Abstract
Phloem fibers are important elements of plant architecture and the target product of many fiber crops. A key stage in fiber development is intrusive elongation, the mechanisms of which are largely unknown. Integrated analysis of miRNA and mRNA expression profiles in intrusivelygrowing fibers obtained by laser microdissection from flax (Linum usitatissimum L.) stem revealed all 124 known flax miRNA from 23 gene families and the potential targets of differentially expressed miRNAs. A comparison of the expression between phloem fibers at different developmental stages, and parenchyma and xylem tissues demonstrated that members of miR159, miR166, miR167, miR319, miR396 families were down-regulated in intrusively growing fibers. Some putative target genes of these miRNA families, such as those putatively encoding growth-regulating factors, an argonaute family protein, and a homeobox-leucine zipper family protein were up-regulated in elongating fibers. miR160, miR169, miR390, and miR394 showed increased expression. Changes in the expression levels of miRNAs and their target genes did not match expectations for the majority of predicted target genes. Taken together, poorly understood intrusive fiber elongation, the key process of phloem fiber development, was characterized from a miRNA-target point of view, giving new insights into its regulation.
1. Introduction
Phloem fibers are important elements of plant architecture by providing mechanical strength and support to a plant in general and to phloem in particular. These fibers are abundantly formed in the long but narrow stems of many fiber-crops. The specific mechanical properties of the phloem fibers are based on extraordinary length of their cells (many centimeters) and their very thick cell walls (up to 15 µm) [1]. Two key processes have a major impact on the specialization of the plant fibers: intrusive growth and cell wall thickening. They both are promising points for genetic manipulations to improve the yield and the quality of bast fibers [2]. In flax stems, these two stages of phloem fiber development are separated in time and space, allowing for the analysis of tissue and stage-specific components [2,3]. Flax stems contain only primary phloem fibers that originate from the procambium close to the stem apex [4,5]. Their development is more advanced towards the stem base. The complete cessation of intrusive growth and the onset of cell wall thickening are marked by the so-called “snap point” (SP) in flax phloem fibers, which is easy to find manually by stepwise increasing the effort required to break the stem [3].
Intrusive growth occurs when a growing cell increases in size faster than neighboring ones, thereby intruding between them [1,6,7]. Later, plant fiber specialization may advance by formation of tertiary cell wall, which is deposited after the primary and secondary cell walls in fibers of many species, including flax [8,9]. Tertiary cell walls are cellulose-enriched, and their cellulose microfibril orientation is close to axial. Entrapment of aggregated rhamnogalacturonan I molecules by laterally interacting cellulose microfibrills leads to tension of the latter and provides contractile properties to the fibers with tertiary cell walls [8,9].
In our previous work on flax plants based on RNA deep sequencing, we obtained transcript abundance profiles and identified a set of genes that are specifically activated at different stages of flax bast fiber development [10,11]. However, the mechanisms controlling both the intrusive elongation and the synthesis of tertiary cell wall, as well as the functioning of the fibers in the whole plant, are still poorly understood.
The discovery of small RNAs (miRNA), a class of low molecular-weight, non-coding, regulatory RNAs of 19–24 nucleotides in size that act at the post-transcriptional level, added another level of complexity to the multi-level program for fine-tuning of gene expression [12,13]. miRNAs are able to regulate a whole range of biological processes, including developmental regulation, growth control, cell differentiation, and biotic and abiotic stresses. In fact, they may be the “master” non-protein regulators of plant and animal development [14,15,16]. The relative conservatism of miRNAs among plants of various taxonomic groups allows one to identify novel homologs of miRNA in different species using a database of already identified small RNA sequences and in silico methods [17,18,19,20].
124 miRNA sequences belonging to 23 families were identified in the flax genome, and more than a hundred of their target genes were predicted [18,21,22,23]. Prior miRNA research identified the roles of flax miRNAs and their potential targets in various stress responses, including nutrient excess or deficiency [24,25,26]. The samples used in these studies were complex mixtures of tissues that included many cell types and cells at differing developmental stages. Taking the advantage of the model system that has been well characterized in our previous works [3,5], we used high-throughput sequencing technology (Illumina) and bioinformatics tools to identify and analyze flax miRNAs and check the expression of their predicted targets in specific cell types and at certain stages of development. For the first time, miRNA and mRNA expression was analyzed during the poorly understood process of fiber intrusive elongation, giving new insights into the regulation of phloem fiber development.
2. Results
2.1. Deep-Sequencing of Flax Fiber Small RNAs
Ten individual libraries of small RNAs from two biological replicates of five different samples were analyzed by high throughput sequencing. The five samples included intrusively growing phloem fibers with only primary cell walls (iFIBa and iFIBb), symplastically growing cortex parenchyma (cPAR), phloem fibers at the stage of tertiary cell wall deposition (tFIB), and xylem (sXYL) that contained mainly cells with secondary cell walls (Figure 1).
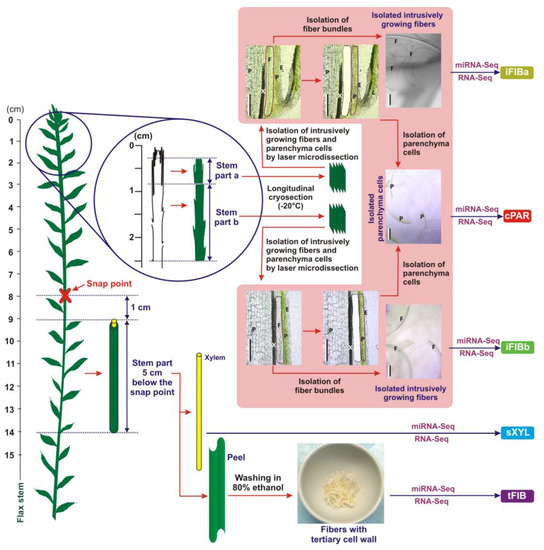
Figure 1.
A scheme of plant material collection to obtain iFIBa, iFIBb, cPAR, sXYL and tFIB samples for subsequent RNA-Seq and miRNA-Seq analysis. Bundles of intrusively (i) growing fibers (the samples iFIBa and iFIBb) were isolated from longitudinal cryosections of the stem part a (0.3–0.8 cm from the stem apex) and b (0.8–2.5 cm from the stem apex) by laser microdissection. Cortex (c) parenchyma (cPAR) was isolated from longitudinal cryosections of the stem parts a and b by laser microdissection, and these two tissue fractions were combined. Fibers depositing tertiary (t) cell walls (tFIB) and xylem enriched in secondary (s) cell walls (sXYL) were sampled 1 cm below the snap point. E—epidermis, F—fiber bundles, L—leaf, P—parenchyma, and X—xylem; Bar—200 µm.
A total of 99,241,346 cleaned and filtered reads (79.98% of the raw miRNA reads) were used for further analysis (Table 1). miRNA expression levels from the 10 libraries datasets were visualized with the Principal Component Analysis (PCA) and analyzed for length distribution (Figure 2). PCA demonstrated that the two samples of intrusively growing fibers (iFIBa and iFIBb, differing in the stage of intrusive elongation; see a Section 2.5) were close to each other, but well separated from the other samples (cPAR, tFIB, sXYL); the variance of replicates within each sample was always much lower than that between the samples. The bulk of reads were 21–26 nt in length, with 24 nt being the most abundant group of small RNAs. Recently updated annotations of plant miRNA demonstrate that 23- or 24-nucleotide miRNAs are very rare and mostly characteristic of heterochromatic siRNAs [27]; nevertheless, the possibility that some plant species generate numerous 23- or 24-nucleotide microRNAs is not excluded [27]. The predominance of 24 nt read length is consistent with the distribution patterns of small RNAs in many plants [28,29,30,31,32], including flax [24].

Table 1.
Summary of reads in miRNA libraries.
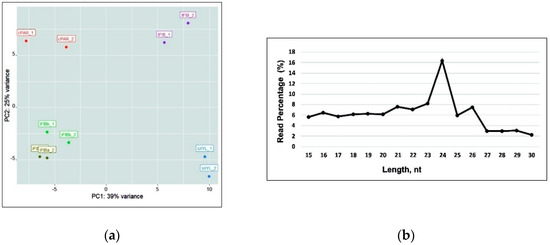
Figure 2.
