Infections of the Xylella fastidiosa subsp. pauca Strain “De Donno” in Alfalfa (Medicago sativa) Elicits an Overactive Immune Response


Abstract
1. Introduction
2. Results
2.1. X. fastidiosa “De Donno” Elicits a Necrotic Response in Alfalfa
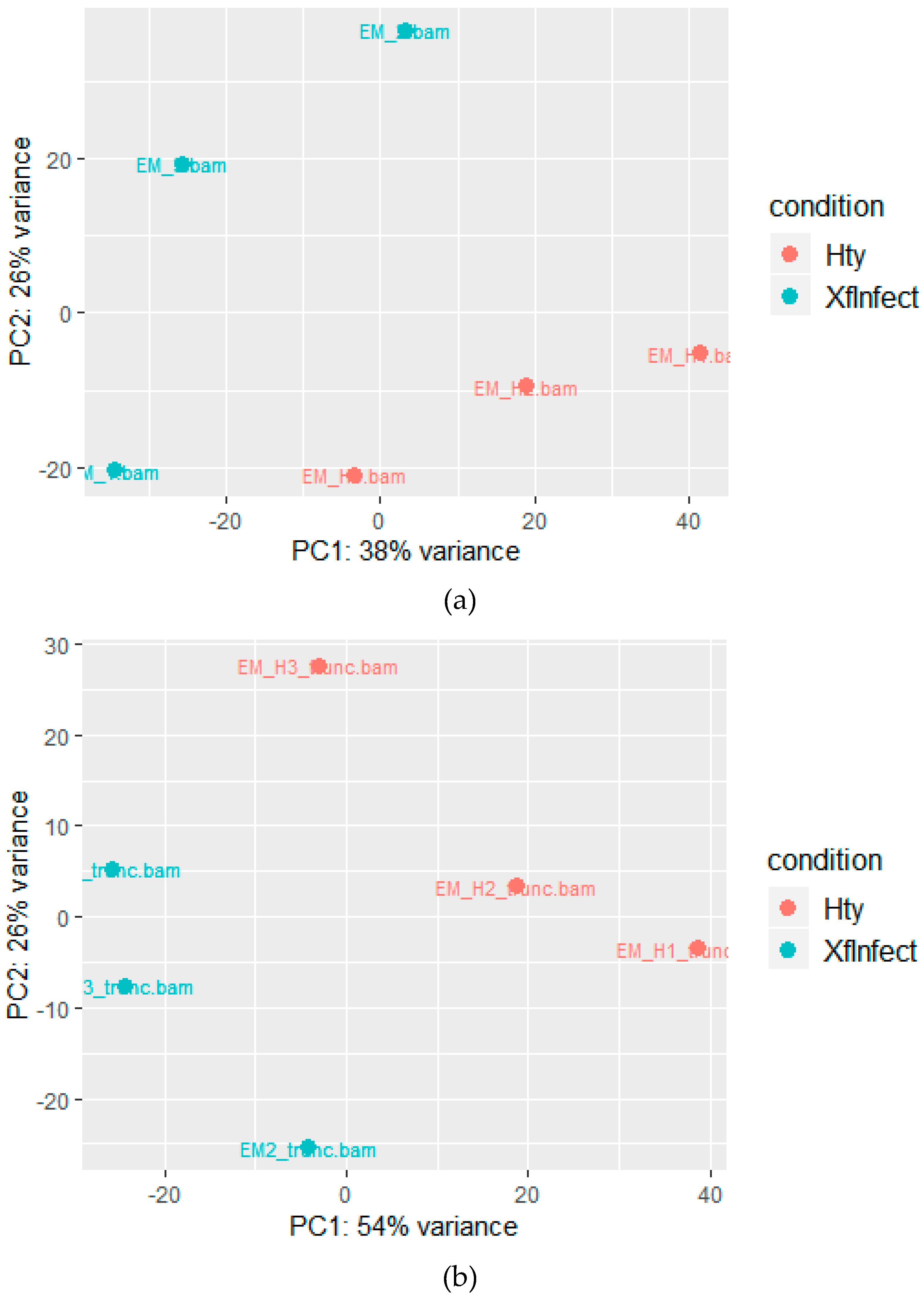
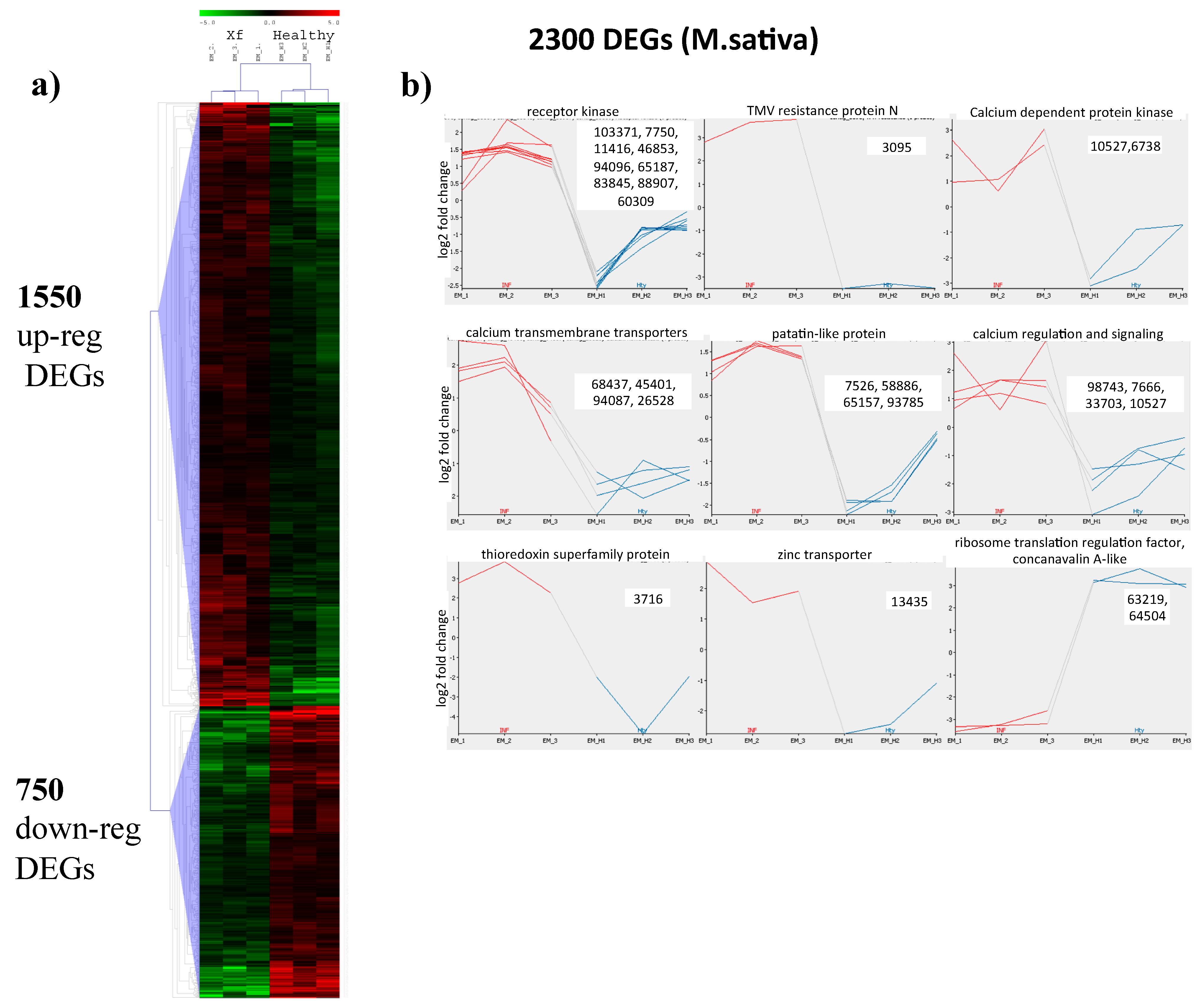
2.2. Mapping to the M. sativa Transcriptome
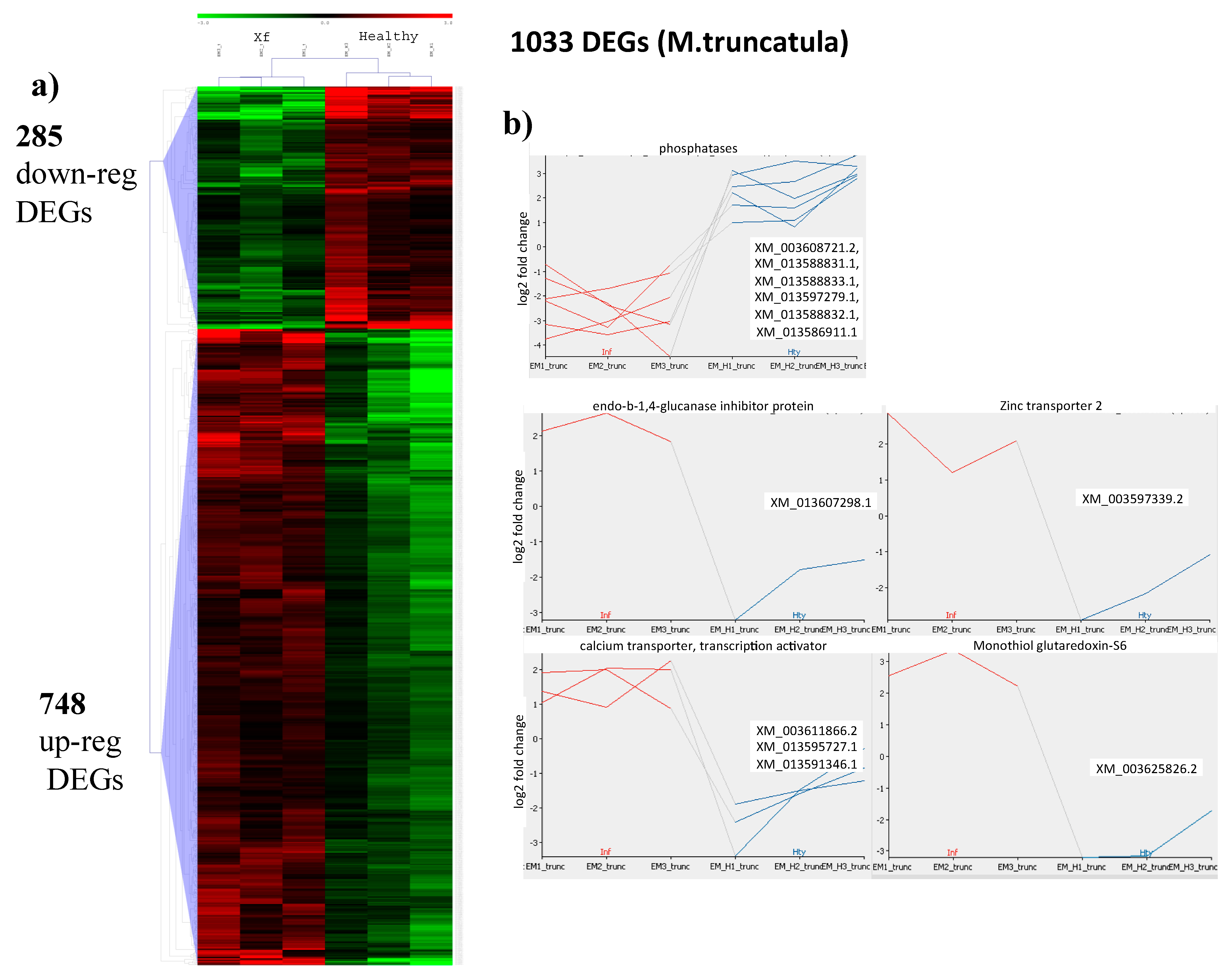
2.3. Mapping to the M. truncatula Genome
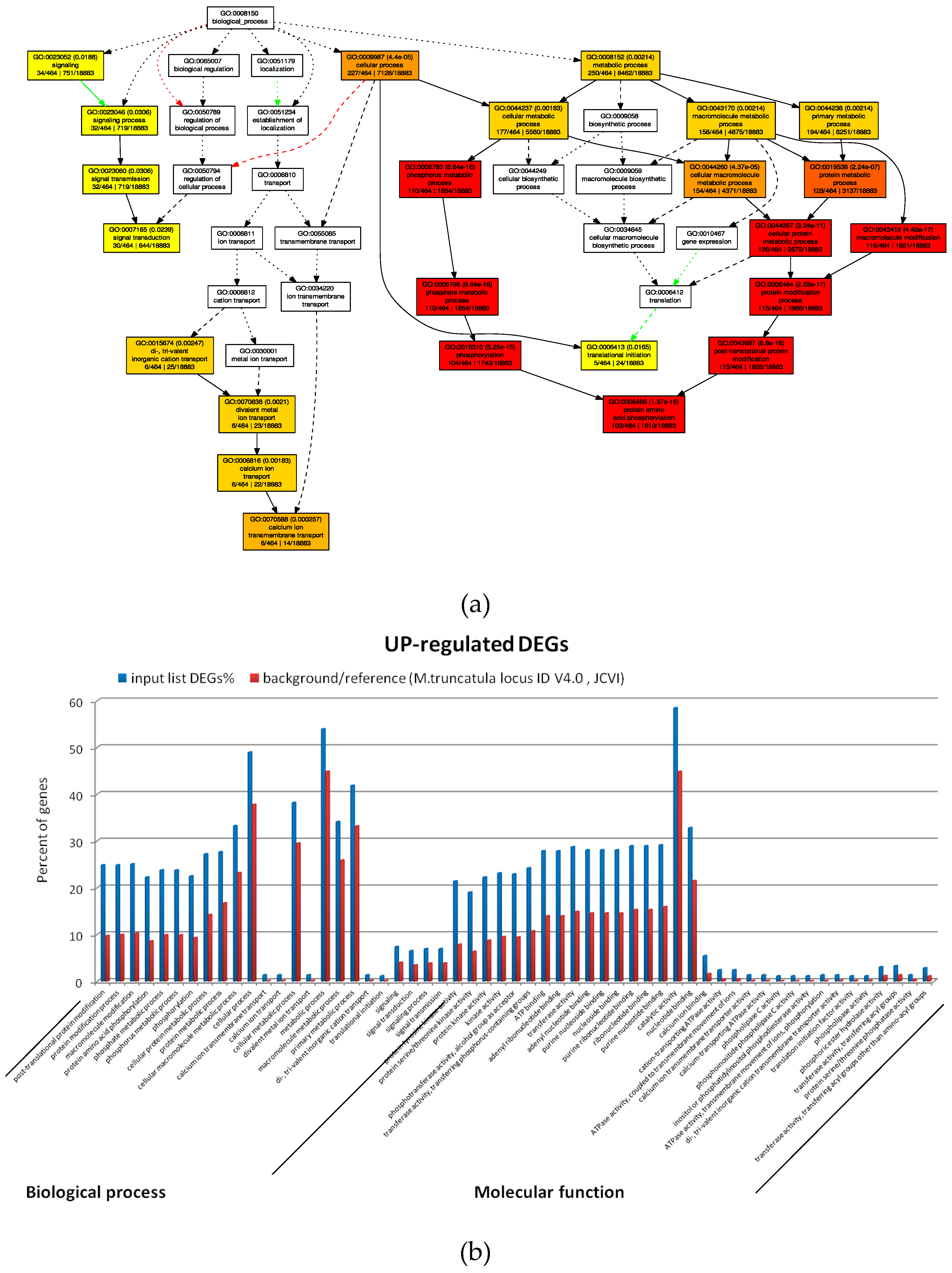
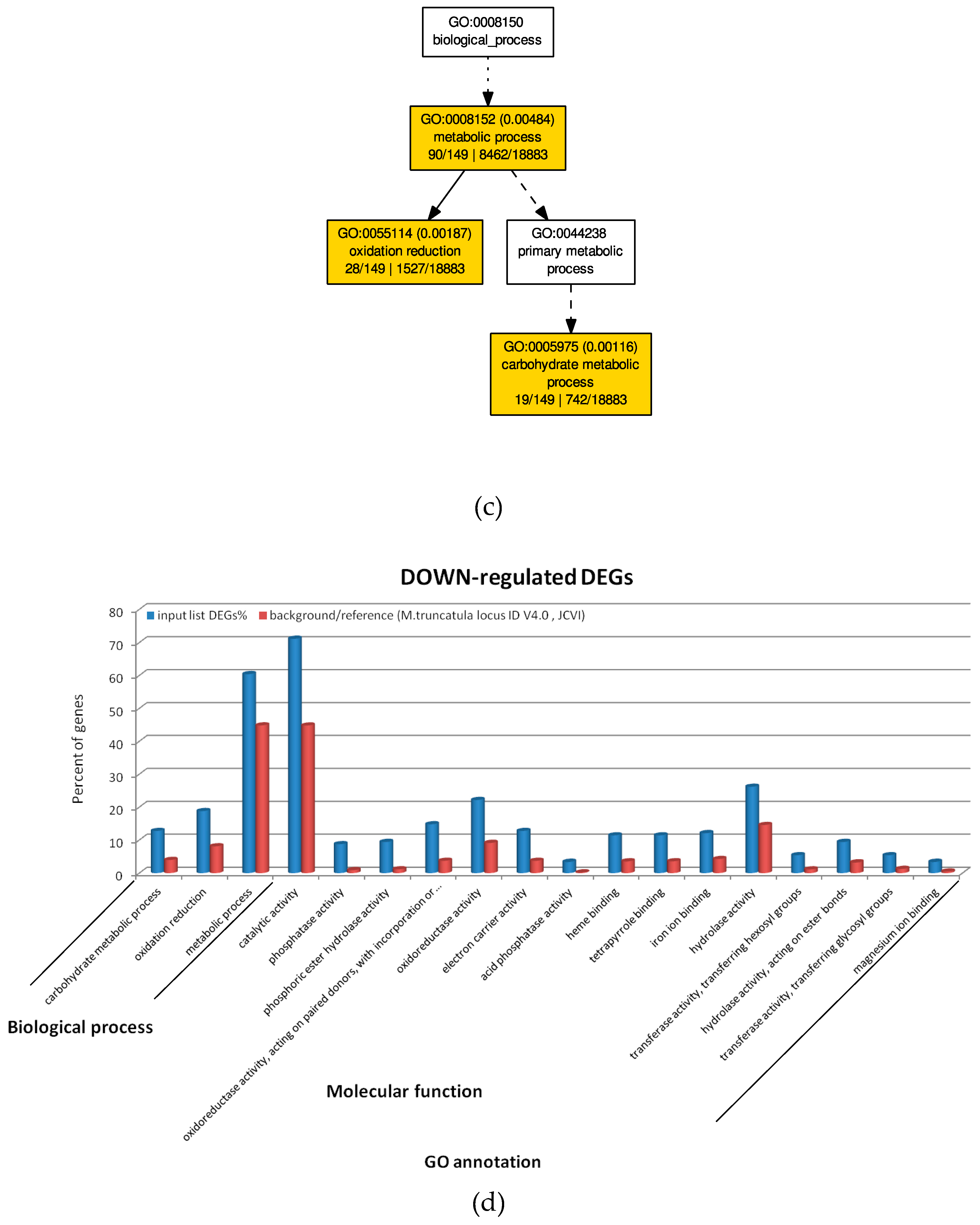
2.4. GO Analysis of M. truncatula by SEA and PAGE Analysis
3. Discussion
4. Materials and Methods
4.1. Test Plants and Bacteria Inoculations
4.2. Diagnostic Tests for Xylella fastidiosa
4.3. Purification of Total RNAs for High Throughput Sequencing
4.4. Analysis of Differential Gene Expression, Pathway Involvement, and Gene Ontology
4.5. Data Records
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jeger, M.; Caffier, D.; Candresse, T.; Chatzivassiliou, E.; Dehnen-Schmutz, K.; Gilioli, G.; Gregoire, J.C.; Miret, J.A.J.; MacLeod, A.; Navajas Navarro, M.; et al. Updated pest categorisation of Xylella fastidiosa. EFSA J. 2018, 16, 5357. [Google Scholar]
- Sicard, A.; Zeilinger, A.R.; Vanhove, M.; Schartel, T.E.; Beal, D.J.; Daugherty, M.P.; Almeida, R.P.P. Xylella fastidiosa: Insights into an emerging plant pathogen. Annu. Rev. Phytopathol. 2018, 56, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Rapicavoli, J.; Ingel, B.; Blanco-Ulate, B.; Cantu, D.; Roper, C. Xylella fastidiosa: An examination of a re-emerging plant pathogen. Mol. Plant. Pathol. 