Current State and Perspectives in Population Genomics of the Common Bean




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
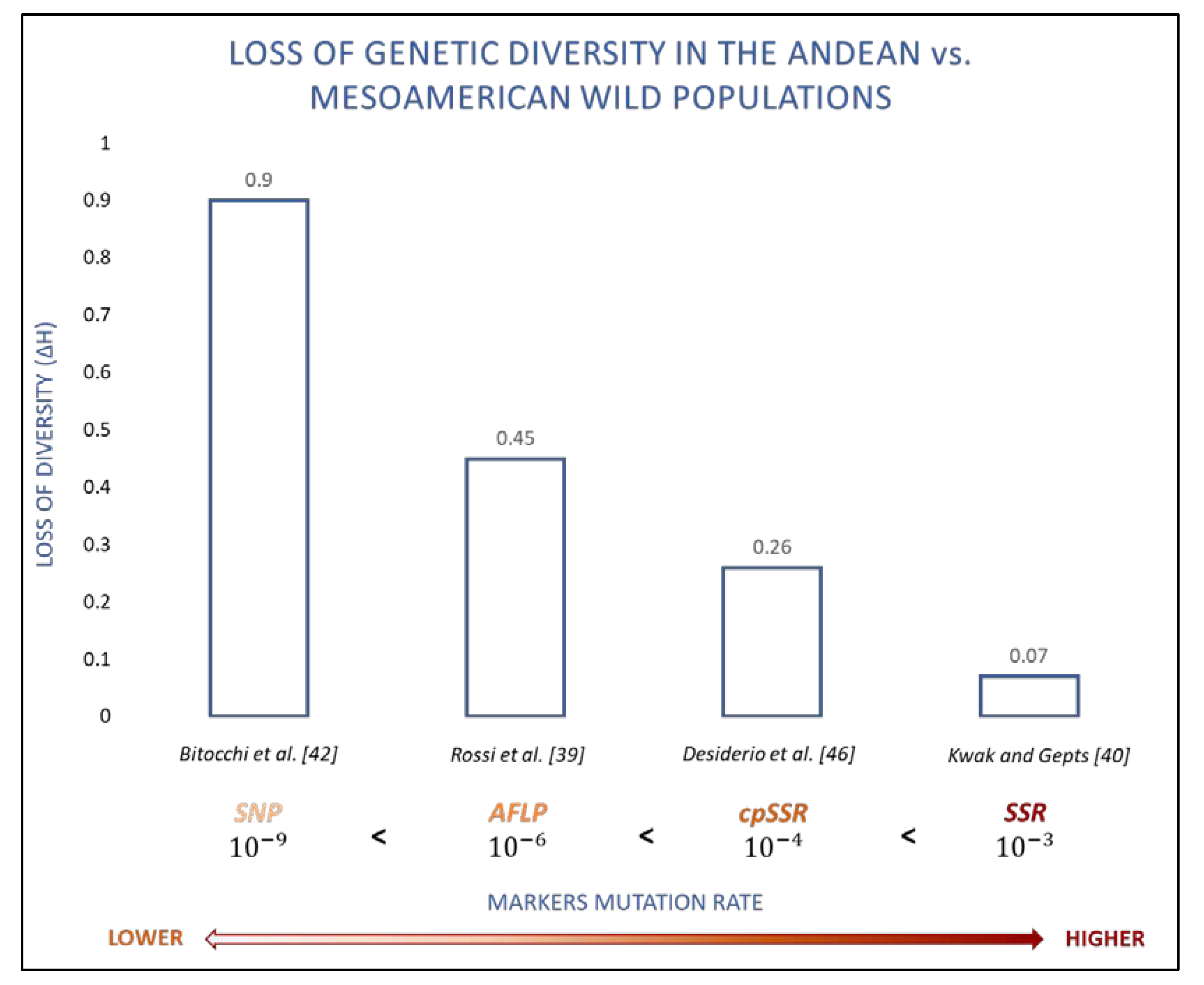
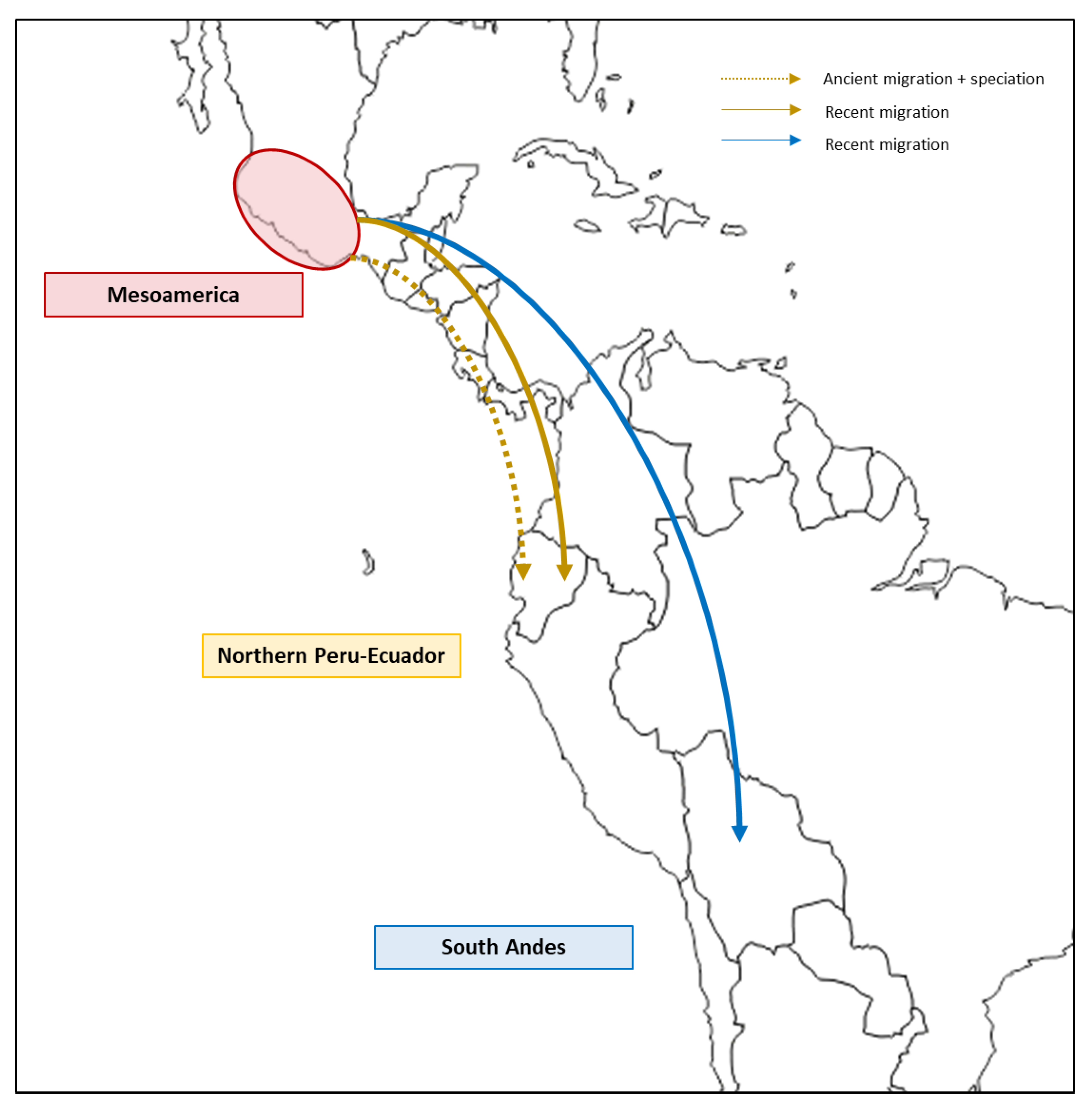
2. Origin of the Common Bean in the Light of Different Molecular Markers
3. Domestication of the Common Bean
3.1. Mapping Domestication Traits
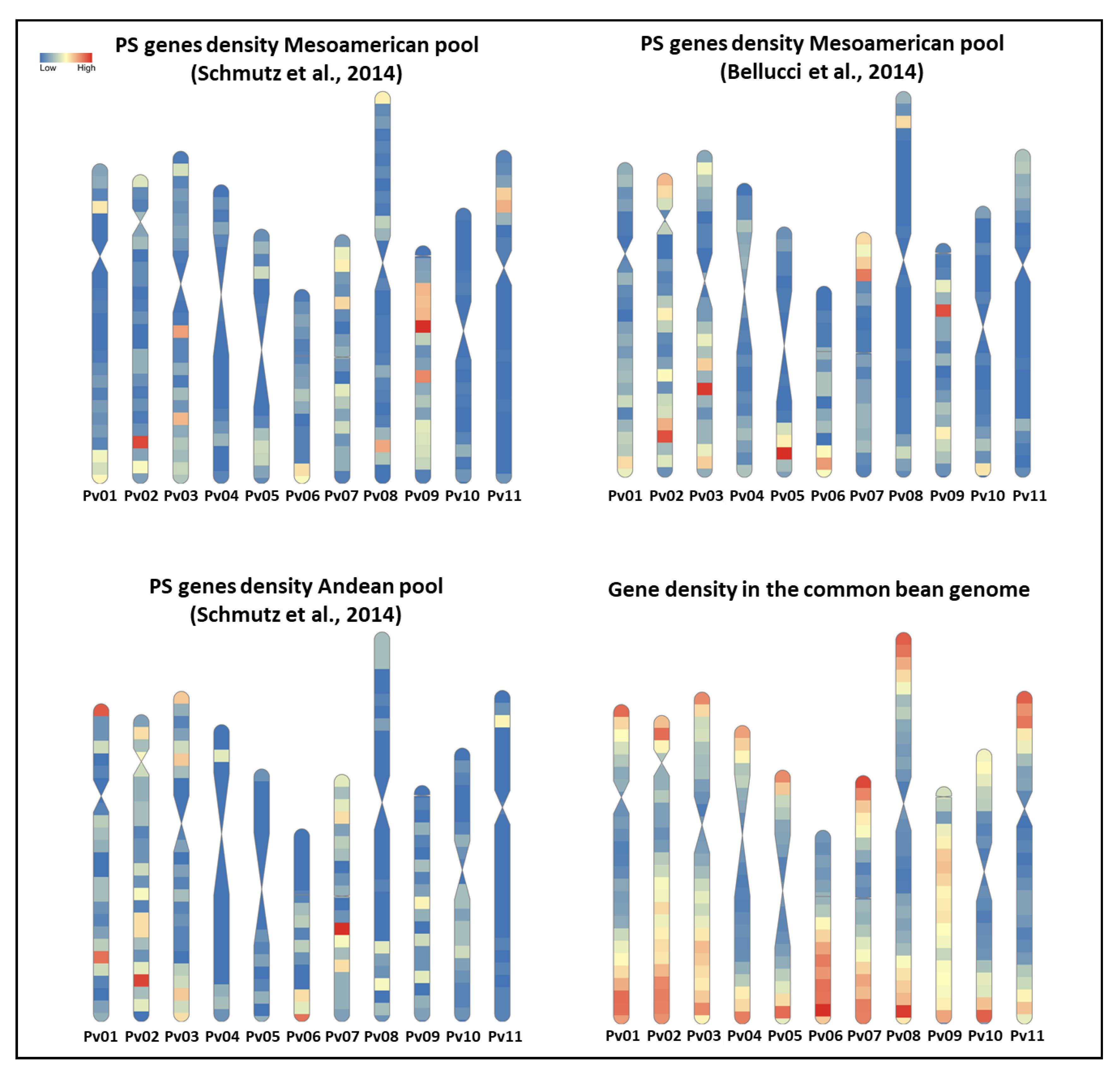
3.2. Signature of Selection
4. Diversification and Adaptation of P. Vulgaris to Different Agro-Ecological Conditions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kimura, M. Evolutionary rate at the molecular level. Nature 1968, 217, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. Recent development of the neutral theory viewed from the wrightian tradition of theoretical population genetics. Proc. Natl. Acad. Sci. USA 1991, 88, 5969–5973. [Google Scholar] [CrossRef]
- Black, W.C.; Baer, C.F.; Antolin, M.F.; DuTeau, N.M. Population genomics: Genome-wide sampling of insect populations. Annu. Rev. Entomol. 2001, 46, 441–469. [Google Scholar] [CrossRef] [PubMed]
- Luikart, G.; England, P.R.; Tallmon, D.; Jordan, S.; Taberlet, P. The power and promise of population genomics: From genotyping to genome typing. Nat. Rev. Genet. 2003, 4, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Hohenlohe, P.A.; Hand, B.K.; Andrews, K.R.; Luikart, G. Population genomics provides key insights in ecology and evolution. In Population Genomics: Concepts, Approaches and Applications; Rajora, O.M.P., Ed.; Publisher: Gewerbestrasse, Switzerland, 2018; pp. 483–510. [Google Scholar] [CrossRef]
- Hoban, S.; Kelley, J.L.; Lotterhos, K.E.; Antolin, M.F.; Bradburd, G.; Lowry, D.B.; Poss, M.L.; Reed, L.K.; Storfer, A.; Whitlock, M.C. Finding the genomic basis of local adaptation: Pitfalls, practical solutions, and future directions. Am. Nat. 2016, 188, 379–397. [Google Scholar] [CrossRef]
- Hendricks, S.; Anderson, E.C.; Antao, T.; Bernatchez, L.; Forester, B.R.; Garner, B.; Hand, B.K.; Hohenlohe, P.A.; Kardos, M.; Koop, B.; et al. Recent advances in conservation and population genomics data analysis. Evol. Appl. 2018, 11, 1197–1211. [Google Scholar] [CrossRef]
