Identification of Markers Associated with Yield Traits and Morphological Features in Maize (Zea mays L.)


 ,
,  ,
, 
Abstract
1. Introduction
2. Results
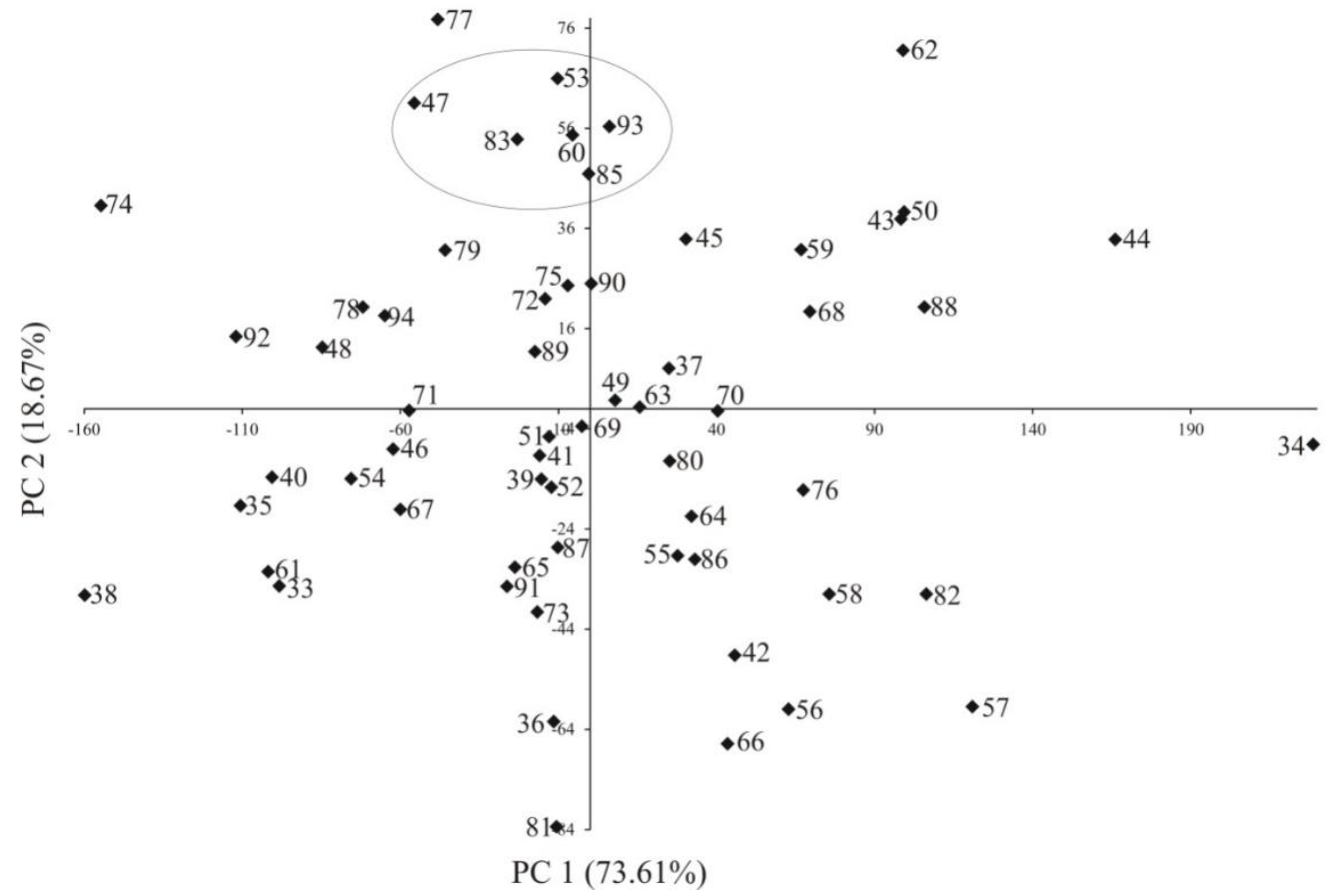
2.1. Phenotyping
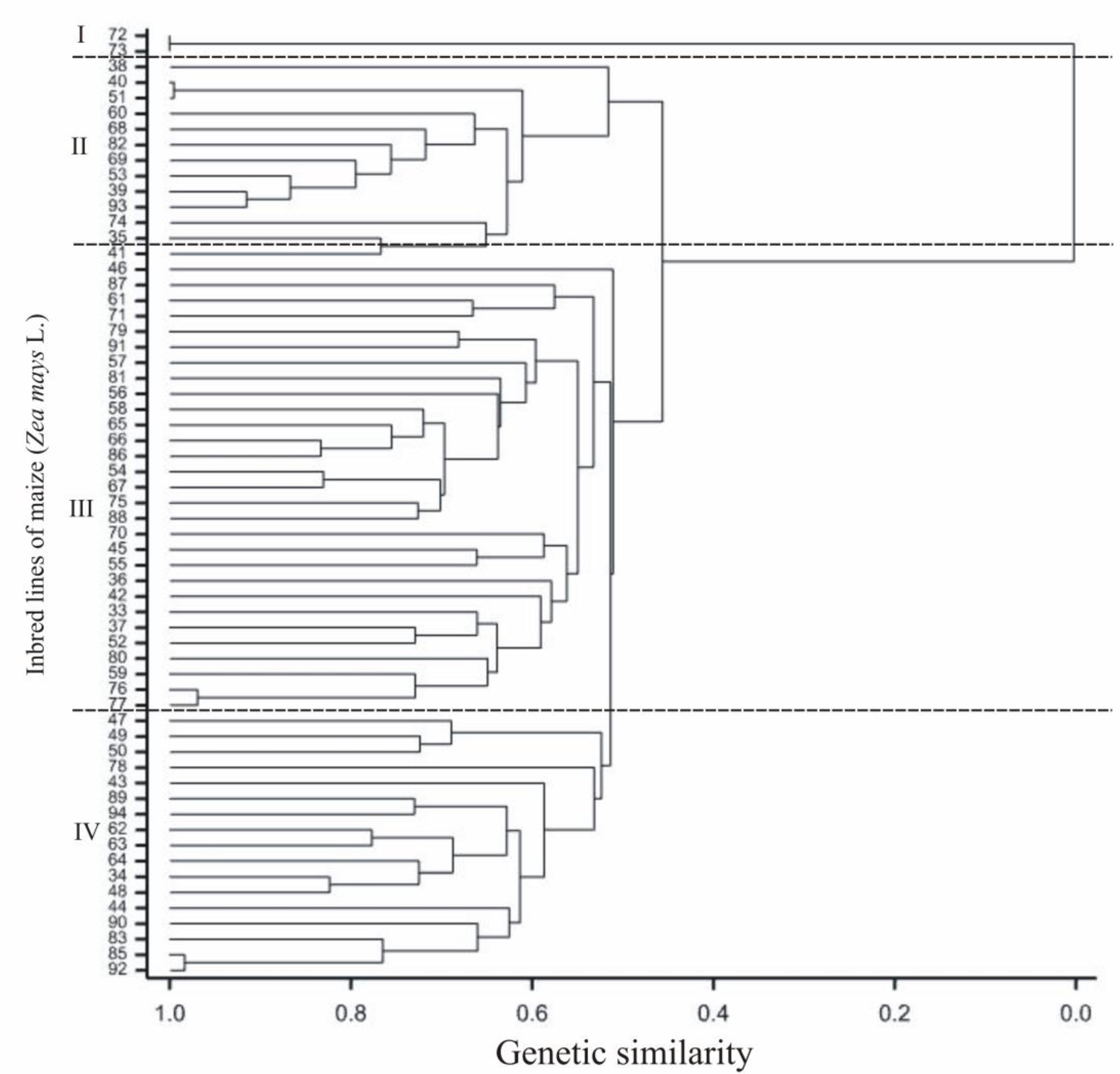
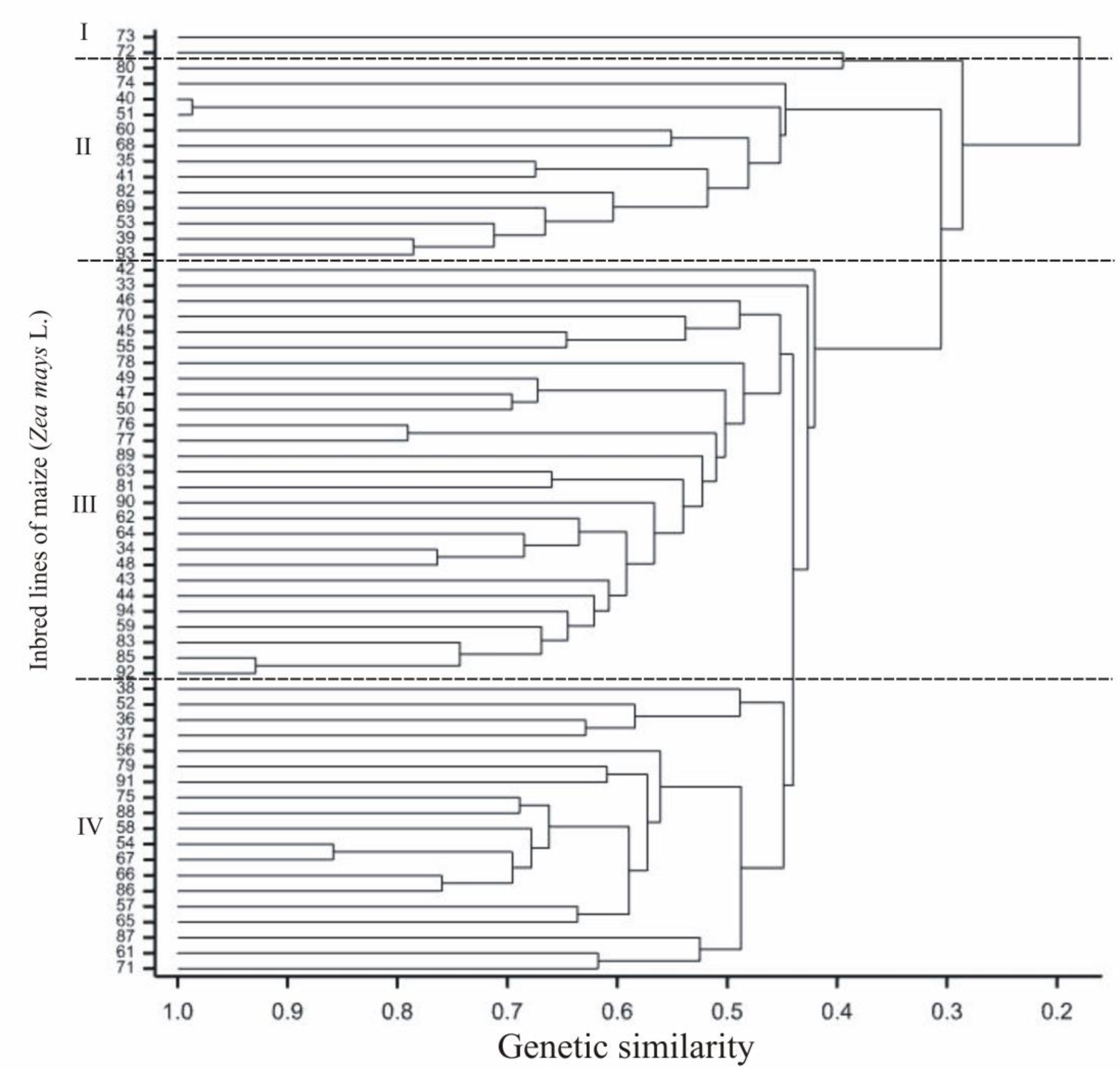
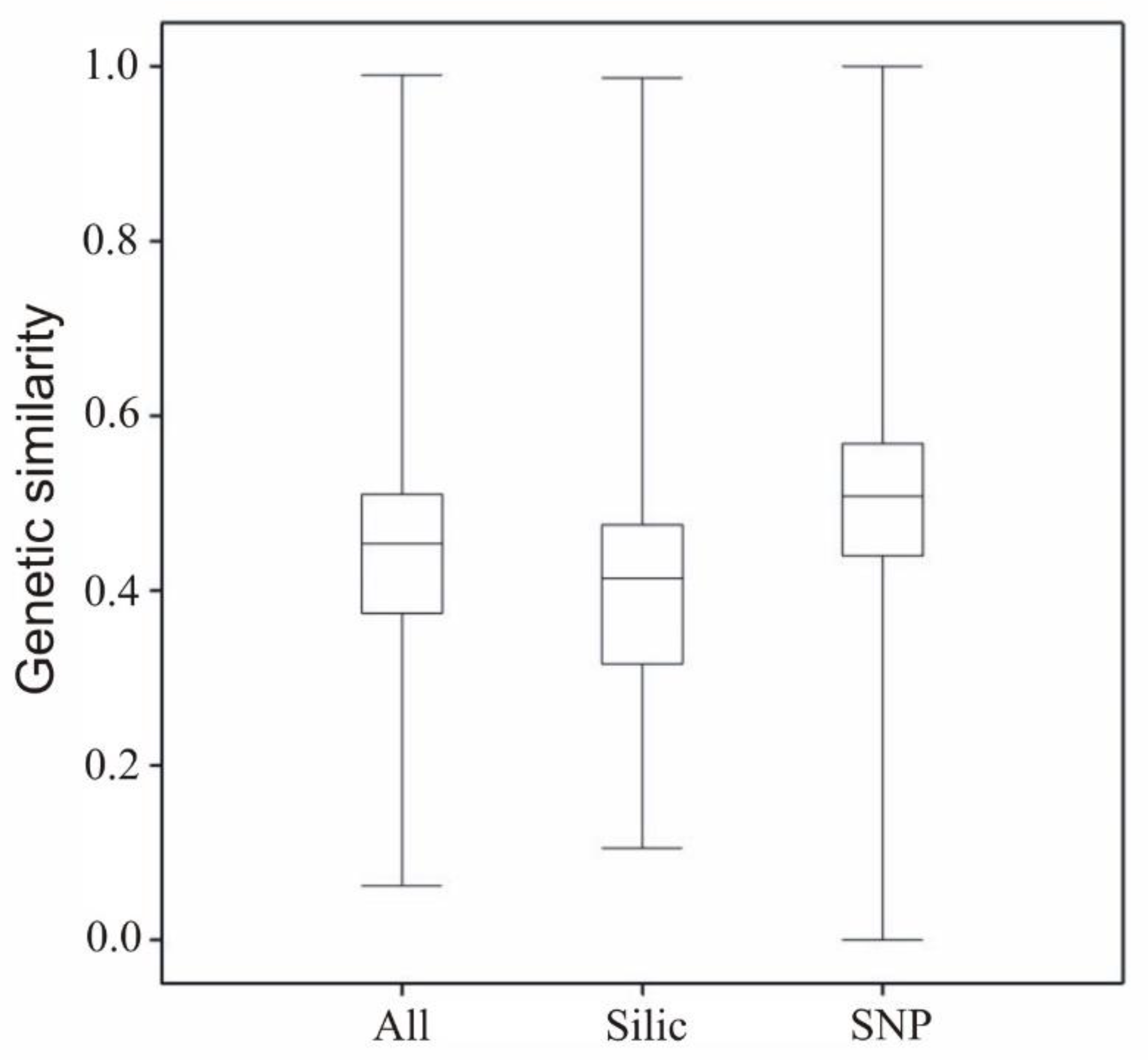
2.2. Genotyping Data (SilicoDArT and SNP)
2.3. Association Mapping
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotyping
4.3. Genotyping and SilicoDArT and SNP Data Processing
4.4. Statistical Analysis and Association Mapping
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schrag, T.A.; Möhring, J.; Melchinger, A.E.; Kusterer, B.; Dhillon, B.S.; Piepho, H.-P.; Frisch, M. Prediction of hybrid performance in maize using molecular markers and joint analyses of hybrids and parental inbreds. Theor. Appl. Genet. 2010, 120, 451–461. [Google Scholar] [CrossRef]
- Liersch, A.; Bocianowski, J.; Woś, H.; Szała, L.; Sosnowska, K.; Cegielska-Taras, T.; Nowosad, K.; Bartkowiak-Broda, I. Assessment of Genetic Relationships in Breeding Lines and Cultivars of and Their Implications for Breeding Winter Oilseed Rape. Crop Sci. 2016, 56, 1540. [Google Scholar] [CrossRef]
- Sosnowska, K.; Cegielska-Taras, T.; Liersch, A.; Karłowski, W.M.; Bocianowski, J.; Szała, L.; Mikołajczyk, K.; Popławska, W. Genetic relationships among resynthesized, semi-resynthesized and natural Brassica napus L. genotypes. Euphytica 2017, 213, 212. [Google Scholar] [CrossRef]
- Szała, L.; Sosnowska, K.; Popławska, W.; Liersch, A.; Olejnik, A.; Kozłowska, K.; Bocianowski, J.; Cegielska-Taras, T. Development of new restorer lines for CMS ogura system with the use of resynthesized oilseed rape (Brassica napus L.). Breed. Sci. 2016, 66, 516–521. [Google Scholar] [CrossRef]
- Gacek, K.; Bayer, P.E.; Bartkowiak-Broda, I.; Szala, L.; Bocianowski, J.; Edwards, D.; Batley, J. Genome-Wide Association Study of Genetic Control of Seed Fatty Acid Biosynthesis in Brassica napus. Front. Plant Sci. 2017, 7. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Andolfatto, P.; Davison, D.; Erezyilmaz, D.; Hu, T.T.; Mast, J.; Sunayama-Morita, T.; Stern, D.L. Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res. 2011, 21, 610–617. [Google Scholar] [CrossRef]