2018, 19, 786–800. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-K.; Iandolino, A.; da Silva, F.G.; Cook, D.R. Water deficit modulates the response of Vitis vinifera to the Pierce’s disease pathogen Xylella fastidiosa. Mol. Plant Microbe. Interact. 2013, 26, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, R.; Gouran, H.; Chakraborty, S.; Gillespie, H.W.; Almeida-Souza, H.O.; Tu, A.; Rao, B.J.; Feldstein, P.A.; Bruening, G.; Goulart, L.R.; et al. The type II secreted lipase/esterase LesA is a key virulence factor required for Xylella fastidiosa pathogenesis in grapevines. Sci Rep. 2016, 6, 18598. [Google Scholar] [CrossRef] [PubMed]
- Gouran, H.; Gillespie, H.; Nascimento, R.; Chakraborty, S.; Zaini, P.A.; Jacobson, A.; Phinney, B.S.; Dolan, D.; Durbin-Johnson, B.P.; Antonova, E.S.; et al. The secreted protease PrtA controls cell growth, biofilm formation and pathogenicity in Xylella fastidiosa. Sci. Rep. 2016, 6, 31098. [Google Scholar] [CrossRef] [PubMed]
- Rapicavoli, J.N.; Blanco-Ulate, B.; Muszynski, A.; Figueroa-Balderas, R.; Morales-Cruz, A.; Azadi, P.; Dobruchowska, J.M.; Castro, C.; Cantu, D.; Roper, M.C. Lipopolysaccharide O-antigen delays plant innate immune recognition of Xylella fastidiosa. Nat. Commun. 2018, 9, 390. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; de Souza, A.A.; Takita, M.A.; Kishi, L.T.; Machado, M.A. RNA-Seq analysis of Citrus reticulata in the early stages of Xylella fastidiosa infection reveals auxin-related genes as a defense response. BMC Genom. 2013, 14, 676. [Google Scholar] [CrossRef]
