Computational Identification and Comparative Analysis of Conserved miRNAs and Their Putative Target Genes in the Juglans regia and J. microcarpa Genomes


Abstract
1. Introduction
2. Material and Methods
2.1. Sequences
2.2. Computational Identification of miRNAs
2.3. Prediction of miRNA Target Genes
2.4. Analysis of GO and KEGG Pathways
3. Results
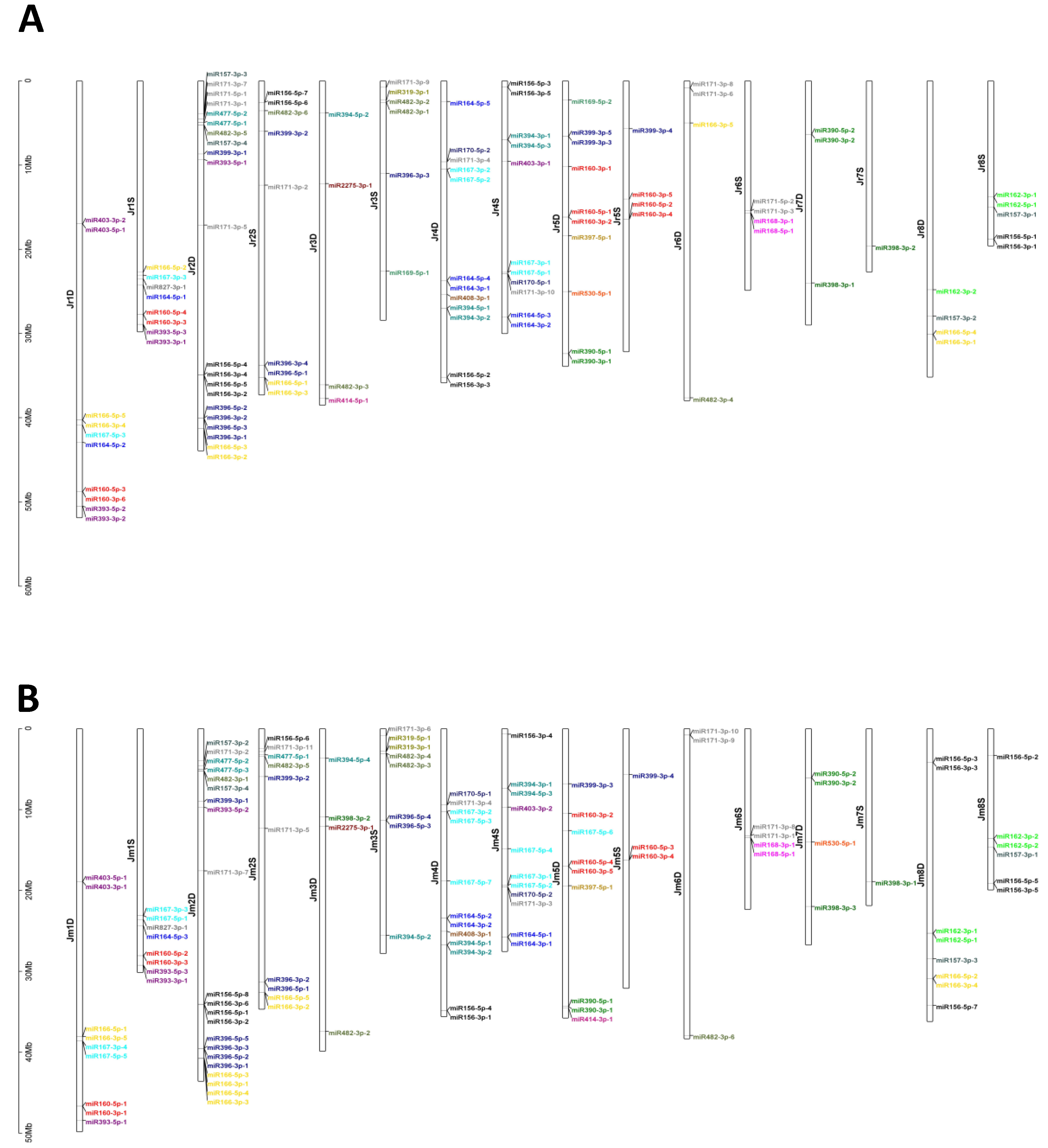
3.1. Computational Identification of miRNAs
3.2. Identification of Target Genes
3.3. Analysis of GO
3.4. KEGG Analysis
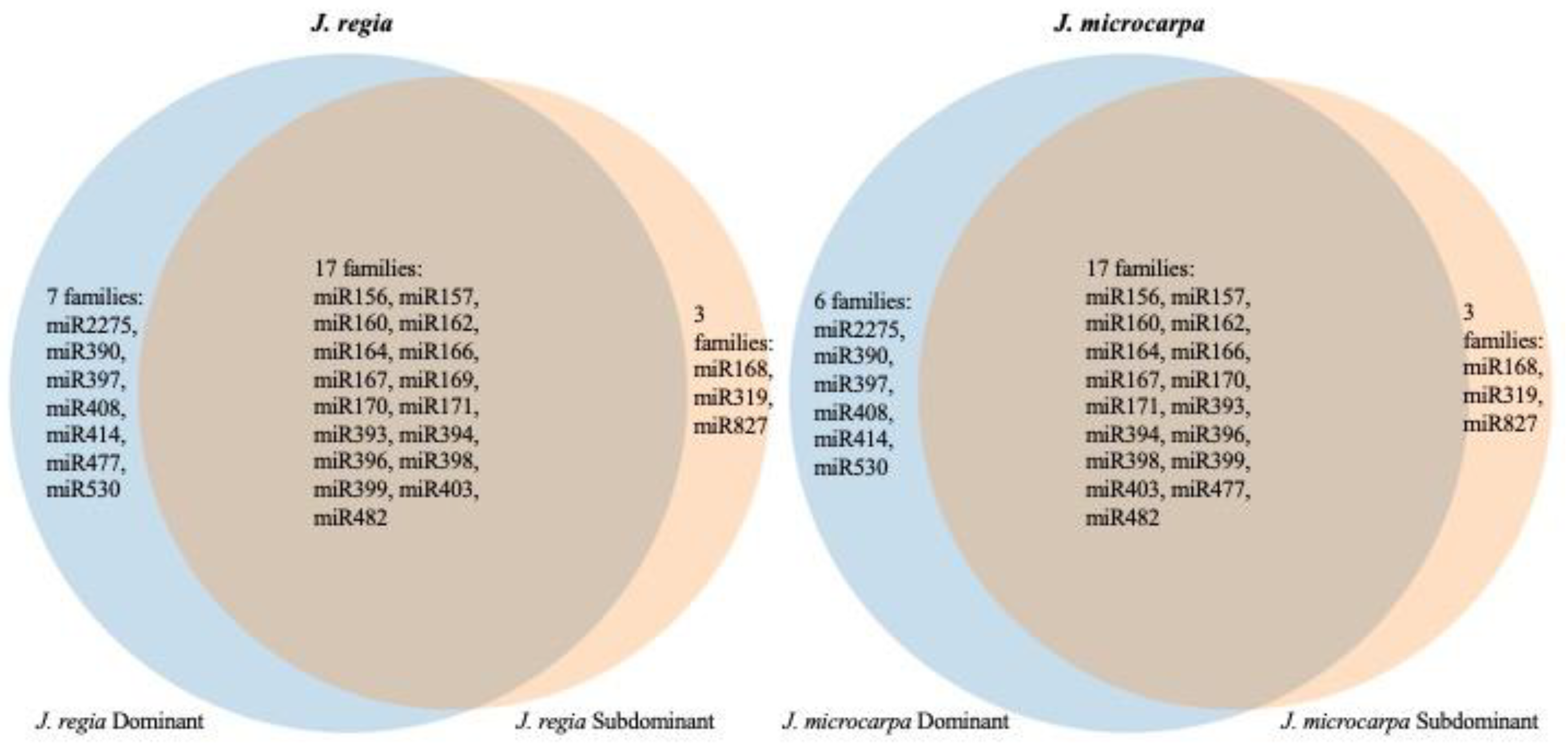
3.5. Comparative Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nat. Cell Biol. 2000, 403, 901–906. [Google Scholar] [CrossRef]
- Aukerman, M.J.; Sakai, H. Regulation of Flowering Time and Floral Organ Identity by a MicroRNA and Its APETALA2-Like Target Genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. bantam Encodes a Developmentally Regulated microRNA that Controls Cell Proliferation and Regulates the Proapoptotic Gene hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar] [CrossRef]
- Carrington, J.C. Role of MicroRNAs in Plant and Animal Development. Science 2003, 301, 336–338. [Google Scholar] [CrossRef]
- Xu, P.; Vernooy, S.Y.; Guo, M.; Hay, B.A. The Drosophila MicroRNA Mir-14 Suppresses Cell Death and Is Required for Normal Fat Metabolism. Curr. Biol. 2003, 13, 790–795. [Google Scholar] [CrossRef]
- Chen, X. A MicroRNA as a Translational Repressor of APETALA2 in Arabidopsis Flower Development. Science 2004, 303, 2022–2025. [Google Scholar] [CrossRef]