- Li, X.; Zhu, C.; Yeh, C.-T.; Wu, W.; Takacs, E.M.; Petsch, K.A.; Tian, F.; Bai, G.; Buckler, E.S.; Muehlbauer, G.J.; et al. Genic and nongenic contributions to natural variation of quantitative traits in maize. Genome Res. 2012, 22, 2436–2444. [Google Scholar] [CrossRef]
- Courtois, B.; Audebert, A.; Dardou, A.; Roques, S.; Ghneim-Herrera, T.; Droc, G.; Frouin, J.; Rouan, L.; Gozé, E.; Kilian, A.; et al. Genome-Wide Association Mapping of Root Traits in a Japonica Rice Panel. PLoS ONE 2013, 8, e78037. [Google Scholar] [CrossRef]
- Cruz, V.M.V.; Kilian, A.; Dierig, D.A. Development of DArT Marker Platforms and Genetic Diversity Assessment of the U.S. Collection of the New Oilseed Crop Lesquerella and Related Species. PLoS ONE 2013, 8, e64062. [Google Scholar] [CrossRef] [PubMed]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C.; et al. Diversity Arrays Technology: A Generic Genome Profiling Technology on Open Platforms. In Data Production and Analysis in Population Genomics; Humana Press: Totowa, NJ, USA, 2012; pp. 67–89. [Google Scholar]
- Raman, H.; Raman, R.; Kilian, A.; Detering, F.; Carling, J.; Coombes, N.; Diffey, S.; Kadkol, G.; Edwards, D.; McCully, M.; et al. Genome-Wide Delineation of Natural Variation for Pod Shatter Resistance in Brassica napus. PLoS ONE 2014, 9, e101673. [Google Scholar] [CrossRef] [PubMed]
- Sansaloni, C.; Petroli, C.; Jaccoud, D.; Carling, J.; Detering, F.; Grattapaglia, D.; Kilian, A. Diversity Arrays Technology (DArT) and next-generation sequencing combined: Genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc. 2011, 5, P54. [Google Scholar] [CrossRef]
- Altshuler, D.; Pollara, V.J.; Cowles, C.R.; Van Etten, W.J.; Baldwin, J.; Linton, L.; Lander, E.S. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 2000, 407, 513–516. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Kilian, B.; Graner, A. NGS technologies for analyzing germplasm diversity in genebanks. Brief. Funct. Genom. 2012, 11, 38–50. [Google Scholar] [CrossRef]
- Abdurakhmonov, I.Y.; Abdukarimov, A. Application of Association Mapping to Understanding the Genetic Diversity of Plant Germplasm Resources. Int. J. Plant Genom. 2008, 2008, 1–18. [Google Scholar] [CrossRef]
