


Impact of Farming System on Soil Microbial Communities Associated with Common Bean in a Region of Northern Spain


Abstract

1. Introduction
2. Results
2.1. Soil Physicochemical Features and Environmental Data
2.2. Prokaryotic (16S) and Fungal (ITS) Sequencing
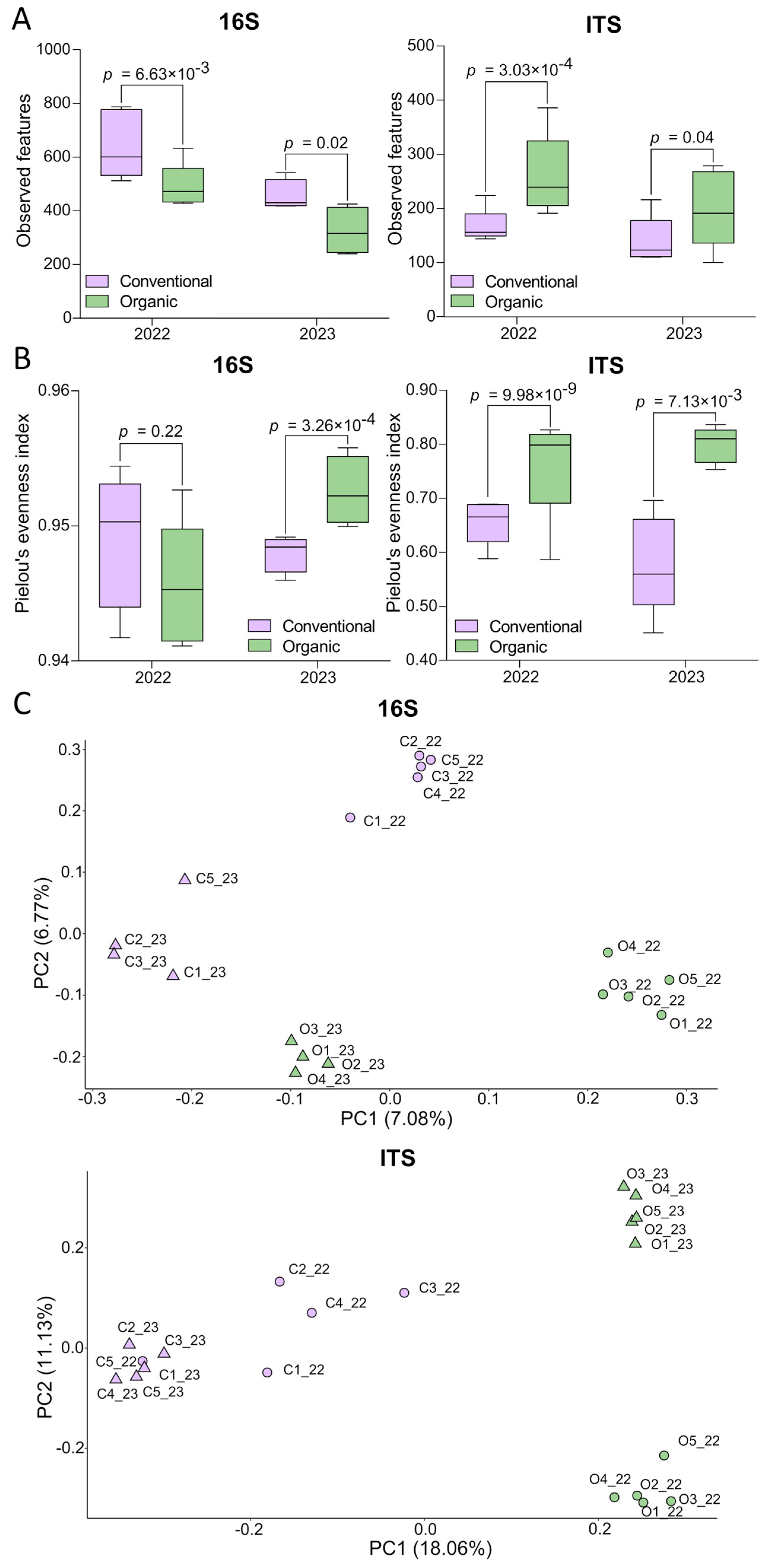
2.3. Alpha and Beta Diversity
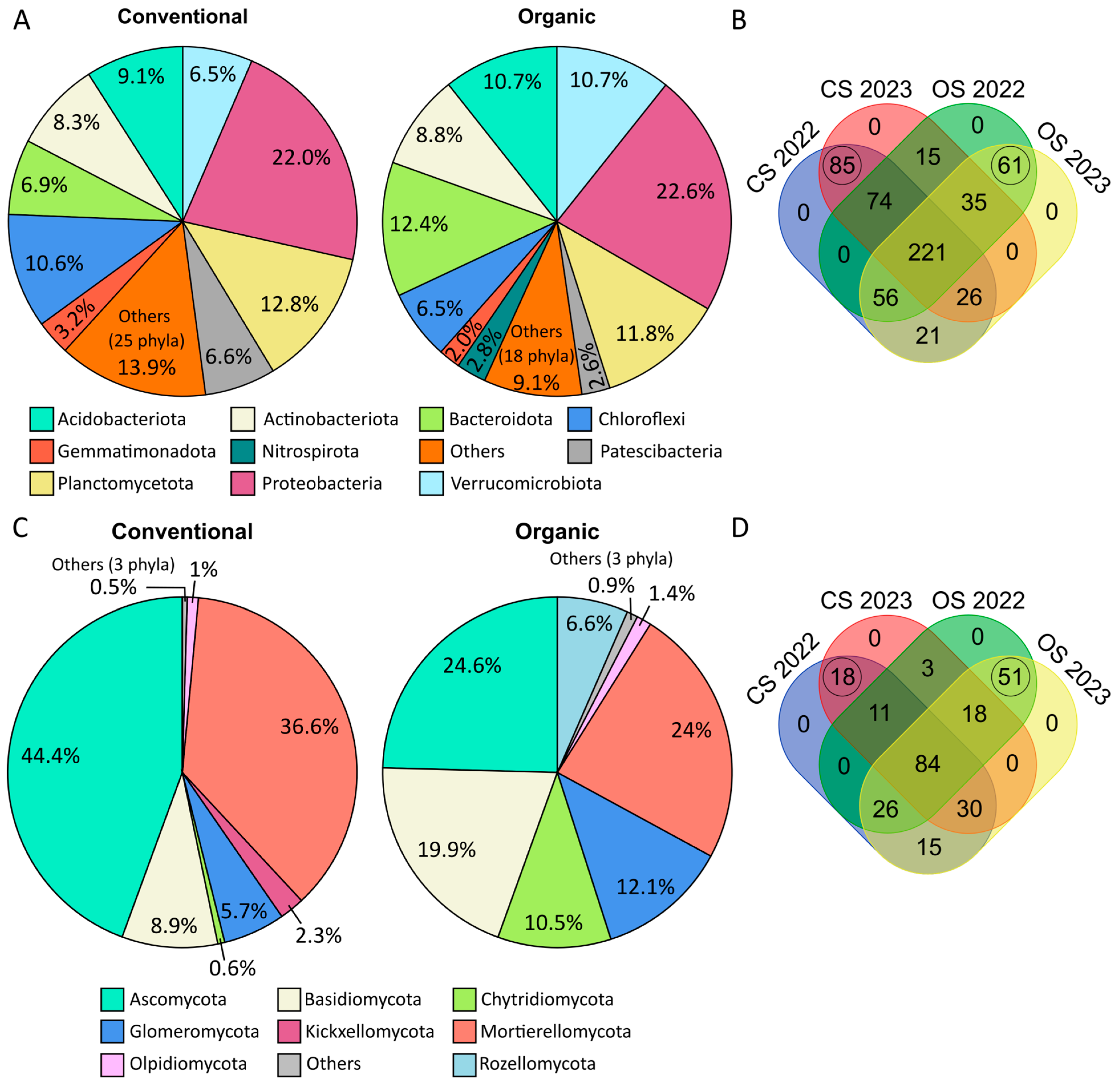
2.4. Taxonomy
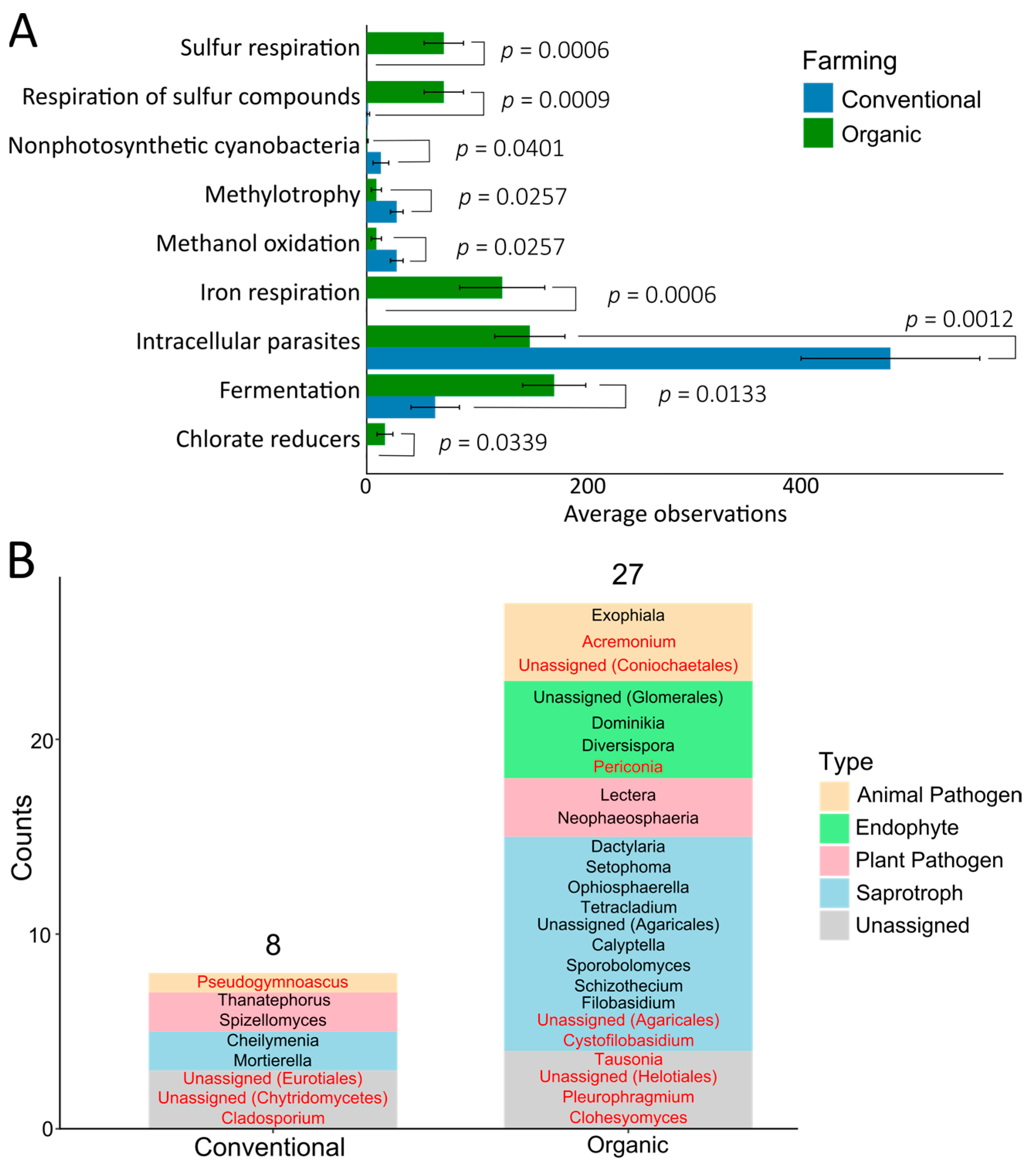
2.5. Differences in Microbial Communities and Functional Analyses
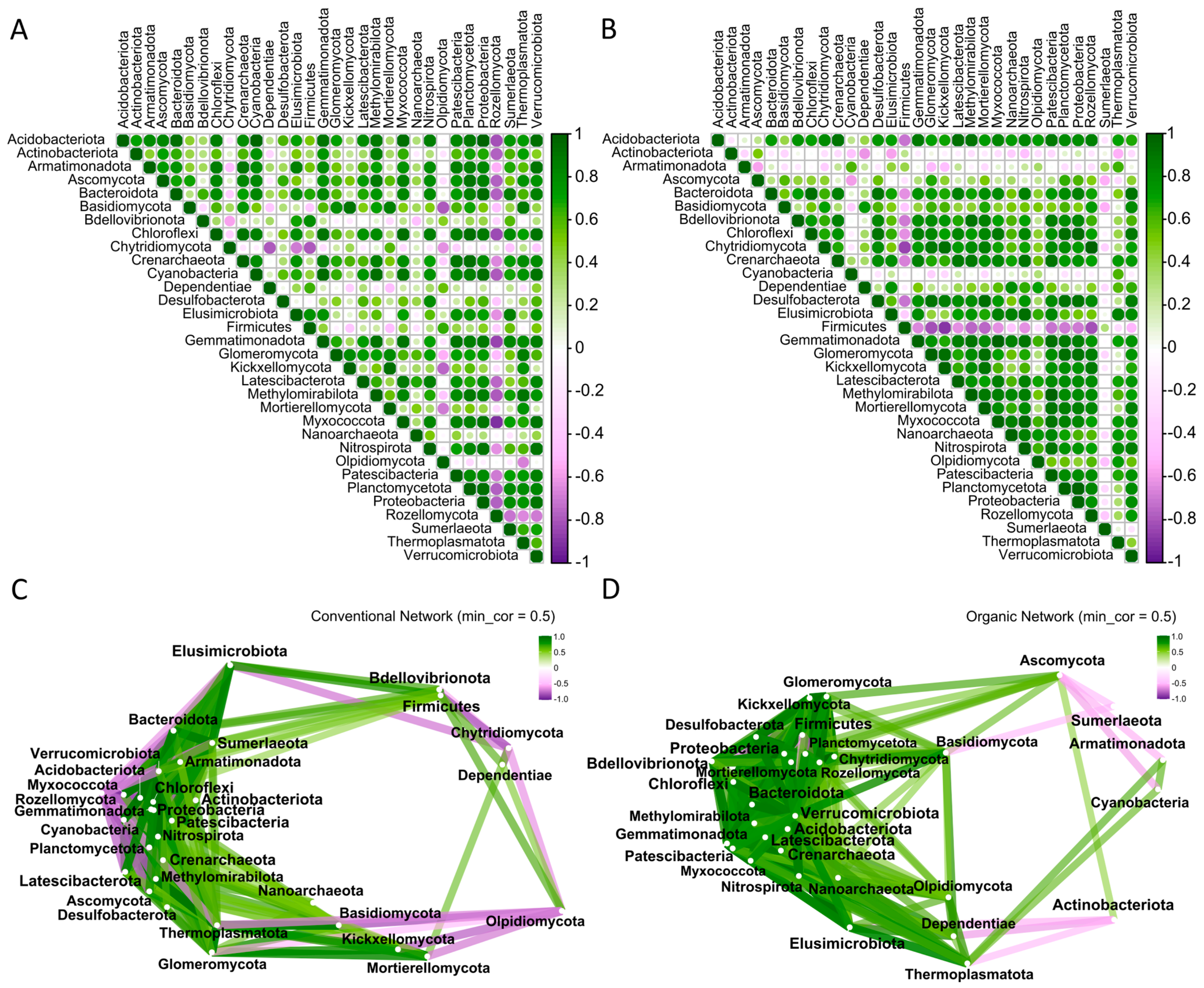
2.6. Interactions Between the Main Fungal and Prokaryotic Phyla
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Soil Sampling for Metabarcoding Analysis
4.3. Soil Physicochemical Analyses and Soil Environmental Data
4.4. Soil DNA Extraction and Sequencing
4.5. Bioinformatics Processing
4.6. Diversity and Statistical Analysis
4.7. Functional Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dixon, J.; Li, L.; Amede, T. A Century of Farming Systems. Part 1: Concepts and Evolution. Farming Syst. 2023, 1, 100055. [Google Scholar] [CrossRef]
- Telo da Gama, J. The Role of Soils in Sustainability, Climate Change, and Ecosystem Services: Challenges and Opportunities. Ecologies 2023, 4, 552–567. [Google Scholar] [CrossRef]
- Bünemann, E.K.; Bongiorno, G.; Bai, Z.; Creamer, R.E.; De Deyn, G.; de Goede, R.; Fleskens, L.; Geissen, V.; Kuyper, T.W.; Mäder, P.; et al. Soil Quality—A Critical Review. Soil Biol. Biochem. 2018, 120, 105–125. [Google Scholar] [CrossRef]
