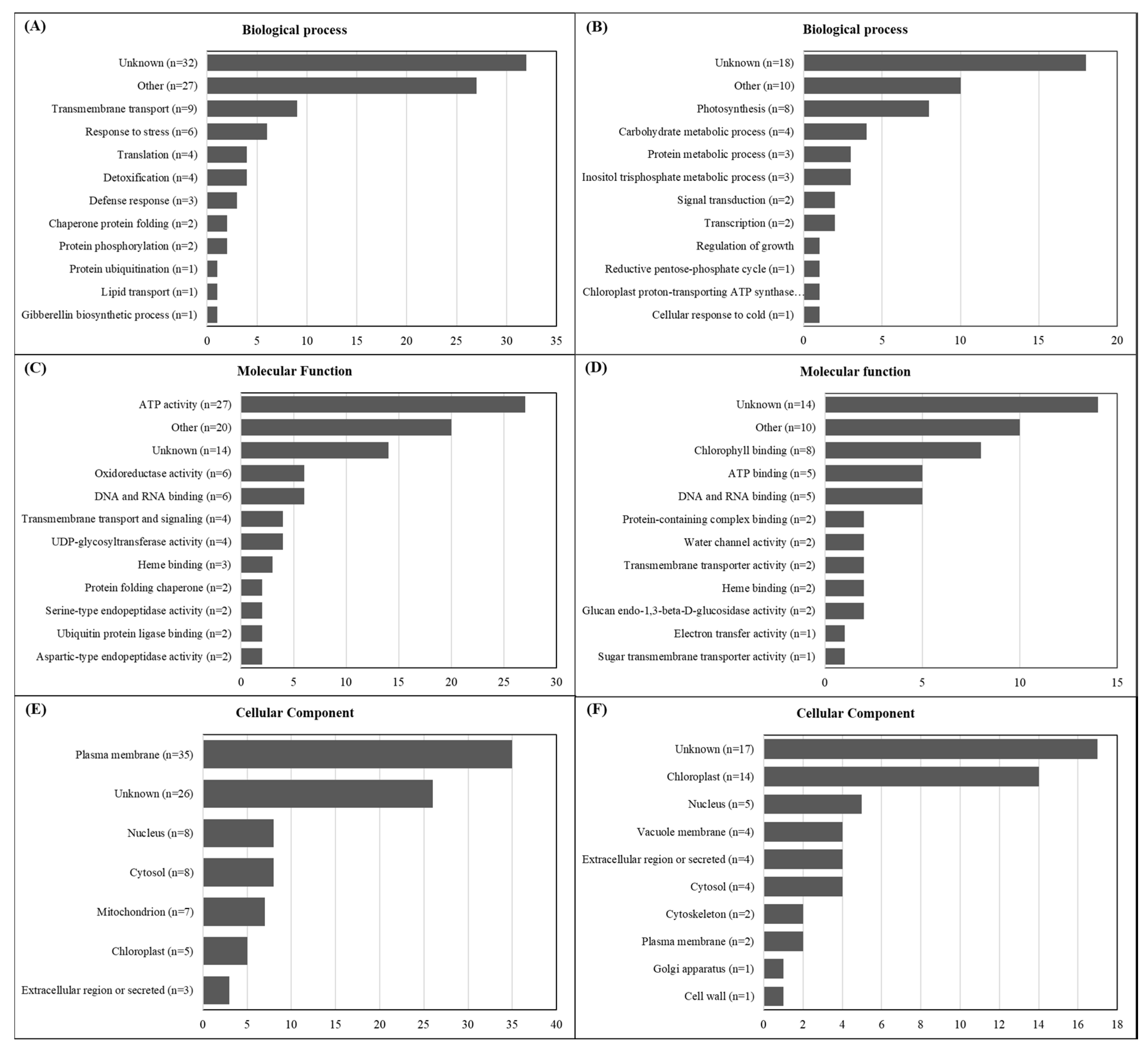
2.2. De Novo Transcriptome Assembly and Functional Annotation
2.3. RNA-Seq Dataset Validation by qRT-PCR Analysis
2.4. EPSPS Sequence Analysis and Expression
2.5. Differentially Expressed Genes in Response to Glyphosate
2.6. Glyphosate Resistance Candidate Gene List
3. Discussion
4. Material and Methods
4.1. Glyphosate Dose–Response and Whole-Plant Shikimic-Acid Bioassay
4.2. RNA-Seq Experimental Design and RNA Extraction
4.3. cDNA Library Construction and Illumina Sequencing
4.4. De Novo Transcriptome Assembly and Functional Annotation
4.5. Differential Gene Expression Analysis and Candidate Target Selection
4.6. RNA-Seq Dataset Validation through qRT-PCR
4.7. EPSPS Transcript Sequence Analysis
4.8. Selection of Differentially Expressed Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Vargas, L.; Roman, E.S.; Rizzardi, M.A.; Silva, V.C. Alteração das características biológicas dos biótipos de azevém (Lolium multiflorum) ocasionada pela resistência ao herbicida glyphosate. Planta Daninha 2005, 23, 153–160. [Google Scholar] [CrossRef][Green Version]
- Preston, C.; Wakelin, A.M.; Dolman, F.C.; Bostamam, Y.; Boutsalis, P. A Decade of glyphosate-resistant Lolium around the world: Mechanisms, genes, fitness, and agronomic management. Weed Sci. 2009, 57, 435–441. [Google Scholar] [CrossRef]
- Vargas, L.; Roman, E.S. Seletividade e eficiência de herbicidas em cereais de inverno. Rev. Bras. Herbic. 2005, 4, 1–10. [Google Scholar] [CrossRef]
- Nandula, V.K. Italian Ryegrass (Lolium perenne ssp. multiflorum) and corn (Zea mays) competition. Am. J. Plant Sci. 2014, 5, 3914–3924. [Google Scholar] [CrossRef]
- Bond, J.A.; Eubank, T.W.; Bond, R.C.; Golden, B.R.; Edwards, H.M. Glyphosate-resistant Italian ryegrass (Lolium perenne ssp. multiflorum) control with fall-applied residual herbicides. Weed Technol. 2014, 28, 361–370. [Google Scholar] [CrossRef]
- Roman, E.S.; Vargas, L.; Rizzardi, M.A.; Mattei, R.W. Resistance of Italian ryegrass (Lolium multiflorum) to glyphosate. Planta Daninha 2004, 22, 301–306. [Google Scholar] [CrossRef]
- Paula, J.; Agostinetto, D.; Schaedler, C.; Vargas, L.; Silva, D.R. Competição de trigo com azevém em função de épocas de aplicação e doses de nitrogênio. Planta Daninha 2011, 29, 557–563. [Google Scholar] [CrossRef]
- Adegas, F.; Vargas, L.; Gazziero, D.; Karam, D.; Silva, A.; Agostinetto, D. Impacto econômico da resistência de plantas daninhas a herbicidas no Brasil. EMBRAPA 2017, 132, 1–12. [Google Scholar]
- Vargas, L.; Adegas, F.; Gazziero, D.; Karam, D.; Agostinetto, D.; Silva, W. Resistência de plantas daninhas aos herbicidas no Brasil. Histórico, distribuição, impacto econômico, manejo e prevenção. In A Era Glyphosate: Agricultura, Meio Ambiente e Homem; Meschede, D.K., Gazziero, D.L.P., Eds.; Midiograf II: Londrina, Brazil, 2016; pp. 219–239. [Google Scholar]