- Rafalski, J.A. Association genetics in crop improvement. Curr. Opin. Plant Biol. 2010, 13, 174–180. [Google Scholar] [CrossRef]
- Zhu, C.; Gore, M.; Buckler, E.S.; Yu, J. Status and Prospects of Association Mapping in Plants. Plant Genome J. 2008, 1, 5. [Google Scholar] [CrossRef]
- Rakoczy-Trojanowska, M.; Krajewski, P.; Bocianowski, J.; Schollenberger, M.; Wakuliński, W.; Milczarski, P.; Masojć, P.; Targońska-Karasek, M.; Banaszak, Z.; Banaszak, K.; et al. Identification of Single Nucleotide Polymorphisms Associated with Brown Rust Resistance, α-Amylase Activity and Pre-harvest Sprouting in Rye (Secale cereale L.). Plant Mol. Biol. Rep. 2017, 35, 366–378. [Google Scholar] [CrossRef]
- Edwards, D.; Batley, J.; Snowdon, R.J. Accessing complex crop genomes with next-generation sequencing. Theor. Appl. Genet. 2013, 126, 1–11. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Bayer, P.E.; Ruperao, P.; Mason, A.S.; Stiller, J.; Chan, C.-K.K.; Hayashi, S.; Long, Y.; Meng, J.; Sutton, T.; Visendi, P.; et al. High-resolution skim genotyping by sequencing reveals the distribution of crossovers and gene conversions in Cicer arietinum and Brassica napus. Theor. Appl. Genet. 2015, 128, 1039–1047. [Google Scholar] [CrossRef]
- Guo, Z.; Tucker, D.M.; Wang, D.; Basten, C.J.; Ersoz, E.; Briggs, W.H.; Lu, J.; Li, M.; Gay, G. Accuracy of Across-Environment Genome-Wide Prediction in Maize Nested Association Mapping Populations. G3 Genes Genomes Genet. 2013, 3, 263–272. [Google Scholar] [CrossRef]
- Li, L.; Petsch, K.; Shimizu, R.; Liu, S.; Xu, W.W.; Ying, K.; Yu, J.; Scanlon, M.J.; Schnable, P.S.; Timmermans, M.C.P.; et al. Mendelian and Non-Mendelian Regulation of Gene Expression in Maize. PLoS Genet. 2013, 9, e1003202. [Google Scholar] [CrossRef]
- Suwarno, W.B.; Pixley, K.V.; Palacios-Rojas, N.; Kaeppler, S.M.; Babu, R. Genome-wide association analysis reveals new targets for carotenoid biofortification in maize. Theor. Appl. Genet. 2015, 128, 851–864. [Google Scholar] [CrossRef]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
- Falconer, D.S.; Mackay, T.F.C. Introduction to Quantitative Genetics, 4th ed.; Prentice Hall: Essex, UK, 1996. [Google Scholar]