(a) The Principal Component Analysis (PCA) of the samples analyzed (including replicates) based on the lus-miR normalized expression pattern. (b) Size distribution of small RNA sequences in all of the libraries.
2.2. Characterization of the Expressed Flax miRNAs
To identify the miRNAs expressed in the samples, the data from high-throughput small RNA sequencing were aligned against 124 flax miRNA precursor sequences. After estimating the expression values, representatives of the 23 known flax miRNA families were identified (Table S1). Representatives of 5 families were the most abundant in all the samples analyzed, with total gene reads (TGR) over 1000 in each sample, namely miR159, miR319, miR397, miR398, miR408 (Figure 3a).
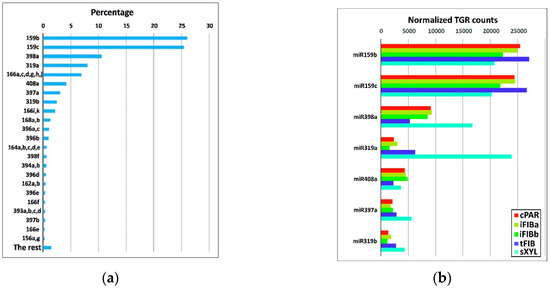
Figure 3.
(a) The most abundant known miRNA families. The indicated miRNA flax families are represented by more than 1000 normalized TGR counts, and “The rest” have fewer than 1000 normalized TGR counts across all the samples as a whole. (b) Normalized TGR counts of the 5 most abundant miRNA families across the samples.
miR159b and miR159c together accounted for over 50% of all detected miRNAs (Figure 3a). The miR159 family is highly conserved and widespread in plants, being expressed in the course of plant development and stress adaptation [17,24,33]. Expression of lus-miR159b and lus-miR159c was detected previously in various parts of flax plants, including stem, and suggested to be involved in the regulation and fine-tuning of the expression of some housekeeping genes [23].
The xylem samples were especially enriched in miR319a and miR398a compared with the other samples (Figure 3b). miR319 regulates transcription factors of the TCP family affecting multiple biological pathways: from hormone biosynthesis and signaling to cell proliferation and differentiation [34]. The three other families of most abundant miRNAs (miR408, miR398, and miR397) are Cu-miRNAs that are relatively conserved among plants and regulate a large number of Cu proteins [35].
2.3. Identification of Differentially Expressed miRNAs during Intrusive Elongation of Phloem Fibers in Flax Stems
To figure out small RNAs that are especially abundant at certain developmental stages of flax phloem fiberes, the miRNA normalized TGR counts for all samples were used to generate a miR-expression heatmap and clustering (Figure 4). A dendrogram of miRNA expression derived from hierarchical clustergram analysis using the Pearson’s correlation was divided into 11 clusters (Figure 4a, the vertical color bar).
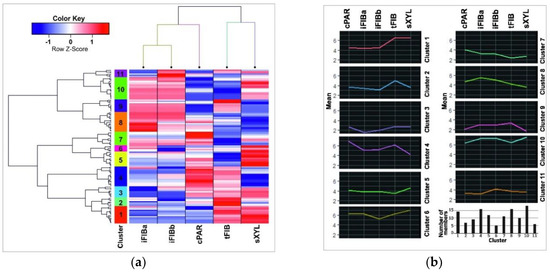
Figure 4.
(a) A heatmap with dendrograms of hierarchical clustering of the data (the lus-miR subset and the samples). Columns represent samples, while rows represent 124 lus-miRs. The 11 clusters are coded by the vertical color bar. The Z-score is the number of standard deviations from the mean. The normalized expression value for each miRNA is depicted by color intensity, with red indicating up-regulated and blue indicating down-regulated lus-miRs. (b) Average expression profiles for each miRNA expression cluster and numbers of cluster members. An average cluster profile was calculated as the mean of normalized and scaled TGRs counts of all miRNAs in each cluster.
A mean cluster profile for each cluster as the mean of TGR counts of all miRNAs in a cluster for each sample was calculated (Figure 4b). Clusters 1, 2, 4, and 6 contained miRNAs that were down-regulated in iFIB samples as compared to the other samples, whereas clusters 5, 7, 8, and 10 included up-regulated miRNAs.
To find out which miRNAs are differentially expressed during different stages of phloem fiber development (intrusive elongation and tertiary cell wall formation), pairwise comparisons between iFIBa vs. tFIB, and iFIBb vs. tFIB were performed. These comparisons revealed 19 down-regulated miRNAs during intrusive elongation that belonged to six families (miR156, 159, 166, 167, 319, 396) and 14 up-regulated miRNAs from five families (miR160, 169, 390, 394, and 399) (Table 2). A member of the miR396 family (miR396c) was down-regulated during intrusive fiber elongation in both a tissue- and stage-specific manner: the expression of this miRNA in both iFIBa and iFIBb was lower than that in the other tissues (cPAR and sXYL) and in the same tissue at the later stage of development (tFIB) (adjusted p-value (padj) < 0.05). Four different members of the miR396 family (a, b, d, e) were significantly down-regulated in intrusively growing fibers as compared with all other samples, except cPAR. In Arabidopsis, members of the miR396 family control cell proliferation and elongation [36]. Several members of miR166 were down-regulated in iFIB samples as compared to tFIB, cPAR, but not sXYL. As for miRNAs significantly up-regulated in iFIB, two members of miR394 had higher transcript abundance as compared to all other samples, except sXYL (Table 2).
Table 2.
The differentially expressed flax miRNAs the expression of which was significantly up- and down-regulated in intrusively growing flax fibers (the cutoff of comparison either iFIBa vs. tFIB, or iFIBb vs. tFIB has a log2FC value ≥2 or ≤−2 with padj < 0.05); FC; fold change.
2.4. Identification of miRNA Targets Up- and Down-Regulated during Intrusive Elongation of Phloem Fibers in Flax Stems
To understand the potential regulatory functions of miRNA that were differentially expressed during intrusive fiber growth, we used the psRNATarget web server that was developed to identify miRNA-target pairs by analyzing the complementary matching between small RNAs and targets and evaluating the accessibility of the target sites from the calculated value of unpaired energy (UPE) [37].
A total of 391,488,774 (97,52%) clean reads were obtained, of which about 88% reads were successfully mapped onto the flax reference genome [38]. To compare changes in gene expression between samples from different experiments, gene expression levels were obtained as normalized TGR using raw counts from HTSeq [39] and input into DESeq2 [40]. From 43,486 protein-coding genes of flax, 30,922 genes with normalized TGR values > 16 in at least one sample were selected for the analysis of differential expression in pairwise comparisons (Table S2). A total of 1942 up-regulated and 3765 down-regulated genes were identified in intrusively growing fibers when pairwise comparisons of iFIBa and iFIBb with tFIB were done (log2FC ≥ 2 or ≤ −2 with a cutoff padj < 0.05, Table S2). Among them, 1495 expressed targets (Table S2) for 33 miRNAs up- and down-regulated in iFIB as compared to tFIB (with a cutoff for fold change (FC) |log2FC| ≥ 2 and padj < 0.05) were predicted (Table 2).
The up-regulated targets for down-regulated miRNAs were enriched in homologs of Arabidopsis genes for transcription factors (SPL9, bZIP5, HB8, HB14, HB15, AP2/B3, and several GRFs) (Table 3). The most significantly down-regulated target genes included those for which Arabidopsis homologs were annotated as auxin-response factors, receptor kinases, transporters, and hormone-related (Table 4).
Table 3.
Intrusive growth-related down-regulated lus-miR families and their up-regulated target genes identified from the comparison of phloem fibers at the stages of intrusive elongation and cell wall thickening (a cutoff of comparison either iFIBa vs. tFIB, or iFIBb vs. tFIB has a log2FC value ≥2 or ≤−2 with padj < 0.05); FC; fold change.
Table 4.
Intrusive growth-related up-regulated lus-miR families and their down-regulated target genes identified from the comparison of phloem fibers at the stages of intrusive elongation and cell wall thickening (a cutoff of comparison either iFIBa vs. tFIB, or iFIBb vs. tFIB has a log2FC value ≥2 or ≤−2 with padj < 0.05).