- Wagg, C.; Schlaeppi, K.; Banerjee, S.; Kuramae, E.E.; van der Heijden, M.G.A. Fungal-Bacterial Diversity and Microbiome Complexity Predict Ecosystem Functioning. Nat. Commun. 2019, 10, 4841. [Google Scholar] [CrossRef]
- Jansson, J.K.; McClure, R.; Egbert, R.G. Soil Microbiome Engineering for Sustainability in a Changing Environment. Nat. Biotechnol. 2023, 41, 1716–1728. [Google Scholar] [CrossRef]
- FAO. Towards a Definition of Soil Health. In Intergovernmental Technical Panel on Soils; Soil Letters; FAO: Rome, Italy, 2020. [Google Scholar]
- Hartmann, M.; Six, J. Soil Structure and Microbiome Functions in Agroecosystems. Nat. Rev. Earth Environ. 2022, 4, 4–18. [Google Scholar] [CrossRef]
- Harrold, L.R.; Field, T.S.; Gurwitz, J.H. Knowledge, Patterns of Care, and Outcomes of Care for Generalists and Specialists. J. Gen. Intern. Med. 1999, 14, 499–511. [Google Scholar] [CrossRef]
- Xiong, W.; Li, R.; Guo, S.; Karlsson, I.; Jiao, Z.; Xun, W.; Kowalchuk, G.A.; Shen, Q.; Geisen, S. Microbial Amendments Alter Protist Communities within the Soil Microbiome. Soil Biol. Biochem. 2019, 135, 379–382. [Google Scholar] [CrossRef]
- Dumack, K.; Fiore-Donno, A.M.; Bass, D.; Bonkowski, M. Making Sense of Environmental Sequencing Data: Ecologically Important Functional Traits of the Protistan Groups Cercozoa and Endomyxa (Rhizaria). Mol. Ecol. Resour. 2020, 20, 398–403. [Google Scholar] [CrossRef]
- Tedersoo, L.; Bahram, M.; Põlme, S.; Kõljalg, U.; Yorou, N.S.; Wijesundera, R.; Ruiz, L.V.; Vasco-Palacios, A.M.; Thu, P.Q.; Suija, A.; et al. Global Diversity and Geography of Soil Fungi. Science (1979) 2014, 346, 1256688. [Google Scholar] [CrossRef]