- Hunter, M.E.; Hoban, S.M.; Bruford, M.W.; Segelbacher, G.; Bernatchez, L. Next-generation, conservation genetics and biodiversity monitoring. Evol. Appl. 2018, 11, 1029–1034. [Google Scholar] [CrossRef]
- Cortinovis, G.; Di Vittori, V.; Bellucci, E.; Bitocchi, E.; Papa, R. Adaptation to novel environments during crop diversification. Curr. Opin. Plant Biol. 2020. [Google Scholar] [CrossRef]
- Harris, H. Enzyme polymorphism in man. Proc. Roy Soc. B 1966, 164, 298–310. [Google Scholar]
- Lewontin, R.C.; Hubby, J.L. A molecular approach to the study of genetic heterozygosity in natural populations. II. Amount of variation and degree of heterozygosity in natural populations of Drosophila pseudoobscura. Genetics 1966, 54, 595–609. [Google Scholar]
- Schlötterer, C. The evolution of molecular markers-just a matter of fashion? Nat. Rev. Genet. 2004, 5, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C.; Lansman, R.A.; Shade, R.O. The use of restriction endonucleases to measure mitochondrial DNA sequence relatedness in natural populations. I. Population structure and evolution in the genus Peromyscus. Genetics 1979, 92, 279–295. [Google Scholar] [PubMed]
- Allendorf, F.W. Genetics and the conservation of natural populations: Allozymes to genomes. Mol. Ecol. 2017, 26, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Khlestkina, E.K.; Salina, E.A. SNP markers: Methods of analysis, ways of development, and comparison on an example of common wheat. Russ. J. Genet. 2006, 42, 585–594. [Google Scholar] [CrossRef]
- Morgante, M.; Olivieri, A.M. PCR-amplified microsatellites as markers in plant genetics. Plant J. 1993, 3, 175–182. [Google Scholar] [CrossRef]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; van de Lee, T.; Hornes, M.; Friters, A.; Pot, J.; Paleman, J.; Kuiper, M.; et al. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef]
- Morin, P.A.; Luikart, G.; Wayne, R.K. The SNP workshop group. SNPs in ecology, evolution and conservation. Trend. Ecol. Evol. 2004, 19, 208–216. [Google Scholar] [CrossRef]
