Water Deficit Transcriptomic Responses Differ in the Invasive Tamarix chinensis and T. ramosissima Established in the Southern and Northern United States

 and
and
Abstract

1. Introduction
2. Results
2.1. Sequence Quality and de Novo Reference Transcriptome Assembly Show Good Gene Coverage
2.2. Tamarix de Novo Transcriptome Functional Annotation Indicated Strong Homology to Beta vulgaris
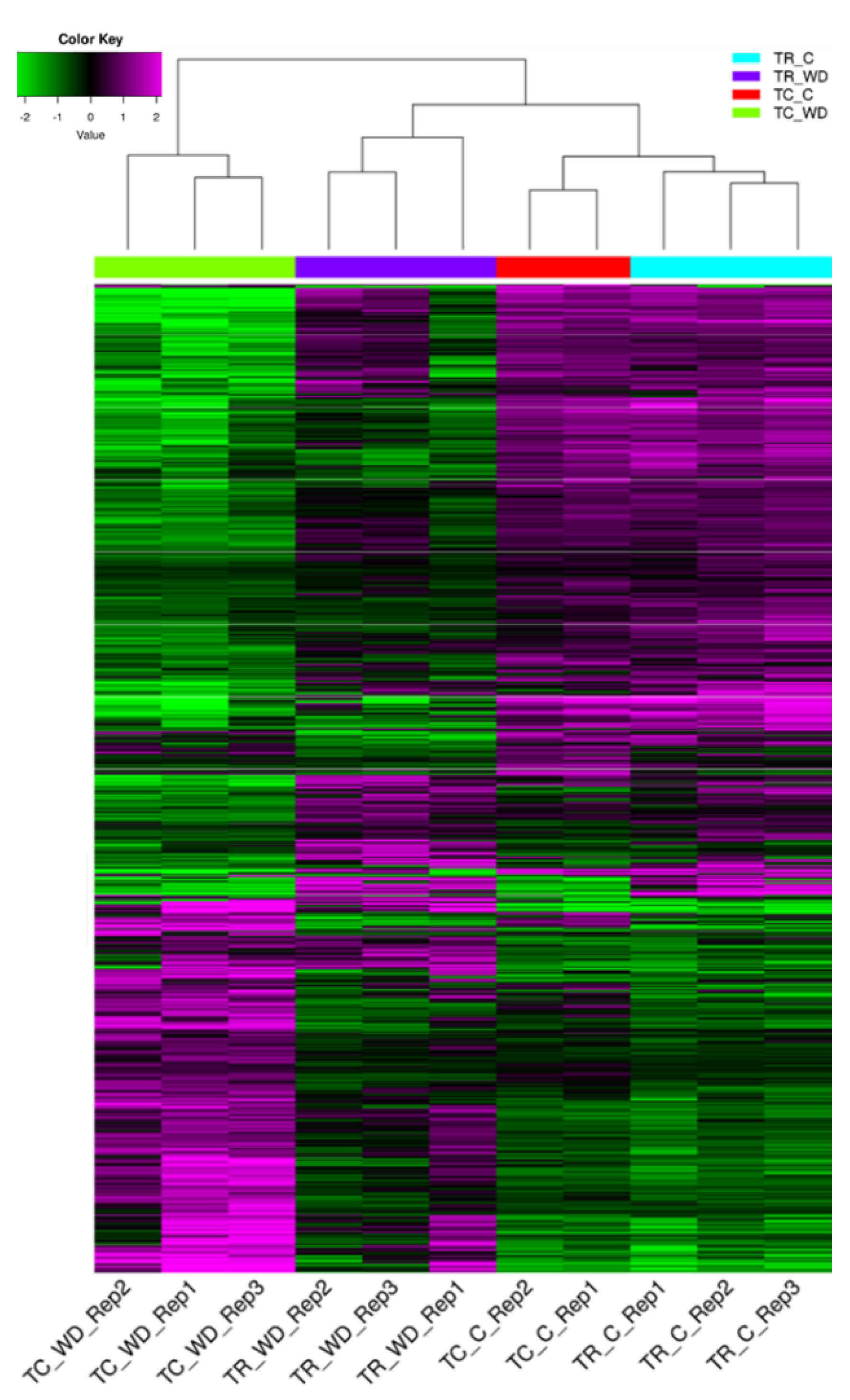
2.3. All Pairwise Comparisons of Species and Water Treatments Show Differential Gene Expression Response Diversity
2.4. Differential Gene Expression in Water Deficit Relative to Control Treatment Indicates a Greater Transcriptomic Response in T. chinensis Than in T. ramosissima
2.5. Gene Ontology Classification Identified Similar Functional Categories for T. chinensis and T. ramosissima
2.6. Tamarix Homologs with B. vulgaris and A. thaliana
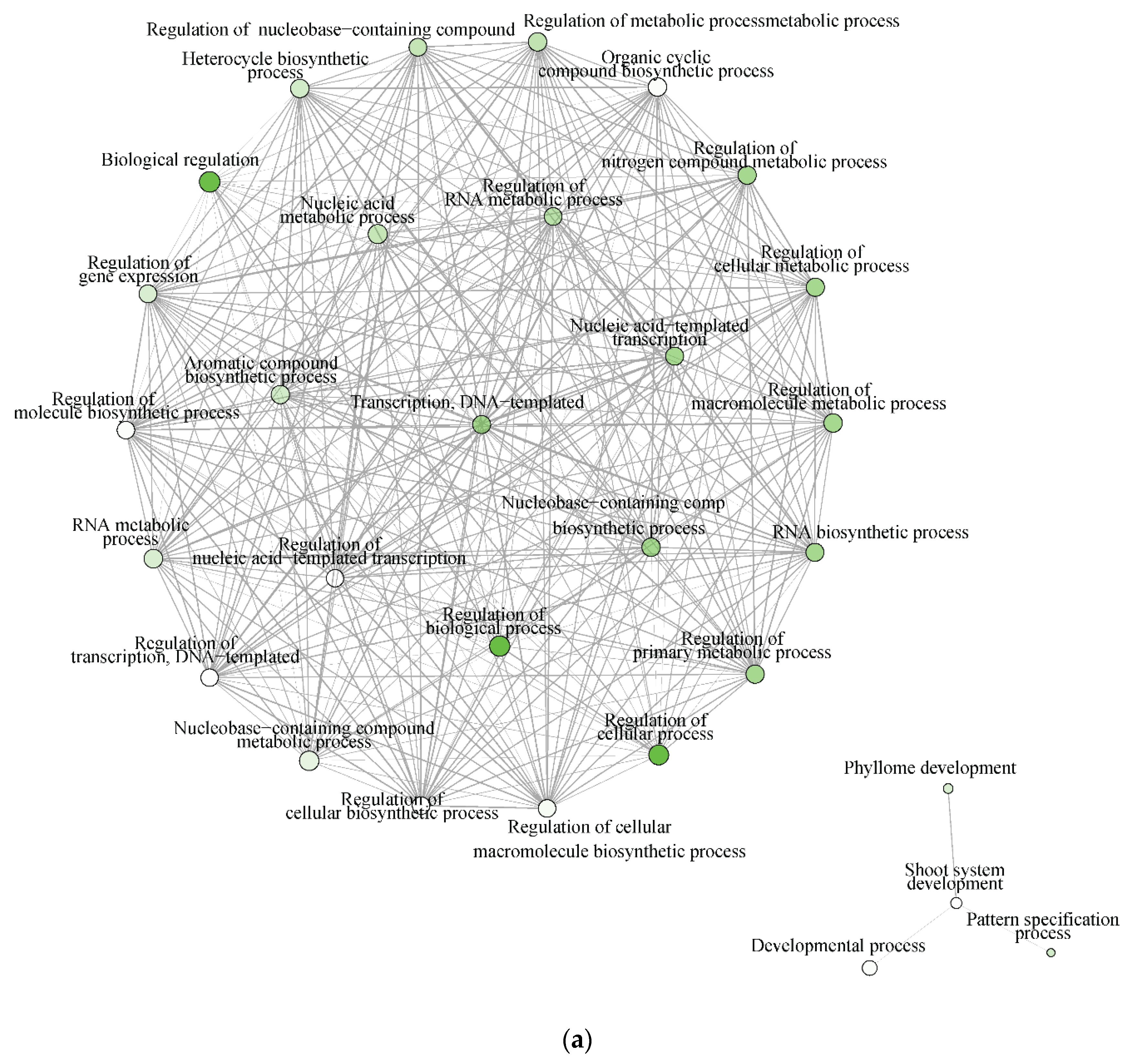
2.7. Biological Processes and Molecular Function Networks Indicated Distinct Differences in the Water Deficit Responses of T. chinensis and T. ramosissima
2.8. Unique Transcription Factors Are DEG in T. chinensis and T. ramosissima in Response to Water Deficit