- Philippot, L.; Chenu, C.; Kappler, A.; Rillig, M.C.; Fierer, N. The Interplay between Microbial Communities and Soil Properties. Nat. Rev. Microbiol. 2023, 22, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Suman, J.; Rakshit, A.; Ogireddy, S.D.; Singh, S.; Gupta, C.; Chandrakala, J. Microbiome as a Key Player in Sustainable Agriculture and Human Health. Front. Soil Sci. 2022, 2, 821589. [Google Scholar] [CrossRef]
- Moreira, H.; Pereira, S.I.A.; Vega, A.; Castro, P.M.L.; Marques, A.P.G.C. Synergistic Effects of Arbuscular Mycorrhizal Fungi and Plant Growth-Promoting Bacteria Benefit Maize Growth under Increasing Soil Salinity. J. Environ. Manag. 2020, 257, 109982. [Google Scholar] [CrossRef] [PubMed]
- Abdelfattah, A.; Malacrinò, A.; Wisniewski, M.; Cacciola, S.O.; Schena, L. Metabarcoding: A Powerful Tool to Investigate Microbial Communities and Shape Future Plant Protection Strategies. Biol. Control 2018, 120, 1–10. [Google Scholar] [CrossRef]
- Semenov, M.V. Metabarcoding and Metagenomics in Soil Ecology Research: Achievements, Challenges, and Prospects. Biol. Bull. Rev. 2021, 11, 40–53. [Google Scholar] [CrossRef]
- Labouyrie, M.; Ballabio, C.; Romero, F.; Panagos, P.; Jones, A.; Schmid, M.W.; Mikryukov, V.; Dulya, O.; Tedersoo, L.; Bahram, M.; et al. Patterns in Soil Microbial Diversity across Europe. Nat. Commun. 2023, 14, 3311. [Google Scholar] [CrossRef]
