Genome-Wide DNA Methylation Profiling in the Lotus (Nelumbo nucifera) Flower Showing its Contribution to the Stamen Petaloid

 ,
, 
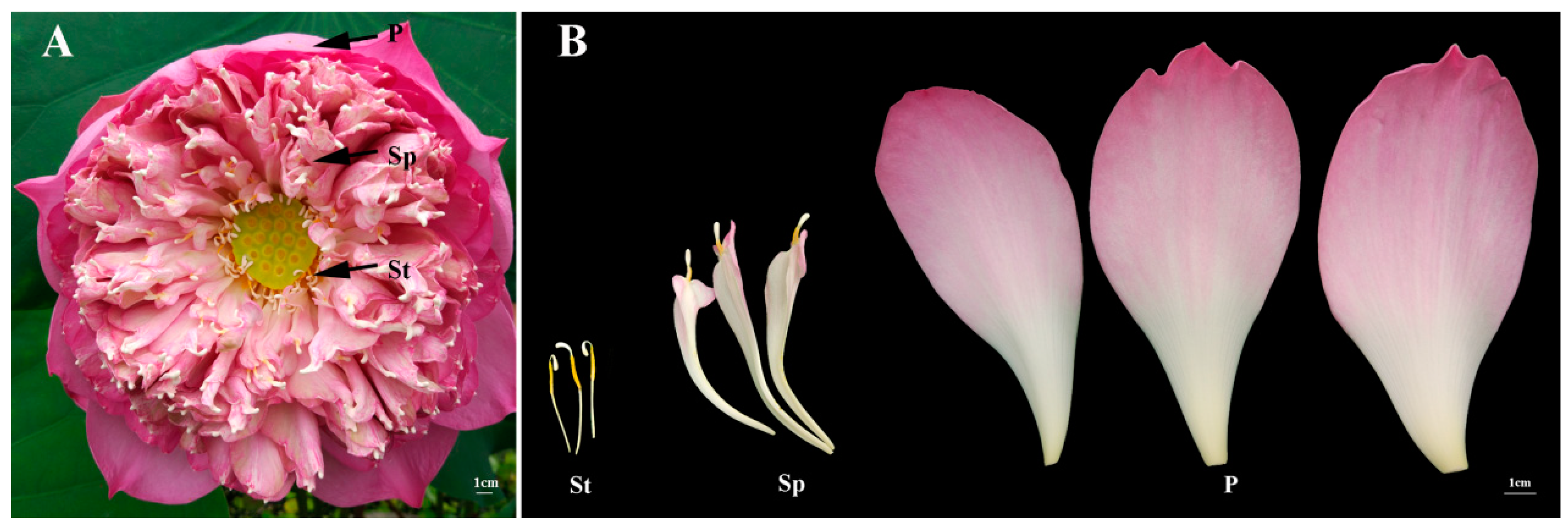{kind=link}
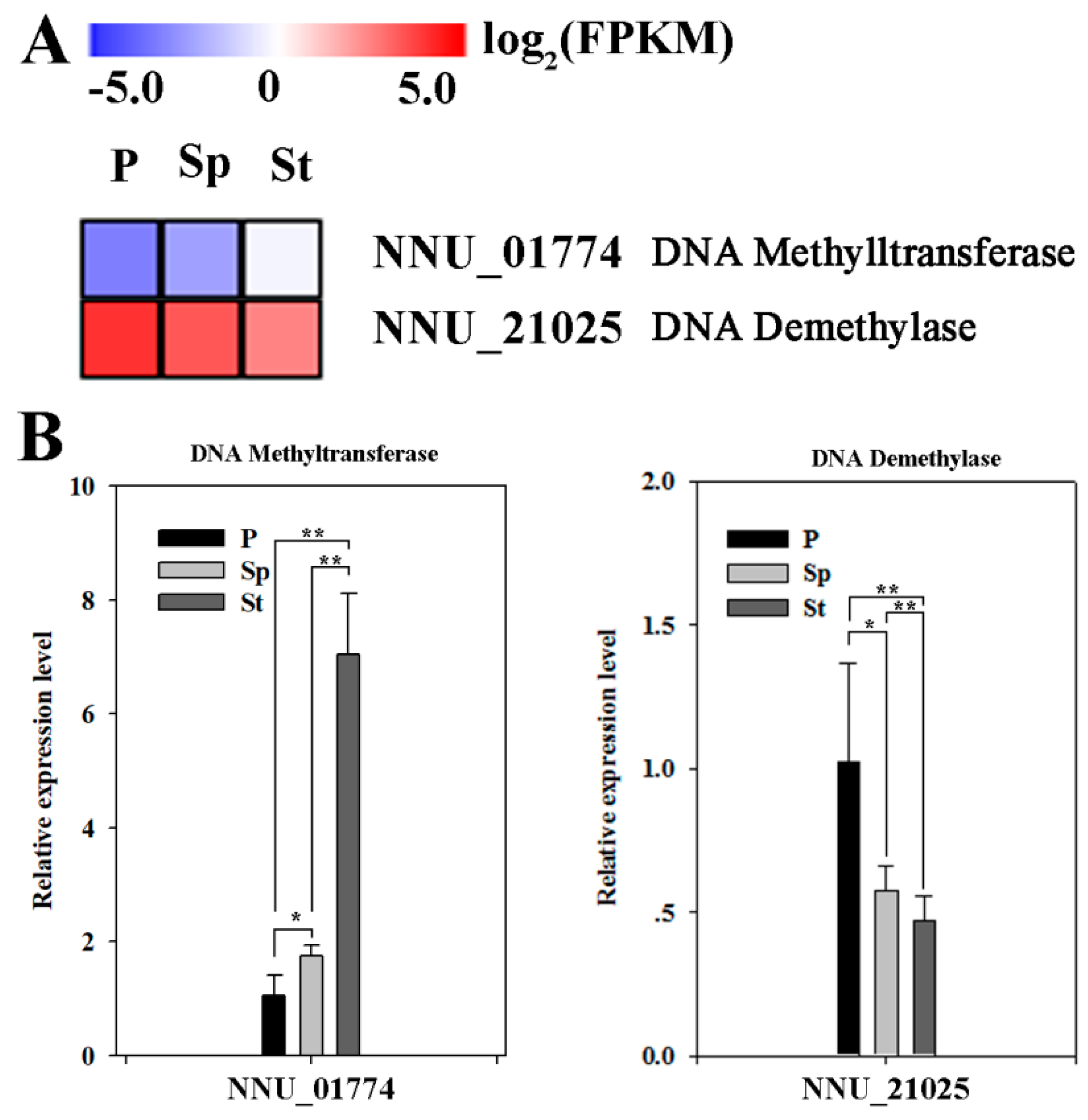{kind=link}
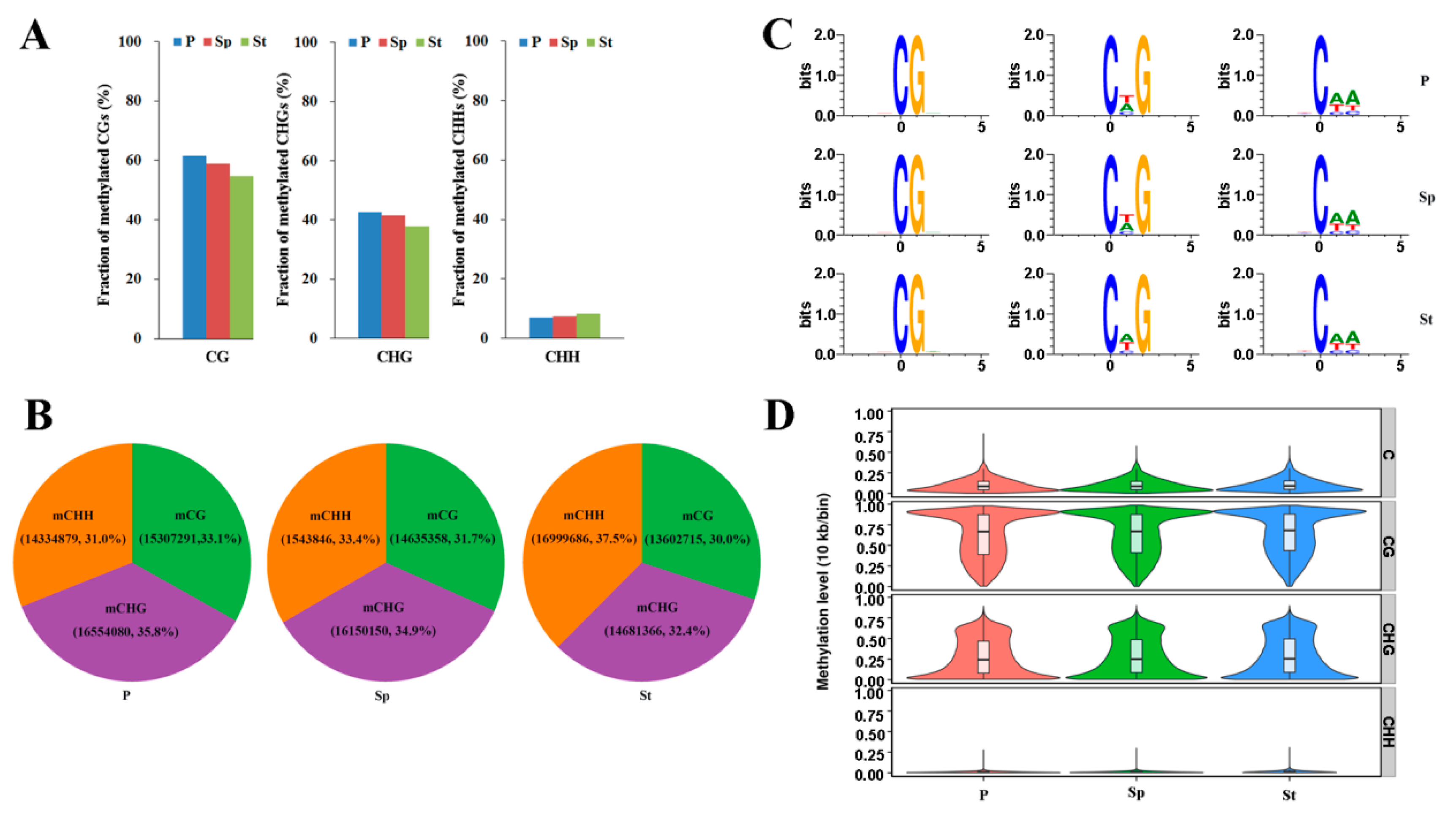{kind=link}
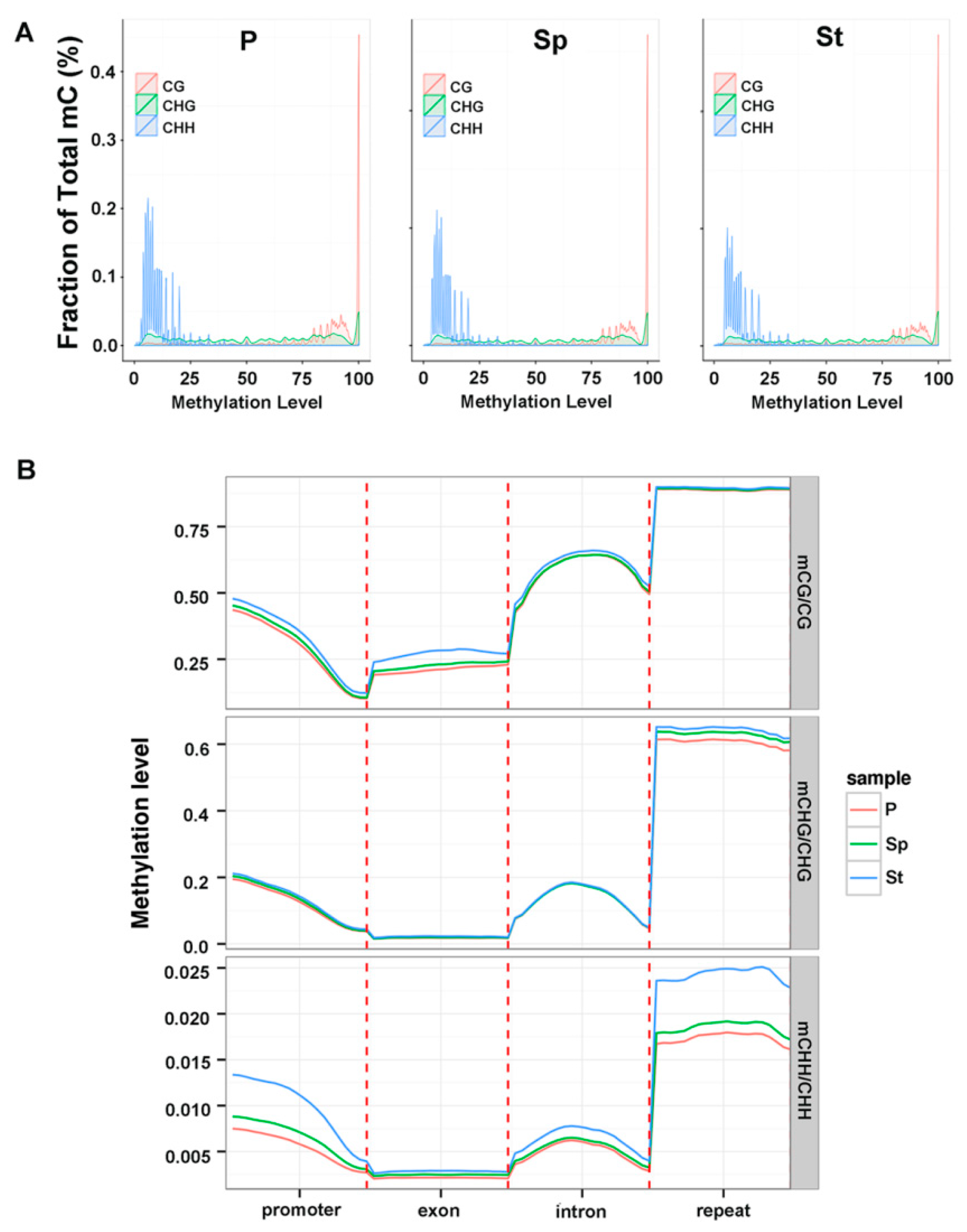{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Expression Levels of Multiple Enzymes Involvement in DNA Methylation
2.2. Whole Methylome Sequencing and Genome Methylation Profiles of the Lotus
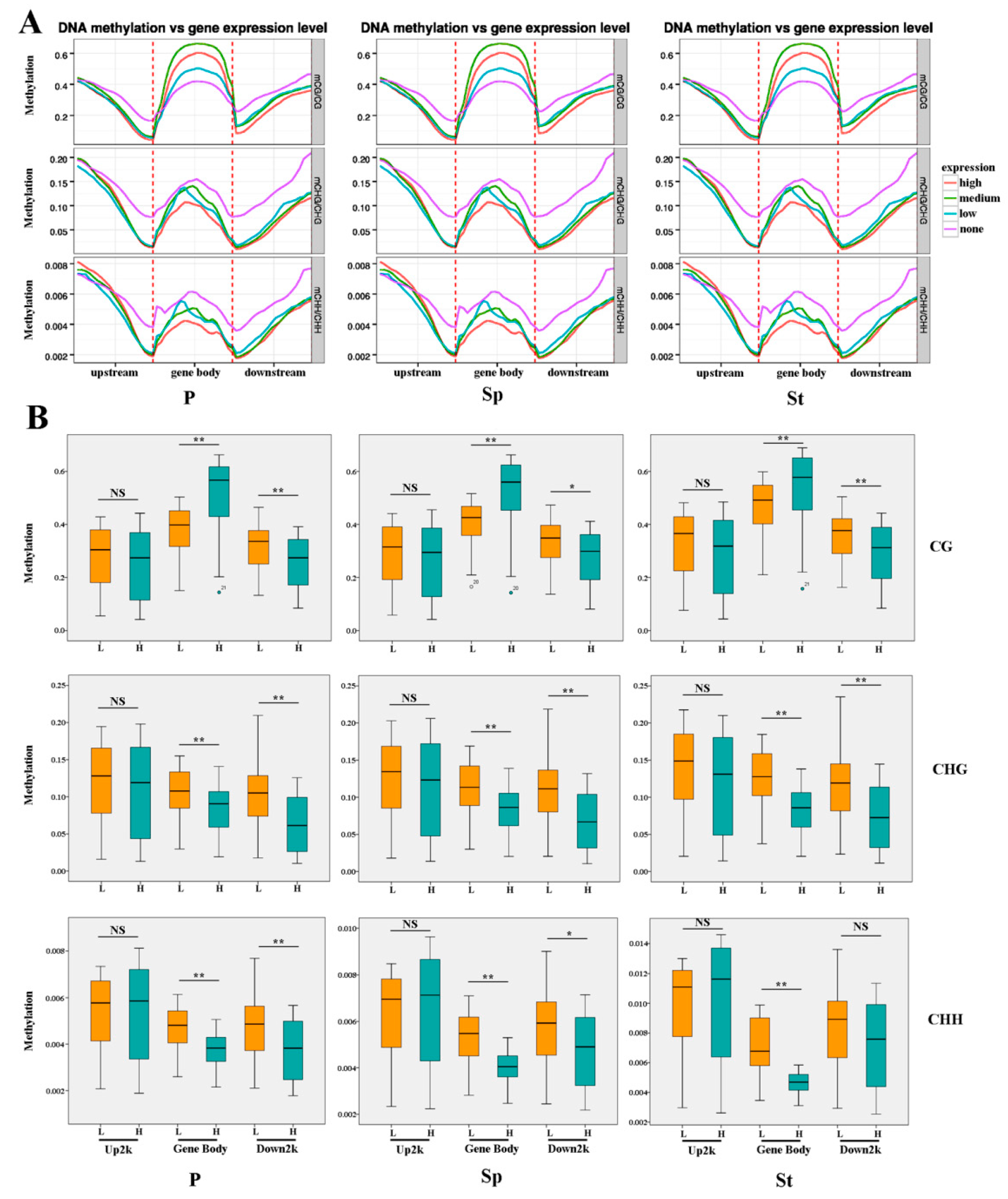
2.3. Methylation Profile of Genes
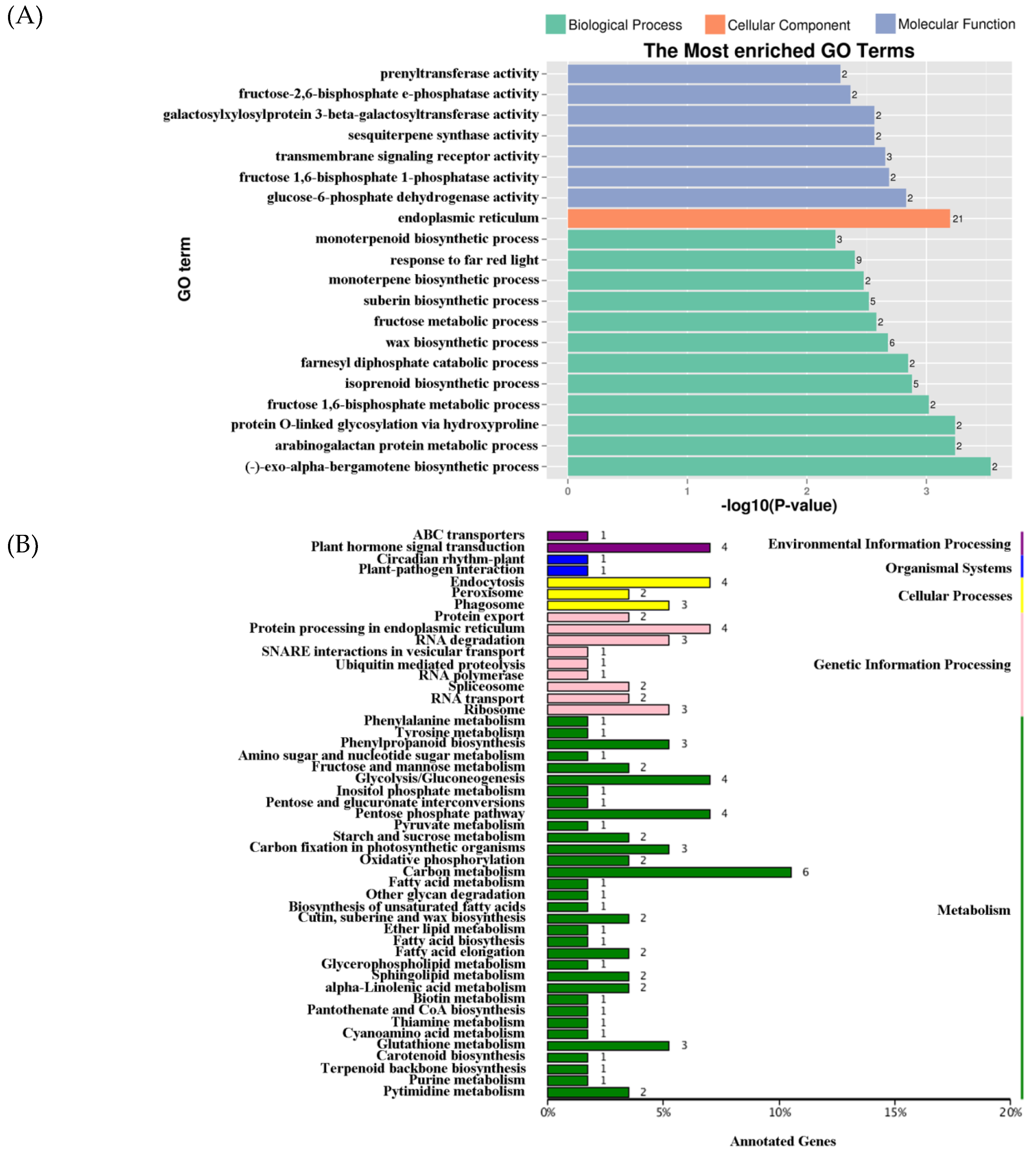
2.4. DEG Expression Associated with DNA Methylation
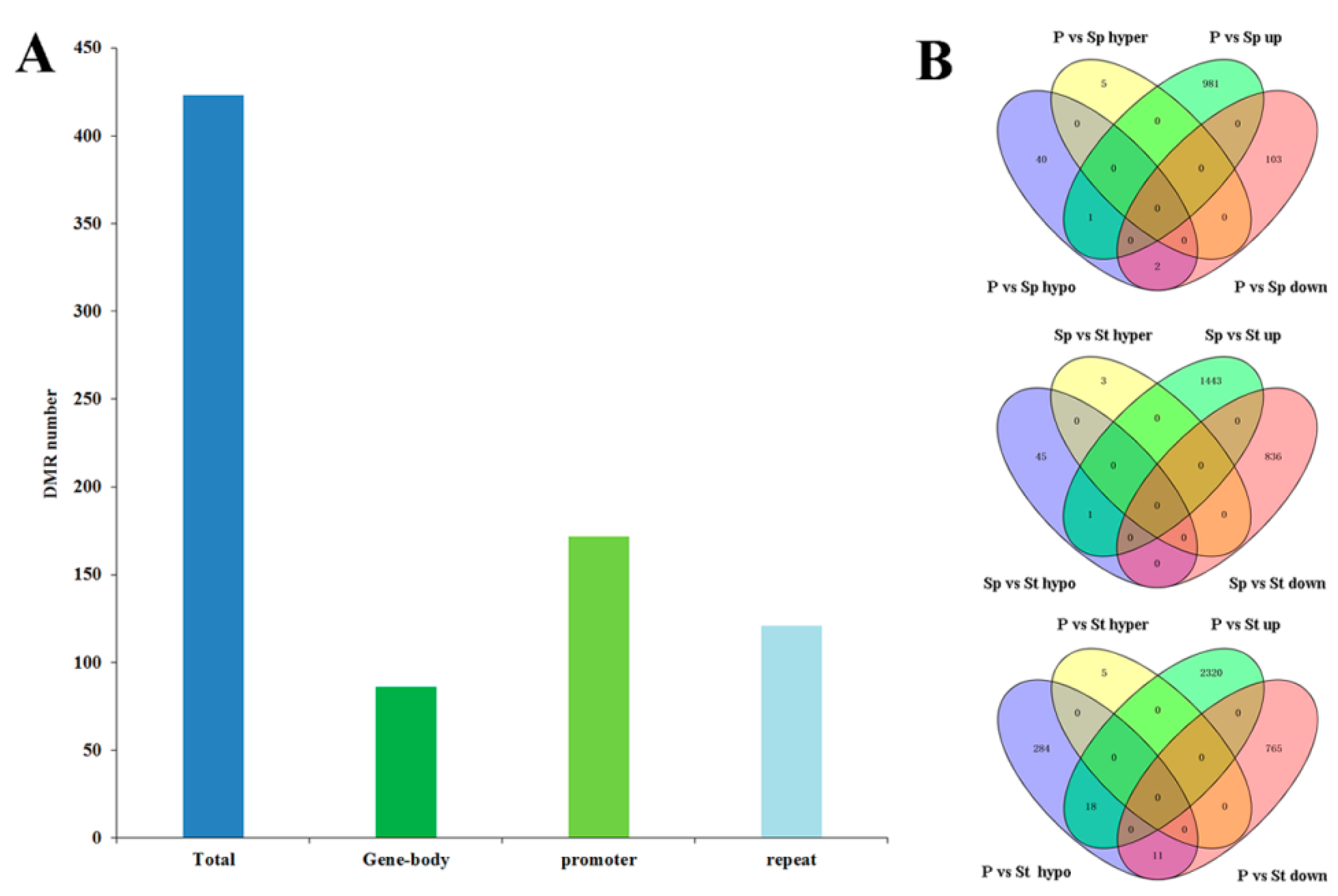
2.5. Distribution of DNA Methylation Variation
3. Conclusions
4. Materials and Methods
4.1. Plant Materials
4.2. Whole-Genome Bisulfite Sequencing
4.3. Bioinformatic Analysis of Whole-Genome Bisulfite Sequencing Data
4.4. Differential Expression Analysis of mRNA Sequencing Data and Confirmation by qRT-PCR
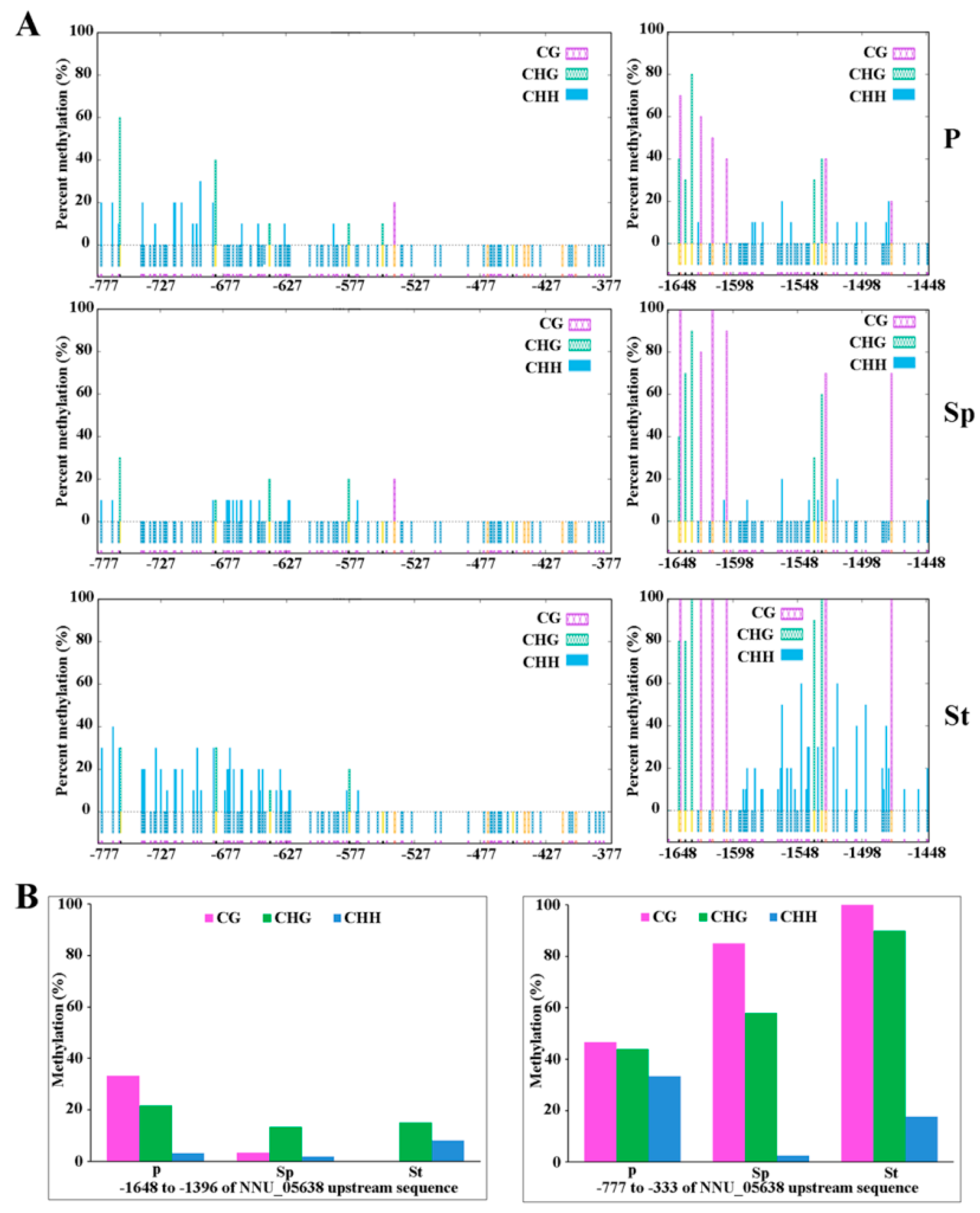
4.5. Validation of the WGBS Result about Methylation of the NNU_05638 Promoter
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BSeq: Bisulfite sequencing; |
| CMT: CHROMOMETHYLASE; |
| CTAB: cetyltrimethylammonium bromide; |
| DEGs: differential expression genes; |
| DMR: differential methylation region; |