- Sunkar, R.; Zhu, J.-K. Novel and Stress-Regulated MicroRNAs and Other Small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Allen, E.; Fahlgren, N.; Calamar, A.; Givan, S.A.; Carrington, J.C. Expression of Arabidopsis MIRNA Genes. Plant Physiol. 2005, 138, 2145–2154. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nat. Cell Biol. 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- German, M.; Pillay, M.; Jeong, D.-H.; Hetawal, A.; Luo, S.; Janardhanan, P.; Kannan, V.; A Rymarquis, L.; Nobuta, K.; German, R.; et al. Global identification of microRNA–target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Djuranovic, S.; Nahvi, A.; Green, R. A Parsimonious Model for Gene Regulation by miRNAs. Science 2011, 331, 550–553. [Google Scholar] [CrossRef]
- Li, C.; Zhang, B. MicroRNAs in Control of Plant Development. J. Cell. Physiol. 2015, 231, 303–313. [Google Scholar] [CrossRef]
- D’Ario, M.; Griffiths-Jones, S.; Kim, M. Small RNAs: Big Impact on Plant Development. Trends Plant Sci. 2017, 22, 1056–1068. [Google Scholar] [CrossRef]
- Swarup, R. and Denyer, T. miRNAs in Plant Development. In Annual Plant Reviews Online; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2019; pp. 689–712. [Google Scholar]
- Llave, C.; Kasschau, K.D.; Rector, M.A.; Carrington, J.C. Endogenous and Silencing-Associated Small RNAs in Plants. Plant Cell 2002, 14, 1605–1619. [Google Scholar] [CrossRef]
- Guo, H.-S.; Xie, Q.; Fei, J.-F.; Chua, N.-H. MicroRNA Directs mRNA Cleavage of the Transcription Factor NAC1 to Downregulate Auxin Signals for Arabidopsis Lateral Root Development. Plant Cell 2005, 17, 1376–1386. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.P.; Wang, Q.L.; Cobb, G.P.; Anderson, T.A. Identification and characterization of new plant microRNAs using EST analysis. Cell Res. 2005, 15, 336–360. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.; Zhang, J.; Wu, L.; Qi, Y.; Zhou, J.-M. Identification of MicroRNAs Involved in Pathogen-Associated Molecular Pattern-Triggered Plant Innate Immunity. Plant Physiol. 