- Sharma, T.R.; Devanna, B.N.; Kiran, K.; Singh, P.K.; Arora, K.; Jain, P.; Tiwari, I.M.; Dubey, H.; Saklani, B.; Kumari, M.; et al. Status and prospects of next-generation sequencing technologies in crop plants. In Next-Generation Sequencing and Bioinformatics for Plant Science; Bhadauria, V., Ed.; Caister Academic Press: Norfolk, UK, 2017; pp. 1–36. [Google Scholar] [CrossRef]
- Akey, J.M.; Zhang, G.; Zhang, K.; Jin, L.; Shriver, M.D. Interrogating a high-density SNP map for signatures of natural selection. Genom. Res. 2002, 12, 1805–1814. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Ellegren, H. Genome sequencing and population genomics in non-model organisms. Trends Ecol. Evol. 2014, 29, 51–63. [Google Scholar] [CrossRef]
- Bellucci, E.; Bitocchi, E.; Ferrarini, A.; Benazzo, A.; Biagetti, E.; Klie, S.; Minio, A.; Rau, D.; Rodriguez, M.; Panziera, A.; et al. Decreased nucleotide and expression diversity and modified co-expression patterns characterize domestication in the common bean. Plant Cell 2014, 26, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Beleggia, R.; Rau, D.; Laidò, G.; Platani, C.; Nigro, F.; Fragasso, M.; De Vita, P.; Scossa, F.; Fernie, A.R.; Nikoloski, Z.; et al. Evolutionary metabolomics reveals domestication-associated changes in tetraploid wheat kernels. Mol. Biol. Evol. 2016, 33, 1740–1753. [Google Scholar] [CrossRef] [PubMed]
- Kingman, J.F.C. The coalescent. Stoch. Process Appl. 1982, 13, 235–248. [Google Scholar] [CrossRef]
- Wright, S.I.; Bi, I.V.; Schroeder, S.G.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S. The effects of artificial selection on the maize genome. Science 2005, 308, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, M.; Tenaillon, M.I.; Bi, I.V.; Schroeder, S.G.; Sanchez-Villeda, H.; Doebley, J.F.; Gaut, B.S.; McMullen, M.D. A large-scale screen for artificial selection in maize identifies candidate agronomic loci for domestication and crop improvement. Plant Cell 2005, 17, 2859–2872. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, M.; Wright, S.I.; McMullen, M.D. Genomic screening for artificial selection during domestication and improvement in maize. Annu. Bot. 2007, 100, 967–973. [Google Scholar] [CrossRef]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef]