- Giampetruzzi, A.; Morelli, M.; Saponari, M.; Loconsole, G.; Chiumenti, M.; Boscia, D.; Savino, V.N.; Martelli, G.P.; Saldarelli, P. Transcriptome profiling of two olive cultivars in response to infection by the CoDiRO strain of Xylella fastidiosa subsp. pauca. BMC Genom. 2016, 17, 475. [Google Scholar] [CrossRef]
- Saponari, M.; Giampetruzzi, A.; Loconsole, G.; Boscia, D.; Saldarelli, P. Xylella fastidiosa in olive in apulia: Where we stand. Phytopathology 2019, 109, 175–186. [Google Scholar] [CrossRef]
- Saponari, M.; Boscia, D.; Altamura, G.; Loconsole, G.; Zicca, S.; D’Attoma, G.; Morelli, M.; Palmisano, F.; Saponari, A.; Tavano, D.; et al. Isolation and pathogenicity of Xylella fastidiosa associated to the olive quick decline syndrome in southern Italy. Sci Rep. 2017, 7, 17723. [Google Scholar] [CrossRef] [PubMed]
- Giampetruzzi, A.; Saponari, M.; Almeida, R.P.P.; Essakhi, S.; Boscia, D.; Loconsole, G.; Saldarelli, P. Complete Genome Sequence of the Olive-Infecting strain Xylella fastidiosa subsp. pauca De Donno. Genome Announc. 2017, 5, e00569-17. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, W.B.; Houston, B.R. Association of pierce’s disease and alfalfa dwarf in California. Plant Dis. Rep. 1941, 25, 475–476. [Google Scholar]