2010, 152, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Shivaprasad, P.V.; Chen, H.-M.; Patel, K.; Bond, D.M.; Santos, B.; Baulcombe, D.C. A MicroRNA Superfamily Regulates Nucleotide Binding Site–Leucine-Rich Repeats and Other mRNAs. Plant Cell 2012, 24, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Kamthan, A.; Chaudhuri, A.; Kamthan, M.; Datta, A. Small RNAs in plants: Recent development and application for crop improvement. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.-N.; Zheng, H.-Q.; Wu, N.-Y.; Chien, C.-H.; Huang, H.-D.; Lee, T.-Y.; Chiang-Hsieh, Y.-F.; Hou, P.-F.; Yang, T.-Y.; Chang, W.-C. PlantPAN 2.0: An update of plant promoter analysis navigator for reconstructing transcriptional regulatory networks in plants. Nucleic Acids Res. 2016, 44, D1154–D1160. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.F.A.; Sajad, M.; Nazaruddin, N.; Fauzi, I.A.; Murad, A.M.A.; Zainat, Z.; Ismail, I. MicroRNA and transcription factor: Key players in plant regulatory network. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for Annotation of Plant MicroRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef]
- Axtell, M.J.; Meyers, B. Revisiting Criteria for Plant MicroRNA Annotation in the Era of Big Data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef]
- Alptekin, B.; Akpinar, B.A.; Budak, H. A Comprehensive Prescription for Plant miRNA Identification. Front. Plant Sci. 2017, 7. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. Mirbase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. Mirbase: From microRNA sequences to function. Nucleic Acids Res. 2018, 47, D155–D162. [Google Scholar] [CrossRef]