- Vlasova, A.; Capella-Gutiérrez, S.; Rendón-Anaya, M.; Hernández-Oñate, M.; Minoche, A.E.; Erb, I.; Camara, F.; Prieto Barja, P.; Corvelo, A.; Sanseverino, W.; et al. Genome and transcriptome analysis of the Mesoamerican common bean and the role of gene duplications in establishing tissue and temporal specialization of genes. Genom. Biol. 2016, 17, 32. [Google Scholar] [CrossRef]
- Bitocchi, E.; Rau, D.; Bellucci, E.; Rodriguez, M.; Murgia, M.L.; Gioia, T.; Santo, D.; Nanni, L.; Attene, G.; Papa, R. Beans (Phaseolus ssp.) as a model for understanding crop evolution. Front. Plant. Sci. 2017, 8, 722. [Google Scholar] [CrossRef]
- Toro, O.; Tohme, J.; Debouck, D.G. Wild bean (Phaseolus vulgaris L.): Description and distribution. In Centro Internacional de Agricultura Tropical (CIAT); International Board for Plant Genetic Resources (IBPGR): Cali, CO, USA, 1990; p. 109, (CIAT publication no. 181). [Google Scholar]
- Debouck, D.G.; Toro, O.; Paredes, O.M.; Johnson, W.C.; Gepts, P. Genetic diversity and ecological distribution of Phaseolus vulgaris in north-western South America. Econ. Bot. 1993, 47, 408–423. [Google Scholar] [CrossRef]
- Gepts, P.; Bliss, F.A. F1 hybrid weakness in the common bean: Differential geographic origin suggests two gene pools in cultivated bean germplasm. J. Hered. 1985, 76, 447–450. [Google Scholar] [CrossRef]
- Gepts, P.; Osborn, T.C.; Rashka, K.; Bliss, F.A. Phaseolin-protein variability in wild forms and landraces of the common bean (Phaseolus vulgaris): Evidence for multiple centers of domestication. Econ. Bot. 1986, 40, 451–468. [Google Scholar] [CrossRef]
- Koenig, R.; Gepts, P. Allozyme diversity in wild Phaseolus vulgaris: Further evidence for two major centers of diversity. Theor. Appl. Genet. 1989, 78, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Velásquez, V.L.; Gepts, P. RFLP diversity in common bean (Phaseolus vulgaris L.). Genome 1994, 37, 256–263. [Google Scholar] [CrossRef]