3. Discussion
3.1. Water Deficit Leaf Reference Transcriptome Shows a Greater Number of Genes Than Salt Stressed Roots
3.2. Differential Gene Expression Differs in Response to Water Deficit in T. chinensis and T. ramosissima
4. Materials and Methods
4.1. Plant Materials
4.2. Genotype Analysis
4.3. Water Deficit Treatment
4.4. RNA Extraction
4.5. De Novo Reference Transcriptome Assembly and Assessment
4.6. Functional Gene Annotation
4.7. GO and KEGG Pathway Enrichment
4.8. Gene Expression Analysis
4.9. Pairwise Comparisons of All Species and Water Treatments
4.10. Differential Gene Expression in Water Deficit Relative to Control Treatment
4.11. Data Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Friedman, J.M.; Auble, G.T.; Shafroth, P.B.; Scott, M.L.; Merigliano, M.F.; Freehling, M.D.; Griffin, E.R. Dominance of non-native riparian trees in western USA. Biol. Invasions 2005, 7, 747–751. [Google Scholar] [CrossRef]
- Chew, M.K. Tamarisk introduction, naturalization, and control in the United States, 1818–1952. In Tamarix: A Case Study of Ecological Change in the American West; Sher, A., Quigley, M.F., Eds.; Oxford University Press: New York, NY, USA, 2013; pp. 269–286. [Google Scholar]