- Crossa, J.; Burgueño, J.; Dreisigacker, S.; Vargas, M.; Herrera-Foessel, S.A.; Lillemo, M.; Singh, R.P.; Trethowan, R.; Warburton, M.; Franco, J.; et al. Association Analysis of Historical Bread Wheat Germplasm Using Additive Genetic Covariance of Relatives and Population Structure. Genetics 2007, 177, 1889–1913. [Google Scholar] [CrossRef]
- Yu, L.-X.; Barbier, H.; Rouse, M.N.; Singh, S.; Singh, R.P.; Bhavani, S.; Huerta-Espino, J.; Sorrells, M.E. A consensus map for Ug99 stem rust resistance loci in wheat. Theor. Appl. Genet. 2014, 127, 1561–1581. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, D.; Guo, X.; Yang, W.; Sun, J.; Wang, D.; Sourdille, P.; Zhang, A. Investigation of genetic diversity and population structure of common wheat cultivars in northern China using DArT markers. BMC Genet. 2011, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R. Relationship between single-cross performance and molecular marker heterozygosity. Theor. Appl. Genet. 1992, 83, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pérez-Rodríguez, P.; Semagn, K.; Beyene, Y.; Babu, R.; López-Cruz, M.A.; San Vicente, F.; Olsen, M.; Buckler, E.; Jannink, J.-L.; et al. Genomic prediction in biparental tropical maize populations in water-stressed and well-watered environments using low-density and GBS SNPs. Heredity 2015, 114, 291–299. [Google Scholar] [CrossRef]
- Gao, Z.-Y.; Zhao, S.-C.; He, W.-M.; Guo, L.-B.; Peng, Y.-L.; Wang, J.-J.; Guo, X.-S.; Zhang, X.-M.; Rao, Y.-C.; Zhang, C.; et al. Dissecting yield-associated loci in super hybrid rice by resequencing recombinant inbred lines and improving parental genome sequences. Proc. Natl. Acad. Sci. USA 2013, 110, 14492–14497. [Google Scholar] [CrossRef]
- Cook, J.P.; McMullen, M.D.; Holland, J.B.; Tian, F.; Bradbury, P.; Ross-Ibarra, J.; Buckler, E.S.; Flint-Garcia, S.A. Genetic Architecture of Maize Kernel Composition in the Nested Association Mapping and Inbred Association Panels. Plant Physiol. 2012, 158, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Benke, A.; Urbany, C.; Stich, B. Genome-wide association mapping of iron homeostasis in the maize association population. BMC Genet. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Dell’Acqua, M.; Gatti, D.M.; Pea, G.; Cattonaro, F.; Coppens, F.; Magris, G.; Hlaing, A.L.; Aung, H.H.; Nelissen, H.; Baute, J.; et al. Genetic properties of the MAGIC maize population: A new platform for high definition QTL mapping in Zea mays. Genome Biol. 2015, 16, 167. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Mol. Plant 2017, 10, 359–374. [Google Scholar] [CrossRef]