| DNMT1: DNA methyltransferase 1; |
| DRM: DOMAINS REARRANGED METHYLTRANSFERASE; |
| FPKM: fragments per kilo base of transcript per million base pairs sequenced; |
| GO: gene ontology; |
| H3K9: histone H3 lysine 9; |
| KEGG: kyoto encyclopedia of genes and genomes; |
| mCs: methylcytosines; |
| MET1: methyltransferase 1; |
| P: petal; |
| PUB33: Plant U-box 33; |
| qRT-PCR: quantitative real time PCR; |
| RdDM: RNA-directed DNA methylation; |
| SAM: shoot apical meristem; |
| SAUR: small auxin up-regulated RNAs; |
| siRNAs: short-interfering RNAs; |
| Sp: petaloid stamen; |
| St: stamen; |
| TSSs: transcription starting sites; |
| WGBS: whole genome bisulfite sequencing. |
References
- Yang, H.; Chang, F.; You, C.; Cui, J.; Zhu, G.; Wang, L.; Zheng, Y.; Qi, J.; Ma, H. Whole-genome DNA methylation patterns and complex associations with gene structure and expression during flower development in Arabidopsis. Plant J. 2015, 81, 268–281. [Google Scholar] [CrossRef]
- Bouyer, D.; Kramdi, A.; Kassam, M.; Heese, M.; Schnittger, A.; Roudier, F.; Colot, V. DNA methylation dynamics during early plant life. Genome Biol. 2017, 18, 179. [Google Scholar] [CrossRef] [PubMed]
- Narsai, R.; Gouil, Q.; Secco, D.; Srivastava, A.; Karpievitch, Y.V.; Liew, L.C.; Lister, R.; Lewsey, M.G.; Whelan, J. Extensive transcriptomic and epigenomic remodelling occurs during Arabidopsis thaliana germination. Genome Biol. 2017, 18, 172. [Google Scholar] [CrossRef]