- Gaskin, J.; Kazmer, D. Introgression between invasive saltcedars (Tamarix chinensis and T. ramosissima) in the USA. Biol. Invasions 2009, 11, 1121–1130. [Google Scholar] [CrossRef]
- Gaskin, J.F.; Schaal, B.A. Hybrid Tamarix widespread in United States invasion and undetected in native Asian range. Proc. Natl. Acad. Sci. USA 2002, 99, 11256–11259. [Google Scholar] [CrossRef] [PubMed]
- Douglass, C.H.; Nissen, S.J.; Hart, C.R. Tamarisk management: Lessons and techniques. In Tamarix: A Case Study of Ecological Change in the American West; Sher, A., Quigley, M.F., Eds.; Oxford University Press: New York, NY, USA, 2013; pp. 333–353. [Google Scholar]
- O’Meara, S.; Larsen, D.; Owens, C. Methods to Control Saltcedar and Russian Olive. In Saltcedar and Russian Olive Control Demonstration Act Science Assessment; Shafroth, P.B., Brown, C.A., Merritt, D.M., Eds.; Scientific Investigations Report: Reston, VA, USA, 2010; pp. 2009–5247. [Google Scholar]
- CABI. Invasive Species Compendium. 2018. Available online: https://www.cabi.org/isc/datasheet/52503 (accessed on 1 September 2019).