- Wallace, J.G.; Bradbury, P.J.; Zhang, N.; Gibon, Y.; Stitt, M.; Buckler, E.S. Association Mapping across Numerous Traits Reveals Patterns of Functional Variation in Maize. PLoS Genet. 2014, 10, e1004845. [Google Scholar] [CrossRef]
- Liu, H.; Luo, X.; Niu, L.; Xiao, Y.; Chen, L.; Liu, J.; Wang, X.; Jin, M.; Li, W.; Zhang, Q.; et al. Distant eQTLs and Non-coding Sequences Play Critical Roles in Regulating Gene Expression and Quantitative Trait Variation in Maize. Mol. Plant 2017, 10, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Malosetti, M.; Ribaut, J.-M.; van Eeuwijk, F.A. The statistical analysis of multi-environment data: Modeling genotype-by-environment interaction and its genetic basis. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- van Eeuwijk, F.A.; Bink, M.C.; Chenu, K.; Chapman, S.C. Detection and use of QTL for complex traits in multiple environments. Curr. Opin. Plant Biol. 2010, 13, 193–205. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | TG | ACGC | TA | TSE | ACSi | ACA | ACBG | ANGLE | CLB | LMA | NPLB | ACSh | ACI | PL | HIP | DC | LC | NRG | NGC | WFGC | DM | WFG15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TG | 1 | |||||||||||||||||||||
| ACGC | 0.61 | 1 | ||||||||||||||||||||
| TA | 0.38 | 0.17 | 1 | |||||||||||||||||||
| TSE | 0.39 | 0.17 | 0.79 | 1 | ||||||||||||||||||
| ACSi | −0.33 | −0.17 | −0.31 | −0.29 | 1 | |||||||||||||||||
| ACA | −0.16 | −0.37 | 0.04 | 0.03 | 0.39 | 1 | ||||||||||||||||
| ACBG | −0.14 | 0.04 | −0.07 | −0.1 | 0.31 | 0.2 | 1 | |||||||||||||||
| ANGLE | −0.13 | 0.07 | −0.21 | −0.17 | −0.01 | −0.09 | 0.18 | 1 | ||||||||||||||
| CLB | −0.25 | −0.32 | −0.27 | −0.19 | −0.04 | 0.11 | 0.1 | 0.51 | 1 | |||||||||||||
| LMA | 0.01 | −0.04 | 0.28 | 0.22 | −0.31 | −0.09 | −0.17 | 0.01 | 0.07 | 1 | ||||||||||||
| NPLB | 0.16 | 0.11 | 0.11 | 0.09 | −0.05 | 0.05 | 0.29 | 0.24 | −0.1 | −0.24 | 1 | |||||||||||