- O’Rourke, J.A.; Fu, F.; Bucciarelli, B.; Yang, S.S.; Samac, D.A.; Lamb, J.F.S.; Monteros, M.J.; Graham, M.A.; Gronwald, J.W.; Krom, N.; et al. The Medicago sativa gene index 1.2: A web-accessible gene expression atlas for investigating expression differences between Medicago sativa subspecies. BMC Genom. 2015, 16, 502. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Krishnakumar, V.; Bidwell, S.; Rosen, B.; Chan, A.; Zhou, S.; Gentzbittel, L.; Childs, K.L.; Yandell, M.; Gundlach, H.; et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genom. 2014, 15, 312. [Google Scholar] [CrossRef] [PubMed]
- Dinesh-Kumar, S.P.; Tham, W.H.; Baker, B.J. Structure-function analysis of the tobacco mosaic virus resistance gene N. Proc. Natl. Acad. Sci. USA 2000, 97, 14789–14794. [Google Scholar] [CrossRef] [PubMed]
- Mata-Perez, C.; Spoel, S.H. Thioredoxin-mediated redox signalling in plant immunity. Plant Sci. 2019, 279, 27–33. [Google Scholar] [CrossRef]
- De La Fuente, L.; Parker, J.K.; Oliver, J.E.; Granger, S.; Brannen, P.M.; van Santen, E.; Cobine, P.A. The bacterial pathogen Xylella fastidiosa affects the leaf ionome of plant hosts during infection. PLoS ONE 2013, 8, e62945. [Google Scholar] [CrossRef]
- Wang, Y.; Bouwmeester, K.; Beseh, P.; Shan, W.; Govers, F. Phenotypic analyses of Arabidopsis T-DNA insertion lines and expression profiling reveal that multiple L-type lectin receptor kinases are involved in plant immunity. Mol. Plant Microbe. Interact. 2014, 27, 1390–1402. [Google Scholar] [CrossRef]
- Lim, C.W.; Yang, S.H.; Shin, K.H.; Lee, S.C.; Kim, S.H. The AtLRK10L1.2, Arabidopsis ortholog of wheat LRK10, is involved in ABA- mediated signaling and drought resistance. Plant Cell Rep. 2015, 34, 447–455. [Google Scholar] [CrossRef]