- Broennimann, O.; Treier, U.A.; Muller-Sharer, H.; Thuiller, W.; Peterson, A.T.; Guisan, A. Evidence of climatic niche shift during biological invasion. Ecol. Lett. 2007, 10, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Garnatje, T.; Hidalgo, O.; Mas de Xaxars, G.; Pellicer, J.; Sanchez-Jimenez, I.; Vitales, D.; Valles, J. First genome size estimations for some eudicot families and genera. Collect. Bot. 2010, 29, 7–16. [Google Scholar] [CrossRef]
- Arianmanesh, R.; Mehregan, I.; Assadi, M.; Nejadsattari, T. Phylogenetic relationships of the Genus Tamarix L. (Tamaricaceae) from Iran based on nuclear and plastid DNA sequences. Asian Conserv. Biol. 2016, 5, 45–50. [Google Scholar]
- Lee, S.R.; Gaskin, J.F.; Kim, Y.D. Molecular diagnosis for a Tamarix species from two reclaimed lands along the Yellow Sea in Korea inferred from genome wide SNP markers. J. Syst. Evol. 2018, 57, 247–255. [Google Scholar] [CrossRef]
- Sun, L.; Yang, R.; Zhang, B.; Zhang, G.; Wu, X.; Zhang, W.; Zhang, B.; Chen, T.; Liu, G. Phylogenetic relationships among species of Tamarix (Tamaricaceae) in China. Biochem. Syst. Ecol. 2016, 69, 213–221. [Google Scholar] [CrossRef]
- Friedman, J.M.; Roelle, J.E.; Gaskin, J.F.; Pepper, A.E.; Manhart, J.R. Latitudinal variation in cold hardiness in introduced Tamarix and native Populus. Evol. Appl. 2008, 1, 598–607. [Google Scholar]
- Friedman, J.M.; Roelle, J.E.; Cade, B.S. Genetic and environmental influences on leaf phenology and cold hardiness of native and introduced riparian trees. Int. J. Biometeorol. 2011, 55, 775–787. [Google Scholar] [CrossRef]
- Lesica, P.; Miles, S. Tamarisk growth at the northern margin of its naturalized range in Montana, USA. Wetlands 2001, 21, 240–246. [Google Scholar] [CrossRef]
- Pearce, C.M.; Smith, D.G. Saltcedar: Distribution, abundance, and dispersal mechanisms, northern Montana, USA. Wetlands 2003, 23, 215–228. [Google Scholar] [CrossRef]
- Sexton, J.P.; McKay, J.K.; Sala, A. Plasticity and genetic diversity may allow saltcedar to invade cold climates in North America. Ecol. Appl. 2002, 12, 1652–1660. [Google Scholar] [CrossRef]
- Sexton, J.P.; Sala, A.; Murray, K. Occurrence, persistence, and expansion of saltcedar (Tamarix spp.) populations in the Great Plains of Montana. Western N. Am. Nat. 2006, 66, 23–44. [Google Scholar] [CrossRef]
- Bray, E. Molecular responses to water deficit. Plant Physiol. 1993, 103, 1035–1040. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K.; Seki, M. Regulatory network of gene expression in the drought and cold stress responses. Curr. Opin. Plant Biol. 2003, 6, 410–417. [Google Scholar] [CrossRef]
- Jarnevich, C.S.; Evangelista, P.; Stohlgren, T.J.; Morisette, J. Improving national-scale invasion maps: Tamarisk in the western United States. West. N. Am. Nat. 2011, 71, 164–175. [Google Scholar] [CrossRef]
- NOAA (National Oceanic and Atmospheric Association). Autumn and November Both Warmer Than Average in the United States. Available online: http://www.noaanews.noaa.gov/stories2011/20111207_novusstats.html (accessed on 4 April 2019).