- Kalyan, K.; Gurjar, D.; Kumar, B.; Kumar, T.V. The Role of Grain Legumes in Enhancing Soil Health and Promoting Sustainable Agricultural Practices: A Review. J. Exp. Agric. Int. 2024, 46, 344–354. [Google Scholar] [CrossRef]
- Añazco, C.; Ojeda, P.G.; Guerrero-Wyss, M. Common Beans as a Source of Amino Acids and Cofactors for Collagen Biosynthesis. Nutrients 2023, 15, 4561. [Google Scholar] [CrossRef]
- Rodríguez Madrera, R.; Negrillo, A.C.; Suárez Valles, B.; Ferreira Fernández, J.J. Phenolic Content and Antioxidant Activity in Seeds of Common Bean (Phaseolus vulgaris L.). Foods 2021, 10, 864. [Google Scholar] [CrossRef]
- Azarbad, H. Conventional vs. Organic Agriculture–Which One Promotes Better Yields and Microbial Resilience in Rapidly Changing Climates? Front. Microbiol. 2022, 13, 903500. [Google Scholar] [CrossRef]
- FAO. Training Manual for Organic Agriculture; Food and Agriculture Organization of the United Nations: Rome, Italy, 2015. [Google Scholar]
- Põlme, S.; Abarenkov, K.; Henrik Nilsson, R.; Lindahl, B.D.; Clemmensen, K.E.; Kauserud, H.; Nguyen, N.; Kjøller, R.; Bates, S.T.; Baldrian, P.; et al. FungalTraits: A User-Friendly Traits Database of Fungi and Fungus-like Stramenopiles. Fungal Divers. 2020, 105, 1–16. [Google Scholar] [CrossRef]
- Nagy, L.G.; Ohm, R.A.; Kovács, G.M.; Floudas, D.; Riley, R.; Gácser, A.; Sipiczki, M.; Davis, J.M.; Doty, S.L.; De Hoog, G.S.; et al. Latent Homology and Convergent Regulatory Evolution Underlies the Repeated Emergence of Yeasts. Nat. Commun 2014, 5, 4471. [Google Scholar] [CrossRef] [PubMed]