- Freyre, R.; Ríos, R.; Guzmán, L.; Debouck, D.G.; Gepts, P. Ecogeographic distribution of Phaseolus spp. (Fabaceae) in Bolivia. Econ. Bot. 1996, 50, 195–215. [Google Scholar] [CrossRef]
- Papa, R.; Gepts, P. Asymmetry of gene flow and differential geographical structure of molecular diversity in wild and domesticated common bean (Phaseolus vulgaris L.) from Mesoamerica. Theor. Appl. Genet. 2003, 106, 239–250. [Google Scholar] [CrossRef]
- Rossi, M.; Bitocchi, E.; Bellucci, E.; Nanni, L.; Rau, D.; Attene, G.; Papa, R. Linkage disequilibrium and population structure in wild and domesticated populations of Phaseolus vulgaris L. Evol. Appl. 2009, 2, 504–522. [Google Scholar] [CrossRef]
- Kwak, M.; Gepts, P. Structure of genetic diversity in the two major gene pools of common bean (Phaseolus vulgaris L., Fabaceae). Theor. Appl. Genet. 2009, 118, 979–992. [Google Scholar] [CrossRef]
- Nei, M.; Maruyama, T.; Chakraborty, R. The bottleneck effect and genetic variability of populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef]
- Bitocchi, E.; Nanni, L.; Bellucci, E.; Rossi, M.; Giardini, A.; Spagnoletti Zeuli, P.; Logozzo, G.; Stougaard, J.; McClean, P.; Attene, G.; et al. Mesoamerican origin of the common bean (Phaseolus vulgaris L.) is revealed by sequence data. Proc. Natl. Acad. Sci. USA 2012, 109, E788–E796. [Google Scholar] [CrossRef]
- Nei, M.; Chakraborty, R.; Fuerst, P.A. Infinite allele model with varying mutation rate. Proc. Natl. Acad. Sci. USA 1976, 73, 4164–4168. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Bottlenecks, genetic polymorphism and speciation. Genetics 2005, 170, 1–4. [Google Scholar] [PubMed]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Desiderio, F.; Bitocchi, E.; Bellucci, E.; Rau, D.; Rodriguez, M.; Attene, G.; Papa, R.; Nanni, L. Chloroplast microsatellite diversity in Phaseolus vulgaris. Front. Plant. Sci. 2013, 3, 312. [Google Scholar] [CrossRef] [PubMed]