| ACSh | −0.37 | −0.2 | −0.45 | −0.4 | 0.35 | 0.17 | 0.28 | 0.03 | 0.18 | −0.17 | 0.08 | 1 | ||||||||||
| ACI | −0.42 | −0.14 | −0.42 | −0.46 | 0.48 | 0.15 | 0.59 | 0.25 | 0.15 | −0.35 | 0.22 | 0.47 | 1 | |||||||||
| PL | 0 | −0.26 | 0.45 | 0.33 | −0.18 | 0.12 | −0.12 | −0.26 | −0.11 | 0.31 | 0.06 | −0.27 | −0.21 | 1 | ||||||||
| HIP | −0.02 | −0.19 | 0.43 | 0.2 | −0.03 | 0.24 | −0.03 | −0.27 | −0.17 | 0.12 | 0.13 | −0.2 | −0.03 | 0.74 | 1 | |||||||
| DC | 0.56 | 0.28 | 0.4 | 0.42 | −0.19 | 0.03 | −0.05 | −0.09 | −0.25 | −0.16 | 0.32 | −0.31 | −0.34 | 0.31 | 0.26 | 1 | ||||||
| LC | −0.04 | −0.09 | 0.07 | 0.13 | −0.31 | −0.29 | 0.06 | 0.14 | 0.25 | 0.33 | −0.08 | −0.05 | −0.05 | 0.27 | 0.04 | −0.03 | 1 | |||||
| NRG | 0.32 | 0.18 | 0.17 | 0.14 | −0.07 | 0.1 | −0.17 | −0.19 | −0.02 | −0.05 | 0.1 | −0.15 | −0.31 | 0.2 | 0.21 | 0.56 | −0.09 | 1 | ||||
| NGC | 0.32 | 0.17 | 0.1 | 0.12 | −0.28 | −0.13 | −0.06 | 0.2 | 0.18 | 0.08 | 0.23 | −0.22 | −0.21 | 0.17 | 0.09 | 0.53 | 0.5 | 0.61 | 1 | |||
| WFGC | 0.37 | 0.18 | 0.27 | 0.27 | −0.23 | −0.04 | 0 | −0.09 | −0.1 | 0.1 | 0.12 | −0.17 | −0.17 | 0.39 | 0.36 | 0.65 | 0.49 | 0.5 | 0.74 | 1 | ||
| DM | −0.12 | −0.16 | −0.36 | −0.37 | −0.11 | −0.06 | −0.11 | 0.33 | 0.26 | −0.04 | −0.02 | −0.08 | −0.06 | −0.26 | −0.34 | −0.28 | −0.13 | −0.24 | −0.07 | −0.53 | 1 | |
| WFG15 | 0.38 | 0.16 | 0.2 | 0.19 | −0.27 | −0.06 | −0.04 | −0.02 | −0.04 | 0.08 | 0.13 | −0.22 | −0.19 | 0.37 | 0.31 | 0.65 | 0.5 | 0.49 | 0.81 | 0.97 | −0.32 | 1 |
| p < 0.05 | p < 0.01 | p < 0.001 |
| Trait Number | Trait | No of Significant Markers | LOD Min | LOD Max | Effect Min | Effect Max | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Silico | SNP | Total | Silico | SNP | Total | Silico | SNP | Total | Silico | SNP | Total | Silico | SNP | Total | ||
| 1 | Type of grain | 40 | 22 | 62 | 2.52 | 2.53 | 2.52 | 7.68 | 4.62 | 7.68 | −0.99 | −0.78 | −0.99 | 0.73 | 0.82 | 0.82 |
| 2 | Anthocyanin coloration of glumes of cob | 101 | 49 | 150 | 2.50 | 2.53 | 2.50 | 9.53 | 10.33 | 10.33 | −1.26 | −1.05 | −1.26 | 1.18 | 1.17 | 1.18 |
| 3 | Time of anthesis (50% of flowering plants) | 30 | 29 | 59 | 2.54 | 2.52 | 2.52 | 5.35 | 6.06 | 6.06 | −2.15 | −2.64 | −2.64 | 2.33 | 1.95 | 2.33 |