- Redecker, D.; Schüßler, A.; Stockinger, H.; Stürmer, S.L.; Morton, J.B.; Walker, C. An Evidence-Based Consensus for the Classification of Arbuscular Mycorrhizal Fungi (Glomeromycota). Mycorrhiza 2013, 23, 515–531. [Google Scholar] [CrossRef]
- Massimo, N.C.; Nandi Devan, M.M.; Arendt, K.R.; Wilch, M.H.; Riddle, J.M.; Furr, S.H.; Steen, C.; U’Ren, J.M.; Sandberg, D.C.; Arnold, A.E. Fungal Endophytes in Aboveground Tissues of Desert Plants: Infrequent in Culture, but Highly Diverse and Distinctive Symbionts. Microb. Ecol. 2015, 70, 61–76. [Google Scholar] [CrossRef]
- Sterkenburg, E.; Bahr, A.; Brandström Durling, M.; Clemmensen, K.E.; Lindahl, B.D. Changes in Fungal Communities along a Boreal Forest Soil Fertility Gradient. New Phytol. 2015, 207, 1145–1158. [Google Scholar] [CrossRef]
- Kirk, P.; Cannon, P.; Stalpers, J.; Minter, D.W. Dictionary of the Fungi, 10th ed.; CABI: Wallingford, UK, 2008. [Google Scholar]
- Harkes, P.; Suleiman, A.K.A.; van den Elsen, S.J.J.; de Haan, J.J.; Holterman, M.; Kuramae, E.E.; Helder, J. Conventional and Organic Soil Management as Divergent Drivers of Resident and Active Fractions of Major Soil Food Web Constituents. Sci. Rep. 2019, 9, 13521. [Google Scholar] [CrossRef]
- Colautti, A.; Civilini, M.; Contin, M.; Celotti, E.; Iacumin, L. Organic vs. Conventional: Impact of Cultivation Treatments on the Soil Microbiota in the Vineyard. Front. Microbiol. 2023, 14, 1242267. [Google Scholar] [CrossRef]
- Maretto, L.; Deb, S.; Ravi, S.; Della Lucia, M.C.; Borella, M.; Campagna, G.; Squartini, A.; Concheri, G.; Nardi, S.; Stevanato, P. 16S Metabarcoding, Total Soil DNA Content, and Functional Bacterial Genes Quantification to Characterize Soils under Long-Term Organic and Conventional Farming Systems. Chem. Biol. Technol. Agric. 2023, 10, 78. [Google Scholar] [CrossRef]