For many combinations of miRNAs and their target genes, an opposite character of differential expression was observed, as expected from the general notion that miRNA marks the complementary mRNA for degradation [41]. For example, when comparing iFIBa and iFIBb versus tFIB, the expression of up- and down-regulated miRNAs was inversely related to the expression of 121 genes predicted as their targets (Table 3 and Table 4). However, these genes accounted for only a small portion of the predicted target sequences. The rest target genes were either not expressed in the samples analyzed (at least with the cutoff used), had no significant changes in expression, or changed their expression in the same direction as their targeting miRNAs. A comparison between iFIBs and tFIB samples revealed that the proportions of up-regulated (5–22%), down-regulated (17–25%), unchanged (53–69%), and non-expressed (5–10%) target mRNAs were rather similar for different miRNA families, independently of their behavior or numbers of their target genes (Figure 5). Notably, the average proportions of these groups of target mRNAs were similar between the up- and down-regulated miRNA families, and constituted 12 ± 4% and 13 ± 6% for the up-regulated, 22 ± 2% and 22 ± 4% for the down-regulated target genes, 58 ± 7% and 61 ± 5% for those with no changes in expression, and 6 ± 1% and 8 ± 2% for the non-expressed target genes, respectively.
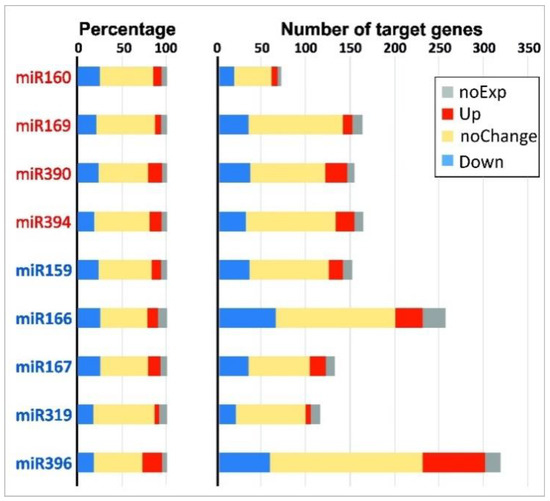
Figure 5.
Proportion (left) and abundance (right) of differentially expressed target genes for up- (miR160, 169, 390 and 394, listed in red) and down-regulated (miR159, 166, 167, 319, 396, listed in blue) miRNA families for pairwise comparisons between iFIBa and tFIB (log2FC ≥ 1 or ≤−1). The up-regulated target genes are presented in red, the down-regulated - in blue, the non-expressed in the samples analyzed—in grey, and the genes without significant changes in transcription - in yellow.
2.5. Comparison of miRNA and mRNA Expression Profiles at the Two Substages of Fiber Intrusive Elongation
To identify genes and miRNAs, the expression of which differs at earlier and later stages of intrusive elongation, we subdivided the zone of intrusive elongation into two parts. The first part (a) was located above the second part (b) (Figure 1), so that (a) contained shorter fibers that were closer to the onset of intrusive elongation. Both samples had very similar mRNA expression patterns. In total, 557 genes were differentially expressed in iFIBa as compared to iFIBb (|log2FC| ≥ 1, padj < 0.05); among them, 114 genes were up-regulated in iFIBa (Table S3). An additional cutoff filtered out genes that were not differentially expressed between cortical parenchyma and intrusively growing fibers (|log2FC| ≥ 1 between iFIBa&b and cPAR), leaving 369 genes, of which 66 were up-regulated.
Fourteen genes (Table 5), the mRNAs of which were enriched at the early stage of intrusive elongation in comparison with all other samples, were revealed. This most intriguing group contained genes for four putative homologs of transcription factors: a Myb domain protein 82 (Lus10038092), a basic helix-loop-helix (bHLH) DNA-binding superfamily protein (Lus10015909), an AT hook motif DNA-binding family protein (Lus10008497), and a growth-regulating factor 5 (GRF5, Lus10020352). In addition, the mRNA of Arabidopsis homolog encoding response regulator (ARR7, Lus10042938) was more abundant in iFIBa than in iFIBb and all other samples (Table 5). The largest difference in mRNA abundance between iFIBa and iFIBb was observed for a putative transducin/WD40 repeat-like protein (Lus10009510) and a dirigent protein 21 (Lus10016231), while the highest TGR value amongst the listed genes was found for a gene encoding putative acyl activating enzyme 12 (AAE12, Lus10020787). Of relevance to miRNA processing, a gene encoding a putative protein argonaute 7 (AGO7, Lus10037136) was up-regulated at the earlier stage of intrusive fiber elongation.
Table 5.
List of genes up-regulated in the iFIBa sample compared to all other samples (a cutoff |log2FC| value iFIBa vs. iFIBb ≥ 1 with padj < 0.05, as well as an additional filter |log2FC| iFIBa&b vs. cPAR ≥ 1; TGR of iFIBa > TGR of any other sample). Log2FC values with padj < 0.05 are highlighted in bold. A heatmap displays normalized gene expression values in the corresponding samples.
Amongst the 124 known flax miRNAs, only one—mi396b—was significantly down-regulated in iFIBa as compared to iFIBb (log2FC = −2.76, padj < 0.05). The predicted targets of this miRNA are growth-regulating factors 1 and 5 (GRF1, GRF5), as well as MAKR2 (a member of the membrane-associated kinase regulator gene family) (Table S2). GRF5 is also targeted by miR390 (up-regulated in iFIBa&b in comparison with the other samples, but without a significant difference between iFIBa and iFIBb), and by miR396a, miR396c, and miR396e (all down-regulated in iFIBa&b, as compared to the other samples, but with no significant difference between iFIBa and iFIBb).
3. Discussion
3.1. miRNAs and Their Predicted Targets That Are Important for Intrusive Growth of Phloem Fibers in Flax Stems
The role of small RNAs in the regulation of plant cell elongation is rather poorly characterized, especially in the case of intrusive growth. This is partly explained by difficulties in obtaining the sample for analysis. Intrusively growing phloem fibers are located deeply inside other stem tissues and are very prone to injury during their isolation procedure: they are already quite long, while still being surrounded by relatively weak primary cell walls only [1,5]. Intrusive growth of plant fibers is the key process of fiber development that determines the final length and width of each individual fiber, the number of cells in fiber bundles and their structure, as well as the strength of fiber connections within a bundle and with other surrounding cells [7,9,42]. The possibility to characterize this process at the cellular and molecular level is significant for fiber crops. The protocol we developed to isolate intrusively growing fibers using cryosectioning with subsequent laser microdissection will help achieve this characterization [11]. To figure out which miRNAs may be important at the stage of phloem fiber intrusive elongation, the expression patterns of all known flax miRNAs in different plant tissues, as well as in phloem fibers at different stages of development, were characterized.
Members of miR396, miR166, miR167, miR159, miR156, and miR319 families were significantly less abundant in phloem fibers during their intrusive growth as compared to the more advanced developmental stage when the thick tertiary cell wall is deposited. It is logical to assume that the up-regulation of these miRNAs at the advanced stage of fiber development could be required for the removal of transcripts which are essential for elongation, and, hence, completed their function. However further functional molecular genetic analysis is needed to check this hypothesis. It may be relevant for the growth regulating factors that were affected by miR396 (Table 3). However, the relationships between miRNAs and their target genes are not always straightforward: the expression of one gene might be regulated by more than one miRNA, and one miRNA is often able to regulate the expression of more than one target gene. For example, the low expression levels of miRNAs from the two families (miR166b and several representatives of miR396 family) in iFIB demonstrated a negative correlation with the high expression level of their predicted target Lus10012048. The putative Arabidopsis homolog of this gene encodes phragmoplast orienting kinesin 2 (Table 3), which was reported to be involved in the spatial control of cytokinesis [43]. The modulation of such control may be related to the development of multinuclearity, which takes place during intrusive elongation and is characteristic for primary phloem fibers [4,42].