- Ariani, A.; Mier y Teran, J.C.; Gepts, P. Spatial and temporal scales of range expansion in wild Phaseolus vulgaris. Mol. Biol. Evol. 2017, 35, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Rendón-Anaya, M.; Montero-Vargas, J.M.; Saburido-Álvarez, S.; Vlasova, A.; Capella-Gutierrez, S.; Ordaz-Ortiz, J.J.; Aguilar, O.M.; Vianello-Brondani, R.P.; Santalla, M.; Delaye, L.; et al. Genomic history of the origin and domestication of common bean unveils its closest sister species. Genome Biol. 2017, 18, 60. [Google Scholar] [CrossRef]
- Bitocchi, E.; Bellucci, E.; Giardini, A.; Rau, D.; Rodriguez, M.; Biagetti, E.; Santilocchi, R.; Spagnoletti Zeuli, P.; Gioia, T.; Logozzo, G.; et al. Molecular analysis of the parallel domestication of the common bean in Mesoamerica and the andes. New Phytol. 2013, 197, 300–313. [Google Scholar] [CrossRef]
- Bellucci, E.; Bitocchi, E.; Rau, D.; Rodriguez, M.; Biagetti, E.; Giardini, A.; Attene, G.; Nanni, L.; Papa, R. Genomics of origin, domestication and evolution of Phaseolus vulgaris. In Genomics of Plant Genetic Resources; Tuberosa, R., Graner, A., Frison, E., Eds.; Springer: Berlin, Germany, 2014; pp. 483–507. [Google Scholar]
- Koinange, E.M.K.; Singh, S.P.; Gepts, P. Genetic control of the domestication syndrome in common bean. Crop Sci. 1996, 36, 1037–1045. [Google Scholar] [CrossRef]
- Di Vittori, V.; Bellucci, E.; Bitocchi, E.; Rau, D.; Rodriguez, M.; Murgia, M.L.; Nanni, L.; Attene, G.; Papa, R. Domestication and crop history. In The Common Bean Genome; Pérez de la Vega, M., Santalla, M., Marsolais, M., Eds.; Compendium of Plant Genomes; Springer International Publishing AG: New York, NY, USA, 2017; pp. 21–55. [Google Scholar] [CrossRef]
- Rau, D.; Murgia, M.L.; Rodriguez, M.; Bitocchi, E.; Bellucci, E.; Fois, D.; Albani, D.; Nanni, L.; Gioia, T.; Santo, D.; et al. Genomic dissection of pod shattering in common bean: Mutations at non-orthologous loci at the basis of convergent phenotypic evolution under domestication of leguminous species. Plant J. 2019, 97, 693–714. [Google Scholar] [CrossRef]
- Parker, T.A.; Berny Mier, Y.; Teran, J.C.; Palkovic, A.; Jernstedt, J.; Gepts, P. Pod indehiscence is a domestication and aridity resilience trait in common bean. New Phytol. 2019, 225, 558–570. [Google Scholar] [CrossRef]
- Di Vittori, V. Common bean domestication (manuscript in preparation).