- Zhu, T.; Wang, L.; You, F.M.; Rodriguez, J.C.; Deal, K.R.; Chen, L.; Li, J.; Chakraborty, S.; Balan, B.; Jiang, C.-Z.; et al. Sequencing a Juglans regia × J. microcarpa hybrid yields high-quality genome assemblies of parental species. Hortic. Res. 2019, 6, 55. [Google Scholar] [CrossRef]
- Bernard, A.; Lheureux, F.; Dirlewanger, E. Walnut: Past and future of genetic improvement. Tree Genet. Genomes 2017, 14, 1. [Google Scholar] [CrossRef]
- Luo, M.-C.; You, F.M.; Li, P.; Wang, J.; Zhu, T.; Dandekar, A.M.; Leslie, C.A.; Aradhya, M.; McGuire, P.E.; Dvorak, J. Synteny analysis in Rosids with a walnut physical map reveals slow genome evolution in long-lived woody perennials. BMC Genom. 2015, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Martínez-García, P.J.; Crepeau, M.; Puiu, D.; Gonzalez-Ibeas, D.; Whalen, J.; Stevens, K.A.; Paul, R.; Butterfield, T.S.; Britton, M.T.; Reagan, R.L.; et al. The walnut (Juglans regia) genome sequence reveals diversity in genes coding for the biosynthesis of non-structural polyphenols. Plant J. 2016, 87, 507–532. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.-N.; Yan, P.-C.; Zhang, B.-W.; Woeste, K.E.; Lin, K.; Zhang, D. Demographically idiosyncratic responses to climate change and rapid Pleistocene diversification of the walnut genus Juglans (Juglandaceae) revealed by whole-genome sequences. New Phytol. 2017, 217, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.A.; Woeste, K.; Chakraborty, S.; Crepeau, M.; Leslie, C.A.; Martínez-García, P.J.; Puiu, D.; Romero-Severson, J.; Coggeshall, M.; Dandekar, A.M.; et al. Genomic Variation Among and Within SixJuglansSpecies. G3 Genes|Genomes|Genetics 2018, 8, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- miRbase. Available online: http://www.mirbase.org (accessed on 15 September 2020).
- psRNATarget. Available online: http://plantgrn.noble.org/psRNATarget (accessed on 15 September 2020).