Moreover, the expression levels of some predicted targets did not have an expected negative correlation with the expression level of the corresponding miRNA (Figure 5). This complex relation could be explained by several mechanisms. It is known that either sequence-specific degradation of target mRNAs or translational repression of the mRNA molecules occur depending on the degree of complementarity of the miRNA sequence with the target sequence [41]. Moreover, miRNAs can specifically activate translation under certain stress conditions, during development, and when cells exit the cell cycle and enter the quiescent stage [44]. The reversed role of miRNA may be coupled to the extent of base-pairing with the target mRNA, which is associated with the properties of the ribonucleoprotein complex [45,46]. In the case of miRNA effect on mRNA translation, the impact may not be visible at the level of transcriptome analysis. Additionally, the abundance of mRNA of a certain gene is the result of a combination of several factors, including activities of corresponding transcriptional regulators, an influence of more than one miRNA (Table 3), differences in the mechanisms, and the rate of mRNA decay [47]. Different combinations and hierarchy of such mechanisms may lead to the diversity of target mRNA abundance upon the specific change in miRNA level. Unidirectional changes in the expression of miRNAs and their target genes were also revealed in other studies, both in Arabidopsis [17] and flax [23]. The relative similarity in the proportion of various effects on the expression of genes targeted by different miRNA families (Figure 5) is to be explained in future studies.
3.2. Members of miR396 Family Are Down-Regulated in Intrusively Growing Phloem Fibers in Flax Stems
Several representatives of miR396 family were down-regulated during intrusive fiber elongation: their expression was the lowest as compared to the other samples (Table 2). This family is known to regulate the expression of genes for growth regulating factors (GRFs). So far, the importance of GRF was mainly reported for leaf development [48,49,50]. Overexpression of miR396 causes a drastic reduction in a cell number in Arabidopsis, while abolishing the regulation of GRF2 by miR396 promotes a moderate increase in organ size [49]. Moreover, miR396-targeted GRFs were required for Arabidopsis leaf adaxial–abaxial polarity and trichome formation, revealing the tight coordination of cell division and differentiation during leaf morphogenesis [50]. Probably, GRFs could also be involved in the regulation of fiber intrusive growth, since putative members of the GRF gene family were up-regulated in the apical part of flax stems. Although their expression subsequently decreased in fibers during intrusive elongation [11], it was rather tissue-specific because GRFs were expressed at a low level in parenchyma cells (Table 3).
Lus10032452 and Lus10042952 were among other predicted targets of miR396; they both are homologous to the gene for ATP binding microtubule motor family protein HINKEL (Table 3). In Arabidopsis, HINKEL is expressed in microspores and developing pollen. Together with TETRASPORE, another microtubule motor family protein, it is responsible for cell plate expansion defects at cytokinesis during pollen mitosis I [51]. HINKEL gene was also up-regulated during Arabidopsis pollen tube growth, which is also characterized as intrusive growth [52].
In flax down-regulation of miR396a/c was reported under saline, alkaline, and saline-alkaline stress, while transgenic rice plants overexpressing miR396c were more sensitive to salt stress [53]. Osmotic stress was suggested to be highly probable in intrusively elongating fibers, since their enlargement is mainly achieved by increasing the vacuole through the accumulation of osmolytes and the resulting inward water flux [11]. Hence, down-regulation of this miRNA and the resulting decreased sensitivity to osmotic stress seems quite reasonable.
A potential target of miR396a and miR396c is a flax homolog of Arabidopsis AGO7 (up-regulated in intrusively growing fibers). These both miRs were down-regulated in intrusively growing fibers as compared to the other tissues. AGO7 is a highly specialized argonaute protein that regulates the auxin-signaling pathway via production of trans-acting siRNAs (ta-siRNAs) by interacting with miR390, which targets TAS3 transcripts for the biogenesis of ta-siRNAs [54,55]. TAS3 ta-siRNAs target several AUXIN RESPONSE FACTOR genes that are involved in the regulation of developmental timing and lateral organ development [56]. Genetic interaction was also shown to exists between the ta-siRNAs biogenesis machinery and the miR396 network during leaf development [57]. The increased expression of miR390 in iFIBs as compared to tFIB suggests a possible role during fiber elongation as well.
3.3. Similar miRNA Families Are Related to Cell Elongation in Various Plant Tissues
Juvenile flax fibers had increased expression of miR160, miR169, miR390, miR394, miR399 compared to fibers with thickened cell walls (Table 2). In cotton seed hairs (trichomes with protrusive growth), miR167, miR396, and miR399 accumulation peaked during rapid elongation, the abundance of miR156 peaked at the end of cotton fiber elongation, and miR160 and miR390 were down-regulated [58]. In rice, miR169g, miR169p, and miR396c were highly accumulated in seedlings with active symplastic growth [59,60].
Several members of miR166 family were significantly down-regulated in phloem fibers during intrusive elongation as compared to the stage of cell wall thickening (Table 2). miR166 is an important regulator of stem apical meristem maintenance [61]. In Arabidopsis, argonaute 10 (AGO10) specifically sequesters miR166/165 to regulate shoot apical meristem (SAM) development [61]. Lus10039386 encoding a homolog of Arabidopsis AGO10 was up-regulated in intrusively growing fibers (Table S1). A similar situation was found for other targets of miR166-HD-ZIP III transcription factors required for the correct specification of the shoot and root apical meristems and procambial specification [61,62,63]. Putative members of HD-ZIP III group-homeobox-leucine zipper family proteins (ATHB8, 14 and 15) were also up-regulated in iFIBa&b (Table 3). Thus, miR166 down-regulation and the increased level of transcripts for some target genes could be the “vestige” from the SAM. However, the difference in expression of these genes with cortical parenchyma cells, which are located at the same stem level and are roughly at the same “age” as intrusively growing fibers, indicates that miR166 family and its targets can be essential during intrusive elongation. In flax, an increase in the expression level of miR166 in the apical part of the stem in comparison with the whole stem was shown previously [22]. However, the part of the flax stem bearing intrusively growing fibers was not considered in this prior study.
Despite some differences in miRNA expression in different samples of elongating plant cells (possibly caused by differences in the type of cell growth), miR396, miR166, miR167, miR169, miR160, miR156, miR319 gene families are likely important players in the process of elongation.
3.4. Genes Important at the Early Stage of Intrusive Elongation
Nothing is known about the mechanisms of intrusive growth initiation. To approach this problem, we compared mRNA and miRNA profiles in the fibers taken at the earlier and the later stage of elongation. Both samples had very similar gene expression patterns and rather limited number of genes and corresponding miRNAs that changed expression significantly (Table 5). Among them were genes, the homologs of which in Arabidopsis encode transducin/WD40, acyl activating enzyme 12 (AAE12), laccase 6, dirigent protein 21, glutamine dumper protein (GDU), transcription factors MYB82 and GRF5, and response regulator (ARR7). Members of the transducin/WD40 protein superfamily often function as molecular “hubs” mediating supramolecular interactions [64,65]. GDU are plant-specific membrane proteins that stimulate amino acid export by activating nonselective amino acid facilitators [66,67]. In Arabidopsis, MYB82 (AT5G52600) is expressed during trichome development [68]. GRF5 is a transcription activator that is strongly expressed in actively growing and developing tissues of Arabidopsis, including the shoot apical meristem [69]. An Arabidopsis homolog of this gene encodes two-component response regulator ARR7 that is involved in the His-to-Asp phosphorelay signal transduction system [70] and acts as a negative regulator of cytokinin signaling [71].
AAE12 has numerous functions and could be involved in the biosynthetic-oxidation of aromatic and cyclic plant hormones, such as jasmonic acid, auxin, and salicylic acid [72], in the accumulation of protective secondary metabolites, such as benzoyloxy glucosinolates [73]. Laccase 6 and dirigent protein 21 are usually associated with lignan and lignin biosynthesis [74,75], which does not take place in elongating cells. However, lac2 mutants showed compromised root elongation in Arabidopsis under PEG-induced dehydration conditions [76].
Other enzymes encoded by the up-regulated genes included homologs of Arabidopsis flavin-binding monooxygenase (probable indole-3-pyruvate monooxygenase YUCCA4 (YUC4, Lus10013125) that is involved in auxin biosynthesis and required for the formation of vascular tissues [77]), and heparan-alpha-glucosaminide N-acetyltransferase (Lus10021514).
Notably, fibers at the earlier stage of intrusive elongation were enriched in the transcripts of AGO7, which is involved in small RNA-mediated post-transcriptional gene silencing. It is the main component of the RNA-induced silencing complex (RISC) that binds to miRNAs for silencing the directed cleavage of homologous mRNAs to repress gene expression. AGO7 associates preferentially with miR390, which guides the cleavage of TAS3 precursor RNA [55]. AGO7 itself is a potential target for miR396a and miR396c. They both were down-regulated in intrusively growing fibers as compared to the other tissues. Thus, flax phloem fibers are enriched in several specific miRNAs at the beginning of intrusive elongation.