- Yuan, J.S.; Tranel, P.J.; Stewart, C.N. Non-target-site herbicide resistance: A family business. Trends Plant Sci. 2007, 12, 6–13. [Google Scholar] [CrossRef]
- Délye, C.; Jasieniuk, M.; Le Corre, V. Deciphering the evolution of herbicide resistance in weeds. Trends Genet. 2013, 29, 649–658. [Google Scholar] [CrossRef]
- Salas, R.A.; Scott, R.C.; Dayan, F.E.; Burgos, N.R. EPSPS gene amplification in glyphosate-resistant Italian Ryegrass (Lolium perenne ssp. multiflorum) populations from Arkansas (United States). J. Agric. Food Chem. 2015, 63, 5885–5893. [Google Scholar] [CrossRef] [PubMed]
- Sammons, R.D.; Gaines, T.A. Glyphosate resistance: State of knowledge. Pest Manag. Sci. 2014, 70, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Délye, C. Unravelling the genetic bases of non-target-site-based resistance (NTSR) to herbicides: A major challenge for weed science in the forthcoming decade. Pest Manag. Sci. 2013, 69, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Unamba, C.I.N.; Nag, A.; Sharma, R.K. Next generation sequencing technologies: The doorway to the unexplored genomics of non-model plants. Front. Plant Sci. 2015, 6, 1074. [Google Scholar] [CrossRef]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef]
- Hereward, J.P.; Werth, J.A.; Thornby, D.F.; Keenan, M.; Chauhan, B.S.; Walter, G.H. Gene expression in response to glyphosate treatment in fleabane (Conyza bonariensis) - glyphosate death response and candidate resistance genes. Pest Manag. Sci. 2018, 74, 2346–2355. [Google Scholar] [CrossRef]
- Piasecki, C.; Yang, Y.; Benemann, D.P.; Kremer, F.S.; Galli, V.; Millwood, R.J.; Cechin, J.; Agostinetto, D.; Maia, L.C.; Vargas, L.; et al. Transcriptomic Analysis Identifies New Non-Target Site Glyphosate-Resistance Genes in Conyza bonariensis. Plants 2019, 8, 157. [Google Scholar] [CrossRef]
- Duhoux, A.; Carrère, S.; Gouzy, J.; Bonin, L.; Délye, C. RNA-Seq analysis of rye-grass transcriptomic response to an herbicide inhibiting acetolactate-synthase identifies transcripts linked to non-target-site-based resistance. Plant Mol. Biol. 2015, 87, 473–487. [Google Scholar] [CrossRef]
- Gardin, J.A.C.; Gouzy, J.; Carrère, S.; Délye, C. ALOMYbase, a resource to investigate non-target-site-based resistance to herbicides inhibiting acetolactate-synthase (ALS) in the major grass weed Alopecurus myosuroides (black-grass). BMC Genomics 2015, 16, 590. [Google Scholar] [CrossRef]
- An, J.; Shen, X.; Ma, Q.; Yang, C.; Liu, S.; Chen, Y. Transcriptome profiling to discover putative genes associated with paraquat resistance in goosegrass (Eleusine indica L.). PLoS ONE 2014, 9, e99940. [Google Scholar] [CrossRef]