- La Camera, S.; Geoffroy, P.; Samaha, H.; Ndiaye, A.; Rahim, G.; Legrand, M.; Heitz, T. A pathogen-inducible patatin-like lipid acyl hydrolase facilitates fungal and bacterial host colonization in Arabidopsis. Plant J. 2005, 44, 810–825. [Google Scholar] [CrossRef]
- Rock, C.; Sukumaran, S.; De La Fuente, L.; Tricoli, D.; Azad, F.; Mamadou, T. Genome editing of TAS4, MIR828 and targets MYBA6/A7: A critical test of Xylella fastidiosa infection and spreading mechanisms in pierce’s disease. Interim Progress Report for CDFA Agreement Number 15-0214SA. 2017. Available online: https://pdfs.semanticscholar.org/c959/50af6093c777158d41a026f842d7a883fc96.pdf?_ga=2.164770813.313564194.1561446455-1572584237.1561446455 (accessed on 29 July 2019).
- Zhao, H.; Sun, R.; Albrecht, U.; Padmanabhan, C.; Wang, A.; Coffey, M.D.; Girke, T.; Wang, Z.; Close, T.J.; Roose, M.; et al. Small RNA profiling reveals phosphorus deficiency as a contributing factor in symptom expression for citrus huanglongbing disease. Mol. Plant 2013, 6, 301–310. [Google Scholar] [CrossRef]
- Edel, K.H.; Marchadier, E.; Brownlee, C.; Kudla, J.; Hetherington, A.M. The evolution of calcium-based signalling in plants. Curr. Biol. 2017, 27, R667–R679. [Google Scholar] [CrossRef]
- Couto, D.; Zipfel, C. Regulation of pattern recognition receptor signalling in plants. Nat. Rev. Immunol 2016, 16, 537–552. [Google Scholar] [CrossRef]