4. Materials and Methods
4.1. Plant Materials and Sample Preparation
Flax plants (Linum usitatissimum L., cultivar Mogilevsky from a collection of All-Russian Flax Research Institute, Torzhok) were grown in the open air under natural photoperiod in pots with 40 cm of soil and received daily watering. Samples for analysis were collected at the period of fast growth, when the stem height was 25–30 cm (40 days after sowing). The tissue sampling scheme for RNA-Seq analysis (Figure 1) was based on the characterized location of fibers in flax stems at different stages of development [3,5]. A set of samples consisted of: 1) intrusively growing phloem fibers with only primary cell walls (iFIBa and iFIBb) isolated from stem parts a (0.3–0.8 cm from the stem apex) and b (0.8–2.5 cm from the stem apex [11]) by laser microdissection; 2) symplastically growing cortex parenchyma (cPAR) isolated from the stem parts a and b by laser microdissection; 3) phloem fibers at the stage of tertiary cell wall deposition (tFIB) collected from a 5 cm long stem portion starting 1 cm below SP; and, 4) a xylem part (sXYL) that contained mainly cells with secondary cell wall, collected from the same stem portion as tFIB. The phloem fibers with tertiary cell walls were isolated from fiber-enriched stem peels by washing several times with 80 % ethanol in a mortar, while gently pressing with a pestle until other tissues and chlorophyll were removed (as described previously [78,79]). All collected samples were frozen in liquid nitrogen and stored at −80 °C until analysis.
4.2. Cryosectioning and Laser Microdissection
Flax stem pieces that included intrusively growing fibers and cortex parenchyma were cut off from the two parts of the stem (a and b) with a razor blade (Figure 1), then immediately frozen in liquid nitrogen and stored at −80 °C. Cryosectioning and fiber bundle isolation by laser microdissection followed the protocol [11]. Briefly, longitudinal stem cryosections (60-μm thick) were taken in a cryostat chamber (CM3050, Leica Microsystems, Wetzlar, Germany) at −20 °C and transferred onto a POL-membrane frame slide (Leica Microsystems). Microdissections were made by using a laser microdissection microscope (LMD7000, Leica Microsystems) at 10× magnification, the laser power of 37, the laser aperture of 11, and the laser speed of 4. Microdissected pieces were collected into the caps of 0.2-mL PCR tubes containing 20 μL of RNA lysis solution (RNAqueous-Micro RNA Isolation Kit, Ambion (Austin, TX, USA)) and stored at −80 °C. The identification of intrusively growing fibers was based on their localization in the stem (between the xylem and 5–6 cell layers of cortex parenchyma and epidermis), elongated cell shape, and their occurrence in bundles. Each final sample for RNA analysis (iFIBa, iFIBb, and cPAR) contained about 700 microdissected pieces obtained from 30 plants.
4.3. RNA Isolation, Library Preparation and Sequencing
All samples were analyzed by next generation sequencing (NGS) in two independent biological replicates. Microdissected pieces from samples iFIBa, iFIBb, and cPAR were transferred from the PCR tube with RNA lysis solution into a microcentrifuge tube with 300 μL of the same lysis buffer for a silica column-based purification. Total RNA was extracted using an RNAqueous-Micro RNA Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Total RNA elution was performed with 2 × 10 μL elution buffer preheated to 95 °C. Total RNA from tFIB and sXYL (5 plants per sample) was isolated using Trizol combined with a mirVana miRNA Isolation Kit (Thermo Fisher Scientific, Lithuania), according to the manufacturer’s instructions.
For all samples, any residual DNA was eliminated with a DNA-free DNA Removal kit (DNA-free DNA Removal kit, Waltham, MA, USA). RNA quantity and quality were analyzed by a Qubit fluorometer (Invitrogen, Carlsbad, CA, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The RNA integrity index (RIN) was 7–8.4, which provides sufficient integrity for NGS. Total RNA of each sample was used to prepare both RNA-library and small RNA library.
cDNA libraries were prepared from the total RNA of iFIBa, iFIBb, cPAR, tFIB, and sXYL samples (up to 1 µg) with a NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA) after selective depletion of ribosomal RNAs using a Ribo-Zero rRNA Removal Kit (Plant) (Illumina, San Diego, CA, USA), according to the manufacturer’s instructions.
Libraries of small RNA were prepared with a NEBNext Multiplex Small RNA Library Prep Set for Illumina (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s protocol. Quality control of the libraries was performed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Further size selection was performed using Agencourt AMPure XP (Beckman Coulter, Indianapolis, Indiana, USA) to select for 143–149 bp fragments.
Sequencing was performed using a high-throughput HiSeq 2500 platform (Illumina, San Diego, CA, USA) in the mode of 60 bp single-end reads using HiSeq SR Cluster Kit v4 cBot and HiSeq SBS Kit v4 50 cycles kits (Illumina, San Diego, CA, USA). The sequences were deposited into the National Center for Biotechnology Information (NCBI) ([9], BioProject: PRJNA475325).
4.4. Bioinformatic Analysis and Data Visualization
Adapter removal, quality trimming, and the removal of rRNAs, tRNAs, snRNAs and snoRNAs found in the RNACentral DB (a non-coding RNA sequence database) [80] were carried out using BBDuk utility of BBToolsv 37.02 [81].
4.4.1. RNA-seq Data Analysis
Clean reads for each sample were mapped onto the flax genome sequence scaffolds by using HISAT2 v2.1.083 [82] with default parameters using the option --rna-strandness R. A reference genome sequence v. 1.0 Linum usitatissimum and an annotation as gff3 file were downloaded from Phytozome v.12 [38,83]. In addition to 43,484 protein-coding genes in the whole-genome assembly of flax, two CESA7 [78] genes were integrated into the existing annotation and numbered as Lus10043485 and Lus10043486. Read count quantification for each gene was generated using HTSeq-count software [39] with default settings. For each gene total gene reads (TGR) was determined as the number of all reads that are mapped to this gene.
4.4.2. miRNA-seq Data Analysis
After preprocessing raw data, we employed cutadapt [84] to remove any reads shorter than 15nt and longer than 30nt. Filtered small RNA reads from given libraries were aligned to 124 flax miRNA precursor sequences downloaded from DB RNACentral (116 out of these were catalogued in miRBase (release 22.1; [85])) using bowtie2 v.2.2.9 [86] and then were counted to measure the expression of identified miRNAs by samtools v.1.3.1 [87].
4.4.3. Differential Expression Analysis
The R package DESeq2 v.1.14.1 [40] was used to perform the differential expression analysis of both mRNA and miRNA from all samples using TGR counts generated for each sample, as described above. DESeq’s estimateSizeFactors and estimateDispersions functions (with default options) were used to obtain normalization factors for each sample and to normalize TGR counts. The normalization was applied to all samples simultaneously to ensure that the expression values were comparable across samples. In addition, for mRNA-seq, we pre-filtered the normalized TGR counts by removing the genes with a TGR < 16 across all samples according to the recommendations of the sequencing quality control project [88]. The resulting mRNA dataset consisted of 30,922 genes that were used for differential expression analysis. Principal component analysis (PCA), as implemented in DESeq2, was performed to investigate similarities between individual samples. Both mRNAs and miRNAs with log2FC ≥ 2 and adjusted p-value < 0.05 (determined according to [40]) were considered as up-regulated; with log2FC ≤ − 2 and adjusted p-value < 0.05- as down-regulated. For comparison of iFIBa and iFIBb, the threshold value was lowered to log2FC ≥ 1. In order to identify the features of gene expression in phloem fibers at the stage of intrusive growth compared to the other flax tissues or phloem fibers at the stage of deposition of the tertiary cell wall, the datasets from iFIBa and iFIBb samples were considered as biological replicates as input to DESeq2 with the name iFIBa&b.
4.4.4. Hierarchical Clustering of Samples and miRNAs
Read counts after pre-filtering were normalized using the regularized-logarithm transformation or rlog [40], that stabilizes the variance across the mean, and the data become approximately homoscedastic [89]. The rlog-transformed values were used directly for computing the distances between samples, for making the PCA plot, and for clustering. The Euclidean distance and the Pearson correlation were used to group the samples and, respectively, to scale and group the genes using the R [90] and the hclust function. Ward.D2’s method was used in both cases. A dendrogram and a heatmap were generated using the R function heatmap.2 in the gplots R package. The cutree function of dendextend R package was used to cut the dendrogram into clusters.