- Stroud, H.; Greenberg, M.V.C.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. The roles of non-CG methylation in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef]
- Kawashima, T.; Berger, F. Epigenetic reprogramming in plant sexual reproduction. Nat. Rev. Genet. 2014, 15, 613–624. [Google Scholar] [CrossRef]
- Matzke, M.A.; Kanno, T.; Matzke, A.J. RNA-Directed DNA Methylation: The Evolution of a Complex Epigenetic Pathway in Flowering Plants. Annu. Rev. Plant Biol. 2015, 66, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hermanson, P.J.; Springer, N.M. Detection of DNA methylation by whole-genome bisulfite sequencing. Methods Mol. Biol. 2018, 1676, 185. [Google Scholar]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulfite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215. [Google Scholar] [CrossRef]
- Chodavarapu, R.K.; Feng, S.; Ding, B.; Simon, S.A.; Lopez, D.; Jia, Y.; Wang, G.L.; Meyers, B.C.; Jacobsen, S.E.; Pellegrini, M. Transcriptome and methylome interactions in rice hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 12040–12045. [Google Scholar] [CrossRef]
- Song, Q.X.; Lu, X.; Li, Q.T.; Chen, H.; Hu, X.Y.; Ma, B.; Zhang, W.K.; Chen, S.Y.; Zhang, J.S. Genome-wide analysis of DNA methylation in soybean. Mol. Plant 2013, 6, 1961–1974. [Google Scholar] [CrossRef]