- Cheng, D.W.; Lin, H.; Takahashi, Y.; Walker, M.A.; Civerolo, E.L.; Stenger, D.C. Transcriptional regulation of the grape cytochrome P450 monooxygenase gene CYP736B expression in response to Xylella fastidiosa infection. BMC Plant Biol. 2010, 10, 135. [Google Scholar] [CrossRef]
- Li, S. Redox Modulation Matters: Emerging Functions for Glutaredoxins in Plant Development and Stress Responses. Plants (Basel) 2014, 3, 559–582. [Google Scholar] [CrossRef]
- Qin, Q.; Bergmann, C.W.; Rose, J.K.C.; Saladie, M.; Kolli, V.S.K.; Albersheim, P.; Darvill, A.G.; York, W.S. Characterization of a tomato protein that inhibits a xyloglucan-specific endoglucanase. Plant J. 2003, 34, 327–338. [Google Scholar] [CrossRef]
- Grotz, N.; Fox, T.; Connolly, E.; Park, W.; Guerinot, M.L.; Eide, D. Identification of a family of zinc transporter genes from Arabidopsis that respond to zinc deficiency. Proc. Natl. Acad. Sci. USA 1998, 95, 7220–7224. [Google Scholar] [CrossRef]
- Silva-Stenico, M.E.; Pacheco, F.T.H.; Pereira-Filho, E.R.; Rodrigues, J.L.M.; Souza, A.N.; Etchegaray, A.; Gomes, J.E.; Tsai, S.M. Nutritional deficiency in citrus with symptoms of citrus variegated chlorosis disease. Braz. J. Biol. 2009, 69, 859–864. [Google Scholar] [CrossRef]
- Malavolta, E.; Prates, N.S. Alteracoes na composicao mineral das folhas de pomares citricos afetados pela anomalia “amarelinho” ou clorose variegada. Laranja 1991, 12, 315–329. [Google Scholar]
- Chen, C.-C.; Chien, W.-F.; Lin, N.-C.; Yeh, K.-C. Alternative functions of Arabidopsis yellow stripe-like3: From metal translocation to pathogen defense. PLoS ONE 2014, 9, e98008. [Google Scholar] [CrossRef]