- Giacomini, D.A.; Gaines, T.; Beffa, R.; Tranel, P.J. Optimizing RNA-seq studies to investigate herbicide resistance. Pest Manag. Sci. 2018, 74, 2260–2264. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.S.; Abercrombie, L.L.G.; Cao, Y.; Halfhill, M.D.; Zhou, X.; Peng, Y.; Hu, J.; Rao, M.R.; Heck, G.R.; Larosa, T.J.; et al. Functional genomics analysis of horseweed (Conyza canadensis) with special reference to the evolution of non–target-site glyphosate resistance. Weed Sci. 2010, 58, 109–117. [Google Scholar] [CrossRef]
- Délye, C.; Duhoux, A.; Pernin, F.; Riggins, C.W.; Tranel, P.J. Molecular mechanisms of herbicide resistance. Weed Sci. 2015, 63, 91–115. [Google Scholar] [CrossRef]
- Peng, Y.; Lai, Z.; Lane, T.; Nageswara-Rao, M.; Okada, M.; Jasieniuk, M.; O’Geen, H.; Kim, R.W.; Sammons, R.D.; Rieseberg, L.H.; et al. De novo genome assembly of the economically important weed horseweed using integrated data from multiple sequencing platforms. Plant Physiol. 2014, 166, 1241–1254. [Google Scholar] [CrossRef] [PubMed]
- Vargas, L.; Ruchel, Q.; Agostinetto, D.; Lamego, F.P.; Langaro, A.C.; Piesanti, S.R. Verificação do mecanismo de resistência ao glyphosate em biótipos de azevém. Planta Daninha 2016, 34, 565–573. [Google Scholar] [CrossRef]
- Novaković, L.; Guo, T.; Bacic, A.; Sampathkumar, A.; Johnson, K.L. Hitting the wall—Sensing and signaling pathways involved in plant cell wall remodeling in response to abiotic stress. Plants 2018, 7, 89. [Google Scholar] [CrossRef]
- Caverzan, A.; Piasecki, C.; Chavarria, G.; Stewart, C.; Vargas, L. Defenses against ROS in crops and weeds: The effects of interference and herbicides. Int. J. Mol. Sci. 2019, 20, 1086. [Google Scholar] [CrossRef]
- Tzin, V.; Galili, G. New insights into the shikimate and aromatic amino acids biosynthesis pathways in plants. Mol. Plant 2010, 3, 956–972. [Google Scholar] [CrossRef]
- Schönbrunn, E.; Eschenburg, S.; Shuttleworth, W.A.; Schloss, J.V.; Amrhein, N.; Evans, J.N.; Kabsch, W. Interaction of the herbicide glyphosate with its target enzyme 5-enolpyruvylshikimate 3-phosphate synthase in atomic detail. Proc. Natl. Acad. Sci. USA 2001, 98, 1376–1380. [Google Scholar] [CrossRef]
- Nol, N.; Tsikou, D.; Eid, M.; Livieratos, I.C.; Giannopolitis, C.N. Shikimate leaf disc assay for early detection of glyphosate resistance in Conyza canadensis and relative transcript levels of EPSPS and ABC transporter genes. Weed Res. 2012, 52, 233–241. [Google Scholar] [CrossRef]