- Li, Q.; Song, J.; West, P.T.; Zynda, G.; Eichten, S.R.; Vaughn, M.W.; Springer, N.M. Examining the causes and consequences of context-specific differential DNA methylation in maize. Plant Physiol. 2015, 168, 1262–1274. [Google Scholar] [CrossRef]
- Yang, J.K.; Bae, A.; Shim, S.; Lee, T.; Lee, J.; Satyawan, D.; Moon, Y.K.; Lee, S.-H. Genome-wide DNA methylation profile in mungbean. Sci. Rep. 2017, 7, 40503. [Google Scholar]
- Kawakatsu, T.; Valdes, M.; Breakfield, N.; Schmitz, R.J.; Nery, J.R.; Urich, M.A.; Han, X.; Lister, R.; Benfey, P.N. Unique cell-type-specific patterns of DNA methylation in the root meristem. Nat. Plants 2016, 2, 16058. [Google Scholar] [CrossRef]
- Deng, J.; Fu, Z.; Chen, S.; Damaris, R.N.; Wang, K.; Li, T.; Yang, P. Proteomic and epigenetic analyses of lotus (Nelumbo nucifera) petals between red and white cultivars. Plant Cell Physiol. 2015, 56, 1546. [Google Scholar] [CrossRef]
- Wang, L.; Fu, J.; Li, M.; Fragner, L.; Weckwerth, W.; Yang, P. Metabolomic and proteomic profiles reveal the dynamics of primary metabolism during seed development of lotus (Nelumbo nucifera). Front Plant Sci. 2016, 7, 750. [Google Scholar] [CrossRef]
- Yang, M.; Zhu, L.; Pan, C.; Xu, L.; Liu, Y.; Ke, W.; Yang, P. Transcriptomic analysis of the regulation of rhizome formation in temperate and tropical lotus (Nelumbo nucifera). Sci. Rep. 2015, 5, 13059. [Google Scholar] [CrossRef]