- Cornara, D.; Cavalieri, V.; Dongiovanni, C.; Altamura, G.; Palmisano, F.; Bosco, D.; Porcelli, F.; Almeida, R.P.P.; Saponari, M. Transmission of Xylella fastidiosa by naturally infected Philaenus spumarius (Hemiptera, Aphrophoridae) to different host plants. J. Appl. Entomol. 2017, 141, 80–87. [Google Scholar] [CrossRef]
- Lopes, J.R.S.; Daugherty, M.P.; Almeida, R.P.P. Strain origin drives virulence and persistence of Xylella fastidiosa in alfalfa. Plant Pathol. 2010, 59, 963–971. [Google Scholar] [CrossRef]
- Ingel, B.; Jeske, D.R.; Sun, Q.; Grosskopf, J.; Roper, C. Xylella fastidiosa endoglucanases mediate the rate of pierce’s disease development in Vitis vinifera in a cultivar-dependent manner. Mol. Plan. Microbe. Interact. 2019. [Google Scholar] [CrossRef]
- Harper, S.J.; Ward, L.I.; Clover, G.R.G. Development of LAMP and real-time PCR methods for the rapid detection of Xylella fastidiosa for quarantine and field applications. Phytopathology 2010, 100, 1282–1288. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Thimm, O.; Blasing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Kruger, P.; Selbig, J.; Muller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Experiment—Inoculation Date: 30/03/2018 | ||||||
|---|---|---|---|---|---|---|
| 3 Months Post Inoculation | 6 Months Post Inoculation * | |||||
| N. inoculated plants | Inoculated shoot n. positive plants (average Cq values) | Non inoculated shoot n. positive plants (average Cq values) | Symptoms | Non inoculated shoot n. positive plants | Roots: n positive plants | Symptoms |