- Cramer, G.R.; Urano, K.; Delrot, S.; Pezzotti, M.; Shinozaki, K. Effects of abiotic stress on plants: A systems biology perspective. BMC Plant Biol. 2011, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Cobb, A.; Reade, P. Herbicides and Plant Physiology, 2nd ed.; Wiley-Blackwell: Shropshire, UK, 2010; p. 286. ISBN 9781405129350. [Google Scholar]
- Pan, L.; Yu, Q.; Han, H.; Mao, L.; Nyporko, A.; Fan, L.; Bai, L.; Powles, S.B. Aldo-keto reductase metabolizes glyphosate and confers glyphosate resistance in Echinochloa colona. Plant Physiol. 2019, 181, 1519–1534. [Google Scholar] [CrossRef] [PubMed]
- Maroli, A.S.; Nandula, V.K.; Dayan, F.E.; Duke, S.O.; Gerard, P.; Tharayil, N. Metabolic profiling and enzyme analyses indicate a potential role of antioxidant systems in complementing glyphosate resistance in an Amaranthus palmeri biotype. J. Agric. Food Chem. 2015, 63, 9199–9209. [Google Scholar] [CrossRef]
- Powles, S.B.; Yu, Q. Evolution in action: Plants resistant to herbicides. Annu. Rev. Plant Biol. 2010, 61, 317–347. [Google Scholar] [CrossRef]
- Kreuz, K.; Tommasini, R.; Martinoia, E. Old enzymes for a new job: Herbicide detoxification in plants. Plant Physiol. 1996, 111, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.; Lim, E.-K.; Poppenberger, B.; Vaistij, F.E. Glycosyltransferases of lipophilic small molecules. Annu. Rev. Plant Biol. 2006, 57, 567–597. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Joshi, D.; Yadav, P.K.; Gupta, A.K.; Bhatt, T.K. Role of ubiquitin-mediated degradation system in plant biology. Front. Plant Sci. 2016, 7, 806. [Google Scholar] [CrossRef]
- Hu, H.; Sun, S.-C. Ubiquitin signaling in immune responses. Cell Res. 2016, 26, 457–483. [Google Scholar] [CrossRef]
- Zulet, A.; Gil-Monreal, M.; Villamor, J.G.; Zabalza, A.; van der Hoorn, R.A.L.; Royuela, M. Proteolytic pathways induced by herbicides that inhibit amino acid biosynthesis. PLoS ONE 2013, 8, e73847. [Google Scholar] [CrossRef]
- Burgos, N.R.; Tranel, P.J.; Streibig, J.C.; Davis, V.M.; Shaner, D.; Norsworthy, J.K.; Ritz, C. Review: Confirmation of resistance to herbicides and evaluation of resistance levels. Weed Sci. 2013, 61, 4–20. [Google Scholar] [CrossRef]
- Singh, B.K.; Shaner, D.L. Rapid determination of glyphosate injury to plants and identification of glyphosate resistant plants. Weed Sci. Soc. Am. 1998, 12, 527–530. [Google Scholar] [CrossRef]
- Perez-Jones, A.; Park, K.W.; Polge, N.; Colquhoun, J.; Mallory-Smith, C.A. Investigating the mechanisms of glyphosate resistance in Lolium multiflorum. Planta 2007, 226, 395–404. [Google Scholar] [CrossRef]