- Zhang, H.M.; Lang, Z.B.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Lin, Z.; Damaris, R.N.; Shi, T.; Li, J.; Yang, P. Transcriptomic analysis identifies the key genes involved in stamen petaloid in lotus (Nelumbo nucifera). BMC Genom. 2018, 19, 554. [Google Scholar] [CrossRef]
- Ming, R.; Vanburen, R.; Liu, Y.; Yang, M.; Han, Y.; Li, L.T.; Zhang, Q.; Kim, M.J.; Schatz, M.C.; Campbell, M. Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.). Genome Biol. 2013, 14, R41. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Su, C.; Wang, C.; He, L.; Yang, C.; Wang, Y. Shotgun bisulfite sequencing of the Betula platyphylla genome reveals the tree’s DNA methylation patterning. Int. J. Mol. Sci. 2014, 15, 22874–22886. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; He, Y.; Valdéslópez, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663. [Google Scholar] [CrossRef]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon Is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef]
- Wang, H.; Beyene, G.; Zhai, J.; Feng, S.; Fahlgren, N.; Taylor, N.J.; Bart, R.; Carrington, J.C.; Jacobsen, S.E.; Ausin, I. CG gene body DNA methylation changes and evolution of duplicated genes in cassava. Proc. Natl. Acad. Sci. USA 2015, 112, 13729–13734. [Google Scholar] [CrossRef]
- Zemach, A.; Mcdaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465. [Google Scholar] [CrossRef]