- Schmidt, J.E.; Kent, A.D.; Brisson, V.L.; Gaudin, A.C.M. Agricultural Management and Plant Selection Interactively Affect Rhizosphere Microbial Community Structure and Nitrogen Cycling. Microbiome 2019, 7, 146. [Google Scholar] [CrossRef]
- Armalytė, J.; Skerniškytė, J.; Bakienė, E.; Krasauskas, R.; Šiugždinienė, R.; Kareivienė, V.; Kerzienė, S.; Klimienė, I.; Sužiedėlienė, E.; Ružauskas, M. Microbial Diversity and Antimicrobial Resistance Profile in Microbiota From Soils of Conventional and Organic Farming Systems. Front. Microbiol. 2019, 10, 892. [Google Scholar] [CrossRef]
- van der Heijden, M.G.A.; Hartmann, M. Networking in the Plant Microbiome. PLoS Biol. 2016, 14, e1002378. [Google Scholar] [CrossRef] [PubMed]
- Dincă, L.C.; Grenni, P.; Onet, C.; Onet, A. Fertilization and Soil Microbial Community: A Review. Appl. Sci. 2022, 12, 1198. [Google Scholar] [CrossRef]
- Smith, M.E.; Vico, G.; Costa, A.; Bowles, T.; Gaudin, A.C.M.; Hallin, S.; Watson, C.A.; Alarcòn, R.; Berti, A.; Blecharczyk, A.; et al. Increasing Crop Rotational Diversity Can Enhance Cereal Yields. Commun. Earth Environ. 2023, 4, 89. [Google Scholar] [CrossRef]
- Yang, X.; Xiong, J.; Du, T.; Ju, X.; Gan, Y.; Li, S.; Xia, L.; Shen, Y.; Pacenka, S.; Steenhuis, T.S.; et al. Diversifying Crop Rotation Increases Food Production, Reduces Net Greenhouse Gas Emissions and Improves Soil Health. Nat. Commun. 2024, 15, 198. [Google Scholar] [CrossRef]