- Pearce, C.M.; Smith, D.G. Invasive saltcedar (Tamarix): Its spread from the American Southwest to the Northern Great Plains. Phys. Geogr. 2007, 28, 507–530. [Google Scholar] [CrossRef]
- Morisette, J.; Jarnevich, T.C.S.; Ullah, A.; Cai, W.; Pedelty, J.A.; Gentle, J.E.; Stohlgren, T.J.; Schnase, J.L. A tamarisk habitat suitability map for the continental United States. Front. Ecol. Environ. 2006, 4, 11–17. [Google Scholar] [CrossRef]
- Bauer, J.T. Invasive species: “back-seat drivers” of ecosystem change? Biol. Invasions 2012, 14, 1295–1304. [Google Scholar] [CrossRef]
- Gao, W.; Bai, S.; Li, Q.; Gao, C.; Liu, G.; Li, G.; Tan, F. Overexpression of TaLEA gene from Tamarix androssowii improves salt and drought tolerance in transgenic poplar (Populus simonii × P. nigra). PLoS ONE 2013, 8, e67462. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhan, L.P.; Zou, X.Z. Improvement of cold tolerance of the half-high bush Northland blueberry by transformation with the LEA gene from Tamarix androssowii. Plant Growth Regul. 2011, 63, 13–22. [Google Scholar] [CrossRef]
- Gao, C.; Zhang, K.; Yang, G.; Wang, Y. Expression Analysis of Four Peroxiredoxin Genes from Tamarix hispidaIi in Response to Different Abiotic Stresses and Exogenous Abscisic Acid (ABA). Int. J. Mol. Sci. 2012, 13, 3751–3764. [Google Scholar] [CrossRef]
- Ji, X.; Nie, X.; Liu, Y.; Zheng, L.; Zhao, H.; Zhang, B.; Huo, L.; Wang, Y. A bHLH gene from Tamarix hispidaIi improves abiotic stress tolerance by enhancing osmotic potential and decreasing reactive oxygen species accumulation. Tree Physiol. 2016, 36, 193–207. [Google Scholar] [PubMed]
- Ji, X.; Wang, Y.; Liu, G. Expression Analysis of MYC genes from Tamarix hispidaIi in response to different abiotic stresses. Int. J. Mol. Sci. 2012, 13, 1300–1313. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gao, C.; Wang, L.; Zheng, L.; Yang, C.; Wang, Y. Comprehensive transcriptional profiling of NaHCO3-stressed Tamarix hispidaIi roots reveals networks of responsive genes. Plant Mol. Biol. 2014, 84, 145–157. [Google Scholar] [CrossRef]
- Wang, L.; Qin, L.; Liu, W.; Zhang, D.; Wang, Y. A novel ethylene-responsive factor from Tamarix hispida, ThERF1, is a GCC-box- and DRE-motif binding protein that negatively modulates abiotic stress tolerance in Arabidopsis. Physiol. Plant. 2014, 152, 84–97. [Google Scholar] [CrossRef]
- Wang, L.; Xu, C.; Wang, C.; Wang, Y. Characterization of a eukaryotic translation initiation factor 5A homolog from Tamarix androssowii involved in plant abiotic stress tolerance. BMC Plant Biol. 2012, 12, 118. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, C.; Liang, Y.; Wang, C.; Yang, C.; Liu, G. A Novel bZIP Gene from Tamarix hispida mediates physiological responses to salt stress in tobacco plants. J. Plant Physiol. 2010, 167, 222–230. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, J.; Zhao, X.; Liu, G.; Yang, C.; Zhan, L. A novel LEA gene from Tamarix androssowii confers drought tolerance in transgenic tobacco. Plant Sci. 2006, 171, 655–662. [Google Scholar] [CrossRef]
- Yang, G.; Yu, L.; Zhang, K.; Zhao, Y.; Guo, Y.; Gao, C. A ThDREB gene from Tamarix hispida improved the salt and drought tolerance of transgenic tobacco and T. hispida. Plant Physiol. Biochem. 2017, 113, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, M.; Gu, Y.; Xu, L.-A. Differentially expressed gene analysis of Tamarix chinensis provides insights into NaCl-stress response. Trees 2017, 31, 645–658. [Google Scholar] [CrossRef]
- Yan, X.; Qian, C.; Yin, X.; Fan, X.; Zhao, X.; Gu, M.; Wang, T.; Ma, X.-F. A whole-transcriptome approach to evaluate reference genes for quantitative diurnal gene expression studies under natural field conditions in Tamarix ramosissima leaves. Electr. J. Biotechnol. 2018, 35, 48–56. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Ohrtman, M.K.; Clay, S.A. Using a pervasive invader for Weed Science education. Weed Technol. 2013, 27, 395–400. [Google Scholar] [CrossRef]