| 4 | Time of silk emergence (50% of flowering plants) | 47 | 24 | 71 | 2.51 | 2.51 | 2.51 | 4.99 | 8.06 | 8.06 | −2.46 | −3.22 | −3.22 | 2.53 | 2.27 | 2.53 |
| 5 | Anthocyanin coloration of silks | 59 | 28 | 87 | 2.51 | 2.54 | 2.51 | 6.23 | 5.50 | 6.23 | −1.17 | −1.46 | −1.46 | 1.51 | 1.38 | 1.51 |
| 6 | Anthocyanin coloration of anthers | 31 | 8 | 39 | 2.50 | 2.52 | 2.50 | 3.67 | 3.63 | 3.67 | −1.07 | −1.08 | −1.08 | 1.29 | 1.25 | 1.29 |
| 7 | Anthocyanin coloration at the base of the glume | 38 | 18 | 56 | 2.60 | 2.53 | 2.53 | 5.57 | 3.63 | 5.57 | −1.30 | −1.09 | −1.30 | 1.22 | 1.11 | 1.22 |
| 8 | Angle between main axis and lateral branches | 40 | 17 | 57 | 2.51 | 2.54 | 2.51 | 6.78 | 4.58 | 6.78 | −0.89 | −0.82 | −0.89 | 1.12 | 0.91 | 1.12 |
| 9 | Curvature of lateral branches | 18 | 10 | 28 | 2.58 | 2.69 | 2.58 | 4.16 | 4.21 | 4.21 | −0.80 | −0.71 | −0.80 | 0.88 | 0.84 | 0.88 |
| 10 | Length of main axis above the highest lateral branch | 30 | 20 | 50 | 2.55 | 2.59 | 2.55 | 5.62 | 4.99 | 5.62 | −0.92 | −0.93 | −0.93 | 0.93 | 0.82 | 0.93 |
| 11 | Number of primary lateral branches | 5 | 1 | 6 | 2.51 | 2.98 | 2.51 | 3.16 | 2.98 | 3.16 | 0.40 | 0.44 | 0.40 | 0.47 | 0.44 | 0.47 |
| 12 | Anthocyanin coloration of sheath | 4 | 9 | 13 | 2.51 | 2.70 | 2.51 | 3.14 | 4.98 | 4.98 | −0.65 | −0.76 | −0.76 | 0.77 | 0.96 | 0.96 |
| 13 | Anthocyanin coloration of internodes | 14 | 6 | 20 | 2.56 | 2.60 | 2.56 | 4.05 | 3.66 | 4.05 | −0.86 | −0.70 | −0.86 | 1.11 | 0.97 | 1.11 |
| 14 | Plant length | 18 | 10 | 28 | 2.64 | 2.65 | 2.64 | 4.45 | 4.48 | 4.48 | −16.29 | 12.55 | −16.29 | 19.27 | 19.02 | 19.27 |
| 15 | Height ratio of peduncle insertion of the upper ear to plant length | 26 | 18 | 44 | 2.54 | 2.62 | 2.54 | 5.67 | 4.82 | 5.67 | −9.25 | −8.74 | −9.25 | 10.87 | 9.76 | 10.87 |
| 16 | Cob diameter | 16 | 13 | 29 | 2.51 | 2.54 | 2.51 | 4.90 | 5.29 | 5.29 | −0.18 | −0.16 | −0.18 | 0.22 | 0.19 | 0.22 |
| 17 | Cob length | 24 | 13 | 37 | 2.51 | 2.52 | 2.51 | 4.70 | 3.93 | 4.70 | −1.07 | −1.22 | −1.22 | 1.63 | 1.27 | 1.63 |
| 18 | Number of rows of grain | 16 | 6 | 22 | 2.51 | 2.54 | 2.51 | 4.22 | 3.66 | 4.22 | −0.91 | −0.88 | −0.91 | 0.93 | 0.81 | 0.93 |
| 19 | Number of grains per cob | 13 | 8 | 21 | 2.57 | 2.53 | 2.53 | 4.31 | 3.22 | 4.31 | −34.25 | −29.05 | −34.25 | 38.10 | 32.47 | 38.10 |