- He, X.J.; Chen, T.; Zhu, J.K. Regulation and function of DNA methylation in plants and animals. Cell Res. 2011, 21, 442. [Google Scholar] [CrossRef]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S.; et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300. [Google Scholar] [CrossRef]
- Meng, D.; Dubin, M.; Zhang, P.; Osborne, E.J.; Stegle, O.; Clark, R.M.; Nordborg, M. Limited contribution of DNA methylation variation to expression regulation in Arabidopsis thaliana. PLoS Genet. 2016, 12, e1006141. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-H.; He, S.; Buttress, T.; Gao, H.; Couchman, M.; Fischer, R.L.; Zilberman, D.; Feng, X. Arabidopsis male sexual lineage exhibits more robust maintenance of CG methylation than somatic tissues. Proc. Natl. Acad. Sci. USA 2016, 113, 15132–15137. [Google Scholar] [CrossRef]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.-F.; An, Y.-Q.C.; et al. Dynamic DNA Methylation in Plant Growth and Development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef] [PubMed]
- Spartz, A.K.; Lee, S.H.; Wenger, J.P.; Gonzalez, N.; Itoh, H.; Inzé, D.; Peer, W.A.; Murphy, A.S.; Overvoorde, P.J.; Gray, W.M. The SAUR19 subfamily of SMALL AUXIN UP RNA genes promote cell expansion. Plant J. 2012, 70, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Chae, K.; Isaacs, C.G.; Reeves, P.H.; Maloney, G.S.; Muday, G.K.; Nagpal, P.; Reed, J.W. Arabidopsis SMALL AUXIN UP RNA63 promotes hypocotyl and stamen filament elongation. Plant J. 2012, 71, 684–697. [Google Scholar] [CrossRef]