- Cleverly, J.R.; Smith, S.D.; Sala, A.; Devitt, D.A. Invasive capacity of Tamarix ramosissima in a Mojave Desert floodplain the role of drought. Oecologia 1997, 111, 12–18. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Narumalani, S.; Mishra, D.R.; Merani, P.; Wilson, R.G. Predicting potential occurrence and spread of invasive plant species along the North Platte River, Nebraska. Invasive Plant Sci. Manag. 2008, 1, 359–367. [Google Scholar] [CrossRef]
- Kerns, B.K.; Naylor, B.J.; Buonopane, M.; Parks, C.G.; Rogers, B. Modeling Tamarisk (Tamarix spp.) habitat and climate change effects in the Northwestern United States. Invasive Plant Sci. Manag. 2009, 2, 200–215. [Google Scholar] [CrossRef]
- Li, J.; Yu, B.; Zhao, C.; Nowak, R.S.; Zhao, Z.; Sheng, Y.; Li, J. Physiological and morphological responses of Tamarix ramosissima and Populus euphratica to altered groundwater availability. Tree Physiol. 2013, 33, 57–68. [Google Scholar] [CrossRef]
- Dohm, J.C.; Minoche, A.E.; Holtgrawe, D.; Capell-Gutierrez, S.; Zakrzewski, F.; Tafer, H.; Rupp, O.; Rosleff, T.; Sorensen, T.R.; Stracke, R.; et al. The genome of the recently domesticated crop plant sugar beet (B. vulgaris). Nature 2014, 505, 546–549. [Google Scholar] [CrossRef]
- Xu, C.; Jiao, C.; Sun, H.; Cai, X.; Wang, X.; Ge, C.; Zheng, Y.; Liu, W.; Sun, X.; Su, Y.; et al. Draft genome of spinach and transcriptome diversity of 120 Spinacia accessions. Nat. Commun. 2017, 8, 15275. [Google Scholar] [CrossRef]
- Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny group classification for the orders and families of flowering plants: APG III. Bot. Gard. Linn. Soc. 2009, 161, 105–121. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Y.; Gao, C. The ethylene response factor (EWRF) genes from Tamarix hispidaIi respond to salt drought and ABA treatment. Trees 2014, 28, 317–327. [Google Scholar] [CrossRef]
- Wang, L.; Wang, C.; Qin, L.; Hu, P.; Wang, Y. ThERF1 from Tamarix hispida confers decreased tolerance to oxidative and drought stresses and is regulated by a WRKY protein. J. For. Res. 2016, 27, 767–772. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, Y.; Wang, Y.; Liu, Z.; Gao, C. Comprehensive analysis of MYB gene family and their expressions under abiotic stresses and hormone treatments in Tamarix hispida. Front. Plant Sci. 2018, 9, 1303. [Google Scholar] [CrossRef]
- Zheng, L.; Liu, G.; Meng, X.; Liu, Y.; Ji, X.; Li, Y.; Nie, X.; Wang, Y.A. WRKY gene from Tamarix hispidaIi, ThWRKY4, mediates abiotic stress responses by modulating reactive oxygen species and expression of stress-responsive genes. Plant Mol. Biol. 2011, 82, 303–320. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, S.; Huang, H.; Jiang, J.; Bai, S.; Liu, G. Improved salt tolerance of Populus davidiana x P. bolleana overexpressed LEA from T. androssowii. J. For. Res. 2014, 25, 813–818. [Google Scholar]
- Yang, Y.; Liao, W.; Yu, X.; Wang, B.; Peng, M.; Ruan, M. Overexpression of MeDREB1D confers tolerance to both drought and cold stresses in transgenic Arabidopsis. Acta Physiol. Plant. 2016, 38, 243. [Google Scholar] [CrossRef]
- Khadka, V.S.; Vaughn, K.; Xie, J.; Swaminathan, P.; Ma, Q.; Cramer, G.R.; Fennell, A.Y. Transcriptomic response is more sensitive to water deficit in shoots than roots of Vitis riparia (Michx.). BMC Plant Biol. 2019, 19, 72. [Google Scholar] [CrossRef]
- Baum, B.R. The Genus Tamarix; Israel Academy of Sciences and Humanities: Jerusalem, Israel, 1978. [Google Scholar]
- Gaskin, J.F.; Kazmer, D.J. Comparison of ornamental and wild saltcedar (Tamarix spp.) along Eastern Montana, USA riverways using chloroplast and nuclear DNA sequence markers. Wetlands 2006, 26, 939–950. [Google Scholar] [CrossRef]
- FastQC. 2018. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 January 2019).