4.5. Computational Prediction of Flax miRNA Targets
The plant small RNA target analysis server (psRNATarget, [91]) with the “user-submitted small RNAs” option, along with the Linum usitatissimum sequences library (200 v1.0) from Phytozome v12 for a reference cDNA library and scoring schema V2 (2017 release), were used to identify the miRNA targets by finding complementary matches between miRNA and target transcripts [37].
5. Conclusions
The miRNAs and their target genes differentially expressed during intrusive elongation of phloem fibers have been identified in samples collected by laser microdissection from flax stems. miR396s and their targets (e.g., GROWTH-REGULATING FACTORs) are especially promising candidates for further study. Several members of miR396 family were down-regulated during intrusive fiber elongation in comparison with the fibers at a more advanced stage of development and different cell types, indicating both tissue- and stage-specific expression. The complex network that involves miR396, AGO7, and miR390 might play an important regulatory role during intrusive elongation. Members of miR166 family that are usually associated with the regulation of shoot apical meristem maintenance and vascular tissue differentiation, may also regulate intrusive growth of phloem fibers, since the specific character of expression for miR166 and its targets (AGO10 and HD-ZIP III) was revealed at this stage of fiber development. The expression levels of miRNAs and their target genes did not match expectations for many target genes.
So far, it is quite difficult to judge about the degree of universality of the regulation network recruiting miRNAs during different types of growth (symplastic, intrusive, protrusive), first of all, due to the scarcity of this kind of information and difficulties in individual cell type sampling. Probably, some miRNAs are more “general” and are involved in elongation of different tissues, acting, for example, on hormone-regulated genes, like miR160 and miR167, that play a role in auxin signaling [92]. However, the involvement of distinct miRNAs in the regulation of certain growth types cannot be excluded as well. Further studies are needed to highlight the universality or specificity of miRNA action in different types of growth.
Supplementary Materials
The following are available online at http://www.mdpi.com/2223-7747/8/2/47/s1, Table S1: List of known flax miRNAs expressed in intrusively growing phloem fibers (iFIBa and iFIBb) compared with cortex parenchyma (cPAR), phloem fibers isolated at the stage of tertiary cell wall deposition (tFIB), and xylem (sXYL); Table S2. List of expressed flax genes in intrusively growing fibers (iFIBa and iFIBb) compared with cortex parenchyma (cPAR), phloem fibers isolated at the stage of tertiary cell wall deposition (tFIB) and xylem samples, enriched in secondary cell walls (sXYL) (a cutoff TGR > 16 in at least one sample); Table S3 List of differentially expressed genes in the iFIBa sample compared to the other samples analyzed (the cutoff |log2FC| values iFIBa vs. iFIBb > 1 with padj < 0.05). A heatmap displays the normalized gene expression values in the corresponding samples.
Author Contributions
Conceptualization, T.G.; formal analysis, O.G. and A.T.; investigation, O.G., T.C. and N.G.; methodology, T.C.; resources, D.S.; supervision, T.G.; visualization, T.C. and N.M.; writing—original draft, O.G. and N.M.; writing—review & editing, T.G.
Funding
This research was partially supported by the Russian Science Foundation [#16–14–10256]. D.S. acknowledges Saint Petersburg State University for the grant 1.40.492.2017.
Acknowledgments
The authors thank the Research park of Saint Petersburg State University: Center for Molecular and Cell Technologies, and Chromas Core Facility, for the access to cryosection and laser microdissection facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gorshkova, T.; Brutch, N.; Chabbert, B.; Deyholos, M.; Hayashi, T.; Lev-Yadun, S.; Mellerowicz, E.J.; Morvan, C.; Neutelings, G.; Pilate, G. Plant Fiber Formation: State of the Art, Recent and Expected Progress, and Open Questions. Crit. Rev. Plant Sci. 2012, 31, 201–228. [Google Scholar] [CrossRef]
- Mokshina, N.; Chernova, T.; Galinousky, D.; Gorshkov, O.; Gorshkova, T. Key Stages of Fiber Development as Determinants of Bast Fiber Yield and Quality. Fibers 2018, 6, 20. [Google Scholar] [CrossRef]
- Gorshkova, T.A.; Sal’nikov, V.V.; Chemikosova, S.B.; Ageeva, M.V.; Pavlencheva, N.V.; van Dam, J.E.G. The snap point: A transition point in Linum usitatissimum bast fiber development. Ind. Crops Prod. 2003, 18, 213–221. [Google Scholar] [CrossRef]
- Esau, K. Vascular differentiation in the vegetative shoot of Linum. III. The origin of bast fibers. Am. J. Bot. 1943, 30, 579–586. [Google Scholar] [CrossRef]
- Ageeva, M.V.; Petrovská, B.; Kieft, H.; Sal’nikov, V.V.; Snegireva, A.V.; van Dam, J.E.; van Veenendaal, W.L.; Emons, A.M.; Gorshkova, T.A.; van Lammeren, A.A. Intrusive growth of flax phloem fibers is of intercalary type. Planta 2005, 222, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Lev-Yadun, S. Intrusive growth—The plant analog of dendrite and axon growth in animals. New Phytol. 2001, 150, 508–512. [Google Scholar] [CrossRef]
- Snegireva, A.; Chernova, T.; Ageeva, M.; Lev-Yadun, S.; Gorshkova, T. Intrusive growth of primary and secondary phloem fibres in hemp stem determines fibre-bundle formation and structure. AoB Plants 2015, 7, plv061. [Google Scholar] [CrossRef]
- Gorshkova, T.; Chernova, T.; Mokshina, N.; Ageeva, M.; Mikshina, P. Plant ‘muscles’: Fibers with a tertiary cell wall. New Phytol. 2018, 218, 66–72. [Google Scholar] [CrossRef]
- Gorshkova, T.; Mikshina, P.; Petrova, A.; Chernova, T.; Mokshina, N.; Gorshkov, O. Plants at Bodybuilding: Development of Plant “Muscles”. In Plant Biomechanics; Geitmann, A., Gril, J., Eds.; Springer: Cham, Switzerland, 2018; pp. 141–163. ISBN 978-3-319-79098-5. [Google Scholar]
- Gorshkov, O.; Mokshina, N.; Gorshkov, V.; Chemikosova, S.; Gogolev, Y.; Gorshkova, T. Transcriptome portrait of cellulose-enriched flax fibers at advanced stage of specialization. Plant Mol. Biol. 2017, 93, 431–449. [Google Scholar] [CrossRef]
- Gorshkova, T.; Chernova, T.; Mokshina, N.; Gorshkov, V.; Kozlova, L.; Gorshkov, O. Transcriptome Analysis of Intrusively Growing Flax Fibers Isolated by Laser Microdissection. Sci. Rep. 2018, 8, 14570. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegansheterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 5, 855–862. [Google Scholar] [CrossRef]
- D’Ario, M.; Griffiths-Jones, S.; Kim, M. Small RNAs: Big Impact on Plant Development. Trends Plant Sci. 2017, 22, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Brant, E.J.; Budak, H. Plant Small Non-coding RNAs and Their Roles in Biotic Stresses. Front. Plant Sci. 2018, 9, 1038. [Google Scholar] [CrossRef]
- Singh, A.; Gautam, V.; Singh, S.; Sarkar Das, S.; Verma, S.; Mishra, V.; Mukherjee, S.; Sarkar, A.K. Plant small RNAs: Advancement in the understanding of biogenesis and role in plant development. Planta 2018, 248, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Bartel, D.P. Antiquity of microRNAs and their targets in land plants. Plant Cell 2005, 17, 1658–1673. [Google Scholar] [CrossRef] [PubMed]