- Wu, J.; Liu, S.; He, Y.; Guan, X.; Zhu, X.; Cheng, L.; Wang, J.; Lu, G. Genome-wide analysis of SAUR gene family in Solanaceae species. Gene 2012, 509, 38–50. [Google Scholar] [CrossRef]
- Mizuno, S.; Osakabe, Y.; Maruyama, K.; Ito, T.; Osakabe, K.; Sato, T.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Receptor-like protein kinase 2 (RPK 2) is a novel factor controlling anther development in Arabidopsis thaliana. Plant J. 2007, 50, 751–766. [Google Scholar] [CrossRef]
- Callis, J. The ubiquitination machinery of the ubiquitin system. Arabidopsis Book 2014, 12, e0174. [Google Scholar] [CrossRef]
- Wang, H.; Lu, Y.; Jiang, T.; Berg, H.; Li, C.; Xia, Y. The Arabidopsis U-box/ARM repeat E3 ligase AtPUB4 influences growth and degeneration of tapetal cells, and its mutation leads to conditional male sterility. Plant J. 2013, 74, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, Y.; Yu, B.; Yin, Z.; Xia, Y. EXTRA-LARGE G PROTEINs Interact with E3 Ligases PUB4 and PUB2 and Function in Cytokinin and Developmental Processes. Plant Physiol. 2017, 173, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, A.; Seo, M.; Kamiya, Y.; Sawa, S. Mystery in genetics: PUB4 gives a clue to the complex mechanism of CLV signaling pathway in the shoot apical meristem. Plant Signal. Behavior. 