- Repinsky, S.L.; Kwak, M.; Gepts, P. The common bean growth habit gene PvTFL1y is a functional homolog of Arabidopsis TFL. Theor. Appl. Genet. 2012, 124, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.E.; Bett, K.E.; Stonehouse, R.; Lee, R.; Pflieger, S.; Moghaddam, S.M.; Geffroy, V.; Miklas, P.; Mamidi, S. White seed colour in common bean (Phaseolus vulgaris) results from convergent evolution in the P (pigment) gene. New Phytol. 2018, 219, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Di Vittori, V.; Gioia, T.; Rodriguez, M.; Bellucci, E.; Bitocchi, E.; Nanni, L.; Attene, G.; Rau, D.; Papa, R. Convergent evolution of the seed shattering trait. Genes 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.L.; Attene, G.; Rodriguez, M.; Bitocchi, E.; Bellucci, E.; Fois, D.; Nanni, L.; Gioia, T.; Albani, D.M.; Papa, R.; et al. A comprehensive phenotypic investigation of the “pod-shattering syndrome” in common bean. Front. Plant Sci. 2017, 8, 251. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, S.; Rossi, M.; Annam, D.; Moghaddam, S.; Lee, R.; Papa, R.; McClean, P. Investigation of the domestication of common bean (Phaseolus vulgaris) using multilocus sequence data. Funct. Plant. Biol. 2011, 38, 953–967. [Google Scholar] [CrossRef]
- Nanni, L.; Bitocchi, E.; Bellucci, E.; Rossi, M.; Rau, D.; Attene, G.; Gepts, P.; Papa, R. Nucleotide diversity of a genomic sequence similar to shatterproof (PvSHP1) in domesticated and wild common bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 2011, 123, 1341–1357. [Google Scholar] [CrossRef]
- Glémin, S.; Bataillon, T. A comparative view of the evolution of grasses under domestication. New Phytol. 2009, 183, 273–290. [Google Scholar] [CrossRef]
- Papa, R.; Acosta, J.; Delgado-Salinas, A.; Gepts, P. A genome-wide analysis of differentiation between wild and domesticated Phaseolus vulgaris from mesoamerica. Theor. Appl. Genet. 2005, 111, 1147–1158. [Google Scholar] [CrossRef]
- Papa, R.; Bellucci, E.; Rossi, M.; Leonardi, S.; Rau, D.; Gepts, P.; Nanni, L.; Gioia, T. Tagging the signatures of domestication in common bean (Phaseolus vulgaris) by means of pooled DNA samples. Annu. Bot. 2007, 100, 1039–1051. [Google Scholar] [CrossRef]
- Mamidi, S.; Rossi, M.; Moghaddam, S.M.; Annam, D.; Lee, R.; Papa, R.; McClean, P. Demographic factors shaped diversity in the two gene pools of wild common bean Phaseolus vulgaris L. Heredity 2013, 110, 267–276. [Google Scholar] [CrossRef]
- Zhaodong, H.; Dekang, L.; Ying, G.; Jisen, S.; Dolf, W.; Guangchuang, Y.; Jinhui, C. RIdeogram: Drawing SVG graphics to visualize and map genome-wide data on idiograms. Peer J. Comput. Sci. 2020, 6, e251. [Google Scholar] [CrossRef]
- Liljegren, S.J.; Roeder, A.H.K.; Kempin, S.A.; Gremski, K.; Østergaard, L.; Guimil, S.; Reyes, D.K.; Yanofsky, M.F. Control of fruit patterning in Arabidopsis by indehiscent. Cell 2004, 116, 843–853. [Google Scholar] [CrossRef]
- Gioia, T.; Logozzo, G.; Attene, G.; Bellucci, E.; Benedettelli, S.; Negri, V. Evidence for introduction bottleneck and extensive inter-gene pool (Mesoamerica x Andes) hybridization in the European common bean (Phaseolus vulgaris L.) germplasm. PLoS ONE 2013, 8, e75974. [Google Scholar] [CrossRef]
- Gepts, P. Origin and evolution of common bean: Past events and recent trends. Hortscience 1998, 33, 1124–1130. [Google Scholar] [CrossRef]
- Cortés, A.J.; Monserrate, F.A.; Ramírez-Villegas, J.; Madriñán, S.; Blair, M.W. Drought tolerance in wild plant populations: The case of common beans (Phaseolus vulgaris L.). PLoS ONE 2013, 8, e62898. [Google Scholar] [CrossRef]
- Porch, T.; Beaver, J.S.; Debouck, D.G.; Jackson, S.A.; Kelly, J.D.; Dempewolf, H. Use of wild relatives and closely related species to adapt common bean to climate change. Agronomy 2013, 3, 433–461. [Google Scholar] [CrossRef]
- Schoville, S.D.; Bonin, A.; François, O.; Lobreaux, S.; Melodelima, C.; Manel, S. Adaptive genetic variation on the landscape: Methods and cases. Annu. Rev. Ecol. Evolut. Syst. 2012, 43, 23–43. [Google Scholar] [CrossRef]
- Manel, S.; Joost, S.; Epperson, B.K.; Holderegger, R.; Storfer, A.; Rosenberg, M.S.; Scribner, K.T.; Bonin, A.; Fortin, M.J. Perspectives on the use of landscape genetics to detect genetic adaptive variation in the field. Mol. Ecol. 2010, 19, 3760–3772. [Google Scholar] [CrossRef]