| 20 | Weight of fresh grains per cob | 19 | 14 | 33 | 2.51 | 2.53 | 2.51 | 3.57 | 5.32 | 5.32 | −15.69 | −13.81 | −15.69 | 12.87 | 18.41 | 18.41 |
| 21 | Dry matter content at harvest time | 12 | 8 | 20 | 2.52 | 2.51 | 2.51 | 5.20 | 2.85 | 5.20 | −2.65 | −2.13 | −2.65 | 2.92 | 2.03 | 2.92 |
| 22 | Weight of fresh grains per cob at 15% moisture | 22 | 15 | 37 | 2.53 | 2.51 | 2.51 | 4.26 | 4.94 | 4.94 | −9.34 | −11.17 | −11.17 | 11.34 | 13.86 | 13.86 |
| Total | 623 | 346 | 969 | |||||||||||||
| Origin Groups of the Lines | Flint | F2 | Inbred Line Numbers | 74 |
| F2/EP1 | 35,39,41,51,53 | |||
| F2/CM7 | 73 | |||
| Flint/BSSS | 38 | |||
| Flint/ID | 40 | |||
| Flint/Lancaster | 60,68 | |||
| German Flint/F2 | 69,82,93 | |||
| Origin unknown | 42,52,36 | |||
| Dent | ID | 33,43,44,56,58,59,61,62,64,65,72,76,79,80,83,85,88,89,90,94 | ||
| BSSS | 47,49,50,87 | |||
| ID/BSSS | 54,57,63,66,67,75,77,78,81,84,86,91,92,45,46 | |||
| Lancaster | 55 | |||
| ID/Lancaster | 50 | |||
| Semident | BSSS | 71 | ||
| Origin unknown | 37,34,48 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomkowiak, A.; Bocianowski, J.; Wolko, Ł.; Adamczyk, J.; Mikołajczyk, S.; Kowalczewski, P.Ł. Identification of Markers Associated with Yield Traits and Morphological Features in Maize (Zea mays L.). Plants 2019, 8, 330. https://doi.org/10.3390/plants8090330
Tomkowiak A, Bocianowski J, Wolko Ł, Adamczyk J, Mikołajczyk S, Kowalczewski PŁ. Identification of Markers Associated with Yield Traits and Morphological Features in Maize (Zea mays L.). Plants. 2019; 8(9):330. https://doi.org/10.3390/plants8090330
Chicago/Turabian StyleTomkowiak, Agnieszka, Jan Bocianowski, Łukasz Wolko, Józef Adamczyk, Sylwia Mikołajczyk, and Przemysław Łukasz Kowalczewski. 2019. "Identification of Markers Associated with Yield Traits and Morphological Features in Maize (Zea mays L.)" Plants 8, no. 9: 330. https://doi.org/10.3390/plants8090330
APA StyleTomkowiak, A., Bocianowski, J., Wolko, Ł., Adamczyk, J., Mikołajczyk, S., & Kowalczewski, P. Ł. (2019). Identification of Markers Associated with Yield Traits and Morphological Features in Maize (Zea mays L.). Plants, 8(9), 330. https://doi.org/10.3390/plants8090330