- Piasecki, C.; Carvalho, I.R.; Cechin, J.; Goulart, F.A.P.; da Maia, L.C.; Agostinetto, D.; Caverzan, A.; Stewart, C.N., Jr.; Vargas, L. Oxidative stress and differential antioxidant enzyme activity in glyphosate-resistant and -sensitive hairy fleabane in response to glyphosate treatment. Bragantia 2019, 78, 379–396. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res. 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Apweiler, R. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 2015, 16, 169. [Google Scholar] [CrossRef]
- Zhang, Z.; Hu, J. Development and Validation of Endogenous Reference Genes for Expression Profiling of Medaka (Oryzias latipes) Exposed to Endocrine Disrupting Chemicals by Quantitative Real-Time RT-PCR. Toxicol. Sci. 2007, 95, 356–368. [Google Scholar] [CrossRef]
- Wan, H.; Zhao, Z.; Qian, C.; Sui, Y.; Malik, A.A.; Chen, J. Selection of appropriate reference genes for gene expression studies by quantitative real-time polymerase chain reaction in cucumber. Anal. Biochem. 2010, 399, 257–261. [Google Scholar] [CrossRef]
- Clarke, B.; Rahman, S. A microarray analysis of wheat grain hardness. Theor. Appl. Genet. 2005, 110, 1259–1267. [Google Scholar] [CrossRef]
- Lee, J.M.; Roche, J.R.; Donaghy, D.J.; Thrush, A.; Sathish, P. Validation of reference genes for quantitative RT-PCR studies of gene expression in perennial ryegrass (Lolium perenne L.). BMC Mol. Biol. 2010, 11, 8. [Google Scholar] [CrossRef]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef]
- Yuan, J.S.; Wang, D.; Stewart, C.N. Statistical methods for efficiency adjusted real-time PCR quantification. Biotechnol. J. 2008, 3, 112–123. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Assembly Statistics * |
|---|---|
| Total assembled bases | 155,613,732 |
| Total transcripts | 146,263 |
| Total contigs at “gene” level | 87,433 |
| Contig N50 | 740 |
| Average contig length (bp) | 575.24 |
| GC (%) | 50.47 |
| Phred score ≥Q30 | 99.95% |
| Contig ID | Uniprot ID | Functional Annotation—Gene | Putative Localization | 1 Log2 FC GR t1/t0 | 1 Log2 FC GS t1/t0 | Fold-Change GR/GS * |
|---|---|---|---|---|---|---|
| Upregulated Genes | ||||||
| DN140736_c3_g2 | Q9STT5 | ABC transporter A family member 7-ABCA7 | Multi-pass membrane protein | 4.8 | 1.9 | 2.5 |
| DN134601_c1_g3 | P53832 | Cell wall integrity and stress response-WSC2 | Plasma membrane | 6.6 | 4.5 | 1.4 |
| DN140896_c3_g3 | Q8GZ17 | COBRA-like protein 7-COBL7 | Cell membrane | 4.6 | 2.8 | 1.7 |
| DN138383_c1_g1 | A0A3B6IQ15 | Fe2OG dioxygenase domain-containing protein | Unknown | 4.0 | 2.4 | 1.7 |
| DN112601_c0_g2 | Q39111 | Gibberellin 20 oxidase 2-GA20OX2 | Unknown | 4.9 | 0.6 | 8.7 |
| DN137545_c0_g2 | P52409 | Glucan endo-1,3-beta-glucosidase-GLC1 | Component of plasma membrane | 3.7 | 2.3 | 1.6 |
| DN130084_c1_g4 | M0Y4P1 | Glycosyltransferase UDP UGT13248 | Intracellular-membrane-bound | 3.5 | 2.7 | 1.3 |