- Barozai, M.Y. In silico identification of micrornas and their targets in fiber and oil producing plant flax (Linum usitatissimum L.). Pak. J. Bot. 2012, 44, 1357–1362. [Google Scholar]
- Zinati, Z.; Shamloo-Dashtpagerdi, R.; Behpouri, A. In silico identification of miRNAs and their target genes and analysis of gene co-expression network in saffron (Crocus sativus L.) stigma. Mol. Biol. Res. Commun. 2016, 5, 233–246. [Google Scholar]
- Samad, A.F.A.; Nazaruddin, N.; Murad, A.M.A.; Jani, J.; Zainal, Z.; Ismail, I. Deep sequencing and in silico analysis of small RNA library reveals novel miRNA from leaf Persicaria minor transcriptome. 3 Biotech 2018, 8, 136. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Zhang, Z.; Ling, Y.; Xu, W.; Su, Z. PNRD: A plant non-coding RNA database. Nucleic Acids Res. 2015, 43, D982–D989. [Google Scholar] [CrossRef]
- Neutelings, G.; Fénart, S.; Lucau-Danila, A.; Hawkins, S. Identification and characterization of mirnas and their potential targets in flax. J. Plant Physiol. 2012, 169, 1754–1766. [Google Scholar] [CrossRef] [PubMed]
- Barvkar, V.T.; Pardeshi, V.C.; Kale, S.M.; Qiu, S.Q.; Rollins, M.; Datla, R.; Gupta, V.S.; Kadoo, N.Y. Genome-wide identification and characterization of microRNA genes and their targets in flax (Linum usitatissimum): Characterization of flax miRNA genes.). Planta 2013, 237, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wu, G.; Yuan, H.; Cheng, L.; Zhao, D.; Huang, W.; Zhang, S.; Zhang, L.; Chen, H.; Zhang, J.; et al. Identification and characterization of miRNAs and targets in flax (Linum usitatissimum) under saline, alkaline, and saline-alkaline stresses. BMC Plant Biol. 2016, 16, 124. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, N.V.; Dmitriev, A.A.; Belenikin, M.S.; Koroban, N.V.; Speranskaya, A.S.; Krinitsina, A.A.; Krasnov, G.S.; Lakunina, V.A.; Snezhkina, A.V.; Sadritdinova, A.F.; et al. Identification, Expression Analysis, and Target Prediction of Flax Genotroph MicroRNAs Under Normal and Nutrient Stress Conditions. Front. Plant Sci. 2016, 7, 399. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, A.A.; Kudryavtseva, A.V.; Bolsheva, N.L.; Zyablitsin, A.V.; Rozhmina, T.A.; Kishlyan, N.V.; Krasnov, G.S.; Speranskaya, A.S.; Krinitsina, A.A.; Sadritdinova, A.F.; et al. miR319, miR390, and miR393 Are Involved in Aluminum Response in Flax (Linum usitatissimum L.). BioMed Res. Int. 2017, 2017, 6915898. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Meyers, B.C. Revisiting criteria for plant microRNA annotation in the era of big data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef]
- Puzey, J.R.; Karger, A.; Axtell, M.; Kramer, E.M. Deep annotation of Populustrichocarpa microRNAs from diverse tissue sets. PLoS ONE 2012, 7, e33034. [Google Scholar] [CrossRef]
- Zhang, R.; Marshall, D.; Bryan, G.J.; Hornyik, C. Identification and characterization of miRNA transcriptome in potato by high-throughput sequencing. PLoS ONE 2013, 8, e57233. [Google Scholar] [CrossRef]
- He, X.; Zheng, W.; Cao, F.; Wu, F. Identification and comparative analysis of the microRNA transcriptome in roots of two contrasting tobacco genotypes in response to cadmium stress. Sci. Rep. 2016, 6, 32805. [Google Scholar] [CrossRef]
- Peng, C.; Chen, X.; Wang, X.; Xu, X.; Wei, W.; Wang, C.; Xu, J. Comparative analysis of miRNA expression profiles in transgenic and non-transgenic rice using miRNA-Seq. Sci. Rep. 2018, 8, 338. [Google Scholar] [CrossRef]
- Ma, C.; Yang, J.; Cheng, Q.; Mao, A.; Zhang, J.; Wang, S.; Weng, Y.; Wen, C. Comparative analysis of miRNA and mRNA abundance in determinate cucumber by high-throughput sequencing. PLoS ONE 2018, 13, e0190691. [Google Scholar] [CrossRef]
- Li, Y.; Alonso-Peral, M.; Wong, G.; Wang, M.B.; Millar, A.A. Ubiquitous miR159 repression of MYB33/65 in Arabidopsis rosettes is robust and is not perturbed by a wide range of stresses. BMC Plant Biol. 2016, 16, 179. [Google Scholar] [CrossRef] [PubMed]
- Schommer, C.; Bresso, E.G.; Spinelli, S.V.; Palatnik, J.F. Role of MicroRNA miR319 in Plant Development. In MicroRNAs in Plant Development and Stress Responses. Signaling and Communication in Plants; Sunkar, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 15, pp. 29–47. ISBN 978-3-642-27383-4. [Google Scholar]
- Pilon, M. The copper microRNAs. New Phytol. 2017, 213, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Ercoli, M.F.; Rojas, A.M.; Debernardi, J.M.; Palatnik, J.F.; Rodriguez, R.E. Control of cell proliferation and elongation by miR396. Plant Signal. Behav. 2016, 11, e1184809. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Wang, Z.W.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef]
- Snegireva, A.V.; Ageeva, M.V.; Amenitskii, S.I.; Chernova, T.E.; Ebskamp, M.; Gorshkova, T.A. Intrusive growth of sclerenchyma fibers. Russ. J. Plant Physiol. 2010, 57, 342–355. [Google Scholar] [CrossRef]
- Lipka, E.; Gadeyne, A.; Stöckle, D.; Zimmermann, S.; De Jaeger, G.; Ehrhardt, D.W.; Kirik, V.; Van Damme, D.; Müller, S. The Phragmoplast-Orienting Kinesin-12 Class Proteins Translate the Positional Information of the Preprophase Band to Establish the Cortical Division Zone in Arabidopsis thaliana. Plant Cell 2014, 26, 2617–2632. [Google Scholar] [CrossRef] [PubMed]
- Rusk, N. When microRNAs activate translation. Res. Highlights 2008, 5, 122–123. [Google Scholar] [CrossRef]
- Vasudevan, S. Posttranscriptional upregulation by microRNAs. Wiley Interdiscip. Rev. RNA 2012, 3, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Saraiya, A.A.; Li, W.; Wang, C.C. Transition of a microRNA from repressing to activating translation depending on the extent of base pairing with the target. PLoS ONE 2013, 8, e55672. [Google Scholar] [CrossRef]
- Narsai, R.; Howell, K.A.; Millar, A.H.; O’Toole, N.; Small, I.; Whelan, J. Genome-wide analysis of mRNA decay rates and their determinants in Arabidopsis thaliana. Plant Cell 2007, 19, 3418–3436. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.M.; Song, Y.; Chen, Z.X.; Yu, D.Q. Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol. Plant. 2009, 136, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.E.; Mecchia, M.A.; Debernardi, J.M.; Schommer, C.; Weigel, D.; Palatnik, J.F. Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development 2010, 137, 103–112. [Google Scholar] [CrossRef]
- Wang, L.; Gu, X.; Xu, D.; Wang, W.; Wang, H.; Zeng, M.; Chang, Z.; Huang, H.; Cui, X. miR396-targeted AtGRF transcription factors are required for coordination of cell division and differentiation during leaf development in Arabidopsis. J. Exp. Bot. 2011, 62, 761–773. [Google Scholar] [CrossRef]
- Oh, S.A.; Bourdon, V.; Das ‘Pal, M.; Dickinson, H.; Twell, D. Arabidopsis kinesins HINKEL and TETRASPORE act redundantly to control cell plate expansion during cytokinesis in the male gametophyte. Mol. Plant 2008, 1, 794–799. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.Z.; Song, L.F.; Zou, J.J.; Su, Z.; Wu, W.H. Transcriptome analyses show changes in gene expression to accompany pollen germination and tube growth in Arabidopsis. Plant Physiol. 2008, 148, 1201–1211. [Google Scholar] [CrossRef]