- Anderson, K.; Gaston, K.J. Lightweight unmanned aerial vehicles will revolutionize spatial ecology. Front. Ecol. Environ. 2013, 11, 138–146. [Google Scholar] [CrossRef]
- Storfer, A.; Murphy, M.A.; Spear, S.F.; Holderegger, R.; Waits, L.P. Landscape genetics: Where are we now? Mol. Ecol. 2010, 19, 3496–3514. [Google Scholar] [CrossRef]
- Rodriguez, M.; Rau, D.; Bitocchi, E.; Bellucci, E.; Biagetti, E.; Carboni, A.; Gepts, P.; Nanni, L.; Papa, R.; Attene, G. Landscape genetics, adaptive diversity, and population structure in phaseolus vulgaris. New Phytol. 2016, 209, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Ariani, A.; Gepts, P. Signatures of environmental adaptation during range expansion of wild common bean (Phaseolus vulgaris). Biorxiv 2019. [Google Scholar] [CrossRef]
- Ortega-Amaro, M.A.; Rodríguez-Hernández, A.A.; Rodríguez-Kessler, M.; Hernández-Lucero, E.; Rosales-Mendoza, S.; Ibáñez-Salazar, A.; Delgado-Sánchez, P.; Jiménez-Bremont, J.F. Overexpression of atgrdp2, a novel glycine-rich domain protein, accelerates plant growth and improves stress tolerance. Front. Plant. Sci. 2015, 5, 782. [Google Scholar] [CrossRef] [PubMed]
- Broughton, W.J.; Hernández, G.; Blair, M.; Beebe, S.; Gepts, P.; Vanderleyden, J. Beans (Phaseolus spp.)—model food legumes. Plant Soil 2003, 252, 55–128. [Google Scholar] [CrossRef]
- Angioi, S.A.; Rau, D.; Attene, G.; Nanni, L.; Bellucci, E.; Logozzo, G.; Negri, V.; Spagnoletti Zeuli, P.L.; Papa, R. Beans in europe: Origin and structure of the european landraces of Phaseolus vulgaris L. Theor. Appl. Genet. 2010, 121, 829–843. [Google Scholar] [CrossRef]
- Santalla, M.; Rodiño, A.P.; De Ron, A.M. Allozyme evidence supporting southwester Europe as a secondary center of genetic diversity for common bean. Theor. Appl. Genet. 2002, 104, 934–944. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Phillips, P.C.; Cresko, W.A. Using population genomics to detect selection in natural populations: Key concepts and methodological considerations. Int. J. Plant Sci. 2010, 171, 1059–1071. [Google Scholar] [CrossRef]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Compar. Funct. Genom. 2001, 2, 155–168. [Google Scholar] [CrossRef]
- De Perez Souza, L.; Scossa, F.; Proost, S.; Bitocchi, E.; Papa, R.; Tohge, T.; Fernie, A.R. Multi-tissue integration of transcriptomic and specialized metabolite profiling provides tools for assessing the common bean (Phaseolus vulgaris) metabolome. Plant J. 2019, 97, 1132–1153. [Google Scholar] [CrossRef]
- Schwahn, K.; Perez de Souza, L.; Fernie, A.R.; Tohge, T. Metabolomics-assisted refinement of the pathways of steroidal glycoalkaloid biosynthesis in the tomato clade. J. Integr. Plant Biol. 2014, 56, 864–875. [Google Scholar] [CrossRef]
- Gerten, D.; Heck, V.; Jägermeyr, J.; Bodirsky, B.L.; Fetzer, I.; Jalava, M.; Kummu, M.; Wolfgang, L.; Rockström, J.; Schaphoff, S.; et al. Feeding ten billion people is possible within four terrestrial planetary boundaries. Nat. Sustain. 2020. [Google Scholar] [CrossRef]
- Exposito-Alonso, M.; Burbano, H.A.; Bossdorf, O.; Nielsen, R.; Weigel, D. Natural selection on the Arabidopsis thaliana genome in present and future climates. Nature 2019, 573, 126–129. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortinovis, G.; Frascarelli, G.; Di Vittori, V.; Papa, R. Current State and Perspectives in Population Genomics of the Common Bean. Plants 2020, 9, 330. https://doi.org/10.3390/plants9030330
Cortinovis G, Frascarelli G, Di Vittori V, Papa R. Current State and Perspectives in Population Genomics of the Common Bean. Plants. 2020; 9(3):330. https://doi.org/10.3390/plants9030330
Chicago/Turabian StyleCortinovis, Gaia, Giulia Frascarelli, Valerio Di Vittori, and Roberto Papa. 2020. "Current State and Perspectives in Population Genomics of the Common Bean" Plants 9, no. 3: 330. https://doi.org/10.3390/plants9030330
APA StyleCortinovis, G., Frascarelli, G., Di Vittori, V., & Papa, R. (2020). Current State and Perspectives in Population Genomics of the Common Bean. Plants, 9(3), 330. https://doi.org/10.3390/plants9030330