- Dai, X.; Zhuang, Z.; Zhao, P.X. Psrnatarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genes 2015, 53, 474–485. [Google Scholar] [CrossRef]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell. Mol. Life Sci. 2006, 63, 246–254. [Google Scholar] [CrossRef]
- Zhu, H.; Zhou, Y.; Castillo-González, C.; Lu, A.; Ge, C.; Zhao, Y.-T.; Duan, L.; Li, Z.; Axtell, M.J.; Wang, X.-J.; et al. Bidirectional processing of pri-miRNAs with branched terminal loops by Arabidopsis Dicer-like1. Nat. Struct. Mol. Biol. 2013, 20, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.C.; Grativol, C.; Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. Computational identification and comparative analysis of miRNA precursors in three palm species. Planta 2016, 243, 1265–1277. [Google Scholar] [CrossRef]
- Singh, R.; Abdullah, M.O.; Low, E.-T.L.; Manaf, M.A.A.; Rosli, R.; Nookiah, R.; Ooi, L.C.-L.; Ooi, S.E.; Chan, K.-L.; Halim, M.A.; et al. Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nat. Cell Biol. 2013, 500, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Guo, Z.; Li, L. Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev. Biol. 2013, 380, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and Functional Diversification of MIRNA Genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef]
- Nozawa, M.; Miura, S.; Nei, M. Origins and Evolution of MicroRNA Genes in Plant Species. Genome Biol. Evol. 2012, 4, 230–239. [Google Scholar] [CrossRef]
- Rhoades, M.W.; Reinhart, B.J.; Lim, L.P.; Burge, C.B.; Bartel, B.; Bartel, D.P. Prediction of plant microRNA targets. Cell 2002, 110, 513–520. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| J. regia | J. microcarpa | |
|---|---|---|
| miRNA family | Number of pre-miRNAs | Number of pre-miRNAs |
| miR156 | 12 | 14 |
| miR157 | 4 | 4 |
| miR160 | 11 | 9 |
| miR162 | 3 | 4 |
| miR164 | 7 | 5 |
| miR166 | 10 | 10 |
| miR167 | 6 | 11 |
| miR168 | 2 | 2 |
| miR169 | 2 | 0 |
| miR170 | 2 | 2 |
| miR171 | 12 | 11 |
| miR2275 | 1 | 1 |
| miR319 | 1 | 2 |
| miR390 | 4 | 4 |
| miR393 | 5 | 4 |
| miR394 | 5 | 6 |
| miR396 | 7 | 8 |
| miR397 | 1 | 1 |
| miR398 | 2 | 3 |
| miR399 | 5 | 4 |
| miR403 | 3 | 3 |
| miR408 | 1 | 1 |
| miR414 | 1 | 1 |
| miR477 | 4 | 3 |
| miR482 | 6 | 6 |
| miR530 | 1 | 1 |
| miR827 | 1 | 1 |
| Total | 119 | 121 |
| Genome | Subgenome | Genes (No.) | Observed and (Expected) Target Genes Regulated by miRNA Located in D Subgenome (No.) | Observed and (Expected) Target Genes Regulated by miRNA Located in S Subgenome (No.) |
|---|---|---|---|---|
| J. regia | D | 18,179 | 202 (201.0) † | 121 (133.6) |
| S | 13,107 | 144 (145.0) | 109 (96.4) | |
| p = 1.00 ** | p = 0.368 | |||
| J. microcarpa | D | 17,093 | 226 (205.2) | 144 (144.2) |
| S | 12,304 | 127 (147.8) | 104 (103.8) | |
| p = 0.092 | p = 1.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhu, T.; Deal, K.R.; Dvorak, J.; Luo, M.-C. Computational Identification and Comparative Analysis of Conserved miRNAs and Their Putative Target Genes in the Juglans regia and J. microcarpa Genomes. Plants 2020, 9, 1330. https://doi.org/10.3390/plants9101330
Wang L, Zhu T, Deal KR, Dvorak J, Luo M-C. Computational Identification and Comparative Analysis of Conserved miRNAs and Their Putative Target Genes in the Juglans regia and J. microcarpa Genomes. Plants. 2020; 9(10):1330. https://doi.org/10.3390/plants9101330
Chicago/Turabian StyleWang, Le, Tingting Zhu, Karin R. Deal, Jan Dvorak, and Ming-Cheng Luo. 2020. "Computational Identification and Comparative Analysis of Conserved miRNAs and Their Putative Target Genes in the Juglans regia and J. microcarpa Genomes" Plants 9, no. 10: 1330. https://doi.org/10.3390/plants9101330
APA StyleWang, L., Zhu, T., Deal, K. R., Dvorak, J., & Luo, M.-C. (2020). Computational Identification and Comparative Analysis of Conserved miRNAs and Their Putative Target Genes in the Juglans regia and J. microcarpa Genomes. Plants, 9(10), 1330. https://doi.org/10.3390/plants9101330