- Gao, P.; Bai, X.; Yang, L.; Lv, D.; Li, Y.; Cai, H.; Ji, W.; Guo, D.; Zhu, Y. Over-expression of osa-MIR396c decreases salt and alkali stress tolerance. Planta 2010, 231, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Vaucheret, H.; Rajagopalan, R.; Lepers, C.; Gasciolli, V.; Mallory, A.C.; Hilbert, J.L.; Bartel, D.P.; Crété, P. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol. Cell 2004, 16, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, T.A.; Howell, M.D.; Cuperus, J.T.; Li, D.; Hansen, J.E.; Alexander, A.L.; Chapman, E.J.; Fahlgren, N.; Allen, E.; Carrington, J.C. Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell 2008, 133, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Qi, Y. RNAi in plants: An argonaute-centered view. Plant Cell 2016, 28, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Mecchia, M.A.; Debernardi, J.M.; Rodriguez, R.E.; Schommer, C.; Palatnik, J.F. MicroRNA miR396 and RDR6 synergistically regulate leaf development. Mech. Dev. 2013, 130, 2–13. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Y.; Liu, J.-Y. Identification and Profiling of microRNAs Expressed in Elongating Cotton Fibers Using Small RNA Deep Sequencing. Front. Plant Sci. 2016, 7, 1722. [Google Scholar] [CrossRef]
- Shao, C.; Ma, X.; Xu, X.; Meng, Y. Identification of the highly accumulated microRNA*s in Arabidopsis (Arabidopsis thaliana) and rice (Oryza sativa). Gene 2012, 515, 123–127. [Google Scholar] [CrossRef]
- Hu, W.; Wang, T.; Yue, E.; Zheng, S.; Xu, J.-H. Flexible microRNA arm selection in rice. Biochem. Biophys. Res. Commun. 2014, 447, 526–530. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, F.; Wang, R.; Zhou, X.; Sze, S.H.; Liou, L.W.; Barefoot, A.; Dickman, M.; Zhang, X. Arabidopsis Argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell 2011, 145, 242–256. [Google Scholar] [CrossRef]
- Miyashima, S.; Honda, M.; Hashimoto, K.; Tatematsu, K.; Hashimoto, T.; Sato-Nara, K.; Okada, K.; Nakajima, K. A comprehensive expression analysis of the Arabidopsis MICRORNA165/6 gene family during embryogenesis reveals a conserved role in meristem specification and a non-cell-autonomous function. Plant Cell Physiol. 2013, 54, 375–384. [Google Scholar] [CrossRef]
- Schuetz, M.; Smith, R.; Ellis, B. Xylem tissue specification, patterning, and differentiation mechanisms. J. Exp. Bot. 2013, 64, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Puranik, S.; Prasad, M. Structure and regulatory networks of WD40 protein in plants. J. Plant Biochem. Biotechnol. 2012, 21 (Suppl. 1), 32–39. [Google Scholar] [CrossRef]
- Guerriero, G.; Hausman, J.-F.L.; Ezcurra, I. WD40-Repeat Proteins in Plant Cell Wall Formation: Current Evidence and Research Prospects. Front. Plant Sci. 2015, 6, 1112. [Google Scholar] [CrossRef] [PubMed]
- Pilot, G.; Stransky, H.; Bushey, D.F.; Pratelli, R.; Ludewig, U.; Wingate, V.P.M.; Frommer, W.B. Overexpression of GLUTAMINE DUMPER1 Leads to Hypersecretion of Glutamine from Hydathodes of Arabidopsis Leaves. Plant Cell 2004, 16, 1827–1840. [Google Scholar] [CrossRef] [PubMed]
- Pratelli, R.; Voll, L.M.; Horst, R.J.; Frommer, W.B.; Pilot, G. Stimulation of nonselective amino acid export by glutamine dumper proteins. Plant Physiol. 2010, 152, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; He, H.; Li, Y.; Ai, Q.; Yu, D. MYB82 functions in regulation of trichome development in Arabidopsis. J. Exp. Bot. 2014, 65, 3215–3223. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, G.; Kim, G.T.; Tsukaya, H. The transcription factor AtGRF5 and the transcription coactivator AN3 regulate cell proliferation in leaf primordia of Arabidopsis thaliana. Plant J. 2005, 43, 68–78. [Google Scholar] [CrossRef]
- Imamura, A.; Hanaki, N.; Umeda, H.; Nakamura, A.; Suzuki, T.; Ueguchi, C.; Mizuno, T. Response regulators implicated in His-to-Asp phosphotransfer signaling in Arabidopsis. Proc. Natl. Acad. Sci. USA 1998, 95, 2691–2696. [Google Scholar] [CrossRef]
- To, J.P.C.; Haberer, G.; Ferreira, F.J.; Deruere, J.; Mason, M.G.; Schaller, G.E.; Alonso, J.M.; Ecker, J.R.; Kieber, J.J. Type-A Arabidopsis response regulators are partially redundant negative regulators of cytokinin signaling. Plant Cell 2004, 16, 658–671. [Google Scholar] [CrossRef]
- Shockey, J.M.; Fulda, M.S.; Browse, J. Arabidopsis Contains a Large Superfamily of Acyl-Activating Enzymes. Phylogenetic and Biochemical Analysis Reveals a New Class of Acyl-Coenzyme A Synthetases. Plant Physiol. 2003, 132, 1065–1076. [Google Scholar] [CrossRef]
- Reichelt, M.; Brown, P.D.; Schneider, B.; Oldham, N.J.; Stauber, E.; Tokuhisa, J.; Kliebenstein, D.J.; Mitchell-Olds, T.; Gershenzon, J. Benzoic acid glucosinolate esters and other glucosinolates from Arabidopsis thaliana. Phytochemistry 2002, 59, 663–671. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Whetten, R.; Bao, W.; Chen, C.-L.; Sederoff, R.R. The role of laccase in lignification. Plant J. 1993, 4, 751–757. [Google Scholar] [CrossRef]
- Paniagua, C.; Bilkova, A.; Jackson, P.; Dabravolski, S.; Riber, W.; Didi, V.; Houser, J.; Gigli-Bisceglia, N.; Wimmerova, M.; Budínská, E.; et al. Dirigent proteins in plants: Modulating cell wall metabolism during abiotic and biotic stress exposure. J. Exp. Bot. 2017, 68, 3287–3301. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Davis, E.J.; Ballif, J.; Liang, M.; Bushman, E.; Haroldsen, V.; Torabinejad, J.; Wu, Y. Mutant identification and characterization of the laccase gene family in Arabidopsis. J. Exp. Bot. 2006, 57, 2563–2569. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Dai, X.; Zhao, Y. Auxin biosynthesis by the YUCCA flavin monooxygenases controls the formation of floral organs and vascular tissues in Arabidopsis. Genes Dev. 2006, 20, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Mokshina, N.; Gorshkova, T.; Deyholos, M.K. Chitinase-like (CTL) and cellulose synthase (CESA) gene expression in gelatinous-type cellulosic walls of flax (Linum usitatissimum L.) bast fibers. PLoS ONE 2014, 9, e97949. [Google Scholar] [CrossRef] [PubMed]
- Mokshina, N.; Gorshkov, O.; Ibragimova, N.; Chernova, T.; Gorshkova, T. Cellulosic fibres of flax recruit both primary and secondary cell wall cellulose synthases during deposition of thick tertiary cell walls and in the course of graviresponse. Funct. Plant Biol. 2017, 44, 820–831. [Google Scholar] [CrossRef]
- Petrov, A.I.; Kay, S.; Kalvari, I.; Howe, K.L.; Gray, K.A.; Bruford, E.A.; Kersey, P.J.; Cochrane, G.; Finn, R.D. RNAcentral: A comprehensive database of non-coding RNA sequences. Nucleic Acids Res. 2017, 45, D128–D134. [Google Scholar] [CrossRef]
- BBTools. Available online: https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk-guide/ (accessed on 21 January 2019).
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- SEQC/MAQC-III Consortium. A comprehensive assessment ofRNA-seq accuracy, reproducibility and information content by thesequencing quality control consortium. Nat. Biotechnol. 2014, 32, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Anders, S.; Kim, V.; Huber, W. RNA-Seq workflow: Gene-level exploratory analysis and differential expression. F1000Research 2015, 4, 1070. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014; ISBN 3-900051-07-0. Available online: http://www.R-project.org (accessed on 12 December 2018).
- psRNATarget: A Plant Small RNA Target Analysis Server (2017 Update). Available online: http://plantgrn.noble.org/psRNATarget/) (accessed on 28 November 2018).
- Havelda, Z. Biogenesis and Function of Plant microRNAs. In Regulation of Gene Expression by Small RNAs, 1st ed.; Gaur, R.K., Rossi, J.J., Eds.; CRC Press: Boca Raton, FL, USA, 2009; pp. 173–196. ISBN 9781138111738. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).