| DN125138_c7_g3 | Q7XT97 | Glycosyltransferase UDP 79 UGT79 | Intracellular-membrane-bound | 3.8 | 2.9 | 1.3 |
| DN123097_c0_g1 | A0A3B6PNF9 | Glycosyltransferase | Unknown | 4.8 | 2.7 | 1.8 |
| DN111587_c0_g1 | P82977 | Subtilisin-chymotrypsin inhibitor-WSCI | Extracellular region or secreted | 5.9 | 3.7 | 1.6 |
| DN135754_c1_g1 | Q9MAX3 | Sulfate transporter 1.2 | Multi-pass membrane protein | 3.6 | 2.1 | 1.7 |
| DN120520_c4_g4 | P69326 | Ubiquitin | Nucleus and cytoplasm | 6.3 | 3.4 | 1.8 |
| DN123915_c1_g2 | P0CH35 | Ubiquitin-60S ribosomal protein L40-2 Ub-CEP52-2 | Nucleus and cytoplasm | 6.4 | N/A | 6.4 |
| DN141149_c4_g5 | A0A3B6THG7 | Uncharacterized protein | Integral component of membrane | 4.9 | 0.5 | 9.9 |
| Downregulated Genes | ||||||
| DN134059_c0_g2 | A0A3B6MSN4 | DAO domain-containing protein | Unknown | −3.1 | −1.2 | 2.5 |
| DN119890_c0_g2 | P17816 | Glycine-rich cell wall structural protein-GRP | Cell wall | −4.5 | −2.0 | 2.2 |
| DN137034_c0_g1 | A0A3B5Y7P6 | RNA polymerase sigma factor | Chloroplast | −2.9 | −1.3 | 2.2 |
| DN136212_c1_g1 | P27626 | Senescence-associated protein-DIN1 | Unknown | −5.2 | N/A | 5.2 |
| DN134440_c0_g3 | Q41558 | Transcription factor-HBP-1b(c1) | Nucleus | −3.5 | −1.5 | 2.4 |
| DN124989_c0_g4 | W5D4P6 | Uncharacterized protein-TRAES_3BF019300240CFD_c1 | Unknown | −5.4 | N/A | 5.4 |
| DN127114_c0_g3 | Q9LPN5 | Vacuolar protein sorting-associated-VPS60-1 | Endosome | −4.1 | −2.1 | 2.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cechin, J.; Piasecki, C.; Benemann, D.P.; Kremer, F.S.; Galli, V.; Maia, L.C.; Agostinetto, D.; Vargas, L. Transcriptome Analysis Identifies Candidate Target Genes Involved in Glyphosate-Resistance Mechanism in Lolium multiflorum. Plants 2020, 9, 685. https://doi.org/10.3390/plants9060685
Cechin J, Piasecki C, Benemann DP, Kremer FS, Galli V, Maia LC, Agostinetto D, Vargas L. Transcriptome Analysis Identifies Candidate Target Genes Involved in Glyphosate-Resistance Mechanism in Lolium multiflorum. Plants. 2020; 9(6):685. https://doi.org/10.3390/plants9060685
Chicago/Turabian StyleCechin, Joanei, Cristiano Piasecki, Daiane P. Benemann, Frederico S. Kremer, Vanessa Galli, Luciano C. Maia, Dirceu Agostinetto, and Leandro Vargas. 2020. "Transcriptome Analysis Identifies Candidate Target Genes Involved in Glyphosate-Resistance Mechanism in Lolium multiflorum" Plants 9, no. 6: 685. https://doi.org/10.3390/plants9060685
APA StyleCechin, J., Piasecki, C., Benemann, D. P., Kremer, F. S., Galli, V., Maia, L. C., Agostinetto, D., & Vargas, L. (2020). Transcriptome Analysis Identifies Candidate Target Genes Involved in Glyphosate-Resistance Mechanism in Lolium multiflorum. Plants, 9(6), 685. https://doi.org/10.3390/plants9060685