| 13 | 6 (29,27) | 0/13 | Leaf scorching | 0 | 0 | 3 plants showed desiccated inoculation shoots (Figure 1) |
| Second Experiment—Inoculation Date: 21/08/2018 | ||||||
| 14 | 7 (21.26) | 0 | Initial shoot apex dieback on 6 plants | 7 (26,66) | 4 ** | 8 plants showed desiccation of the inoculation |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abou Kubaa, R.; Giampetruzzi, A.; Altamura, G.; Saponari, M.; Saldarelli, P. Infections of the Xylella fastidiosa subsp. pauca Strain “De Donno” in Alfalfa (Medicago sativa) Elicits an Overactive Immune Response. Plants 2019, 8, 335. https://doi.org/10.3390/plants8090335
Abou Kubaa R, Giampetruzzi A, Altamura G, Saponari M, Saldarelli P. Infections of the Xylella fastidiosa subsp. pauca Strain “De Donno” in Alfalfa (Medicago sativa) Elicits an Overactive Immune Response. Plants. 2019; 8(9):335. https://doi.org/10.3390/plants8090335
Chicago/Turabian StyleAbou Kubaa, Raied, Annalisa Giampetruzzi, Giuseppe Altamura, Maria Saponari, and Pasquale Saldarelli. 2019. "Infections of the Xylella fastidiosa subsp. pauca Strain “De Donno” in Alfalfa (Medicago sativa) Elicits an Overactive Immune Response" Plants 8, no. 9: 335. https://doi.org/10.3390/plants8090335
APA StyleAbou Kubaa, R., Giampetruzzi, A., Altamura, G., Saponari, M., & Saldarelli, P. (2019). Infections of the Xylella fastidiosa subsp. pauca Strain “De Donno” in Alfalfa (Medicago sativa) Elicits an Overactive Immune Response. Plants, 8(9), 335. https://doi.org/10.3390/plants8090335