2015, 10, e1028707. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. USA 1996, 93, 8449–8454. [Google Scholar] [CrossRef]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Z.; Liu, M.; Damaris, R.N.; Nyong’a, T.M.; Cao, D.; Ou, K.; Yang, P. Genome-Wide DNA Methylation Profiling in the Lotus (Nelumbo nucifera) Flower Showing its Contribution to the Stamen Petaloid. Plants 2019, 8, 135. https://doi.org/10.3390/plants8050135
Lin Z, Liu M, Damaris RN, Nyong’a TM, Cao D, Ou K, Yang P. Genome-Wide DNA Methylation Profiling in the Lotus (Nelumbo nucifera) Flower Showing its Contribution to the Stamen Petaloid. Plants. 2019; 8(5):135. https://doi.org/10.3390/plants8050135
Chicago/Turabian StyleLin, Zhongyuan, Meihui Liu, Rebecca Njeri Damaris, Tonny Maraga Nyong’a, Dingding Cao, Kefang Ou, and Pingfang Yang. 2019. "Genome-Wide DNA Methylation Profiling in the Lotus (Nelumbo nucifera) Flower Showing its Contribution to the Stamen Petaloid" Plants 8, no. 5: 135. https://doi.org/10.3390/plants8050135
APA StyleLin, Z., Liu, M., Damaris, R. N., Nyong’a, T. M., Cao, D., Ou, K., & Yang, P. (2019). Genome-Wide DNA Methylation Profiling in the Lotus (Nelumbo nucifera) Flower Showing its Contribution to the Stamen Petaloid. Plants, 8(5), 135. https://doi.org/10.3390/plants8050135