- Chen, T.; Nomura, K.; Wang, X.; Sohrabi, R.; Xu, J.; Yao, L.; Paasch, B.C.; Ma, L.; Kremer, J.; Cheng, Y.; et al. A Plant Genetic Network for Preventing Dysbiosis in the Phyllosphere. Nature 2020, 580, 653–657. [Google Scholar] [CrossRef]
- Singh, S.P.; Schwartz, H.F. Breeding Common Bean for Resistance to Diseases: A Review. Crop Sci. 2010, 50, 2199–2223. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Miller, H.; Liu, J.; Hou, Z.; Liang, S.; Zhao, X.; Zhang, H.; Borch, T. Fungicide Azoxystrobin Induced Changes on the Soil Microbiome. Appl. Soil. Ecol. 2020, 145, 103343. [Google Scholar] [CrossRef]
- Sun, P.; Wu, J.; Lin, X.; Wang, Y.; Zhu, J.; Chen, C.; Wang, Y.; Jia, H.; Shen, J. Effect of Ozonated Water, Mancozeb, and Thiophanate-Methyl on the Phyllosphere Microbial Diversity of Strawberry. Front. Plant Sci. 2022, 13, 967797. [Google Scholar] [CrossRef]
- Baćmaga, M.; Wyszkowska, J.; Kucharski, J. Response of Soil Microbiota, Enzymes, and Plants to the Fungicide Azoxystrobin. Int. J. Mol. Sci. 2024, 25, 8104. [Google Scholar] [CrossRef]
- Farghaly, M.F.M.; Zayed, S.M.A.D.; Soliman, S.M. Deltamethrin Degradation and Effects on Soil Microbial Activity. J. Environ. Sci. Health Part B 2013, 48, 575–581. [Google Scholar] [CrossRef]
- Qiu, D.; Xu, N.; Zhang, Q.; Zhou, W.; Wang, Y.; Zhang, Z.; Yu, Y.; Lu, T.; Sun, L.; Zhou, N.-Y.; et al. Negative Effects of Abamectin on Soil Microbial Communities in the Short Term. Front. Microbiol. 2022, 13, 1053153. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.V.; Musumeci, M.A. Trichoderma as Biological Control Agent: Scope and Prospects to Improve Efficacy. World J. Microbiol. Biotechnol. 2021, 37, 90. [Google Scholar] [CrossRef] [PubMed]