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Chan, J.; Kishore, R.; Lee, R.; Muller, H.M.; Raciti, D.; Van Auken, K.; Sternberg, P.W. Expansion of the gene ontology knowledge base and resources. Nucleic Acids Res. 2017, 45, 331–338. [Google Scholar]
- The Gene Ontology Consortium. The Gene Ontology Resource. Available online: http://geneontology.org/ (accessed on 29 October 2019).
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, 1040–1045. [Google Scholar] [CrossRef]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, 1182–1187. [Google Scholar] [CrossRef]
- Araport11. Available online: https://www.arabidopsis.org/download/index-auto.jsp?dir=/download_files/Sequences (accessed on 4 April 2019).
- Vulgaris, B. Available online: http://plants.ensembl.org/B._vulgaris/Info/Index (accessed on 4 April 2019).
- Vinifera, V. Available online: http://genomes.cribi.unipd.it/grape (accessed on 4 April 2019).
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Ge, X.S.; Jung, D.; Yao, R. ShinyGo: A graphical enrichment tool for animals and plants. Bioinform. 2019, btz931. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, L.S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinform. 2010, 32, 11.7.1–11.7.14. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.S.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO a web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, 293–297. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, 46, 71–75. [Google Scholar] [CrossRef]
- Grimplet, J.; Cramer, G.; Dickerson, J.; Van Hemert, J.; Mathiason, K.; Fennell, A. VitisNet: Omics Integration through Grapevine Molecular Networks. PLoS ONE 2009, 4, e8365. [Google Scholar] [CrossRef]
- Grimplet, J.; VanHemert, J.; Carbonell-Bejerano, P.; Diaz-Riquelme, J.; Dickerson, J.; Fennell, A.; Pezzotti, M.; Martinez–Zapater, J.M. Comparative analysis of grapevine whole—Genome gene predictions, functional annotation, categorization and integration of the predicted gene sequences. BMC Res. Notes 2012, 5, 213–222. [Google Scholar] [CrossRef]
- Osier, M.V. VitisPathways: Gene pathway analysis for V. vinifera. Vitis 2016, 55, 129–133. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Number |
|---|---|
| Total genes according to Trinity Assembly | 117,867 |
| Total Trinity transcripts | 271,872 |
| Percent Guanine-Cytosine | 40.9 |
| Contig N10 | 4150 |
| Contig N20 | 3234 |
| Contig N30 | 2659 |
| Contig N40 | 2215 |
| Contig N50 | 1824 |
| Median contig length (bp) | 622 |
| Average contig length (bp) | 1058 |
| Total assembled bases | 287,833,633 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swaminathan, P.; Ohrtman, M.; Carinder, A.; Deuja, A.; Wang, C.; Gaskin, J.; Fennell, A.; Clay, S. Water Deficit Transcriptomic Responses Differ in the Invasive Tamarix chinensis and T. ramosissima Established in the Southern and Northern United States. Plants 2020, 9, 86. https://doi.org/10.3390/plants9010086
Swaminathan P, Ohrtman M, Carinder A, Deuja A, Wang C, Gaskin J, Fennell A, Clay S. Water Deficit Transcriptomic Responses Differ in the Invasive Tamarix chinensis and T. ramosissima Established in the Southern and Northern United States. Plants. 2020; 9(1):86. https://doi.org/10.3390/plants9010086
Chicago/Turabian StyleSwaminathan, Padmapriya, Michelle Ohrtman, Abigail Carinder, Anup Deuja, Cankun Wang, John Gaskin, Anne Fennell, and Sharon Clay. 2020. "Water Deficit Transcriptomic Responses Differ in the Invasive Tamarix chinensis and T. ramosissima Established in the Southern and Northern United States" Plants 9, no. 1: 86. https://doi.org/10.3390/plants9010086
APA StyleSwaminathan, P., Ohrtman, M., Carinder, A., Deuja, A., Wang, C., Gaskin, J., Fennell, A., & Clay, S. (2020). Water Deficit Transcriptomic Responses Differ in the Invasive Tamarix chinensis and T. ramosissima Established in the Southern and Northern United States. Plants, 9(1), 86. https://doi.org/10.3390/plants9010086