- Oehl, F.; Sieverding, E.; Mäder, P.; Dubois, D.; Ineichen, K.; Boller, T.; Wiemken, A. Impact of Long-Term Conventional and Organic Farming on the Diversity of Arbuscular Mycorrhizal Fungi. Oecologia 2004, 138, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Khatri, S.; Dubey, S.; Shivay, Y.S.; Jelsbak, L.; Sharma, S. Organic Farming Induces Changes in Bacterial Community and Disease Suppressiveness against Fungal Phytopathogens. Appl. Soil Ecol. 2023, 181, 104658. [Google Scholar] [CrossRef]
- Montgomery, D.R.; Biklé, A. Soil Health and Nutrient Density: Beyond Organic vs. Conventional Farming. Front. Sustain. Food Syst. 2021, 5, 699147. [Google Scholar] [CrossRef]
- Banerjee, S.; Walder, F.; Büchi, L.; Meyer, M.; Held, A.Y.; Gattinger, A.; Keller, T.; Charles, R.; van der Heijden, M.G.A. Agricultural Intensification Reduces Microbial Network Complexity and the Abundance of Keystone Taxa in Roots. ISME J. 2019, 13, 1722–1736. [Google Scholar] [CrossRef]
- Geisen, S.; Wall, D.H.; van der Putten, W.H. Challenges and Opportunities for Soil Biodiversity in the Anthropocene. Curr. Biol. 2019, 29, R1036–R1044. [Google Scholar] [CrossRef]
- Lori, M.; Symnaczik, S.; Mäder, P.; De Deyn, G.; Gattinger, A. Organic Farming Enhances Soil Microbial Abundance and Activity—A Meta-Analysis and Meta-Regression. PLoS ONE 2017, 12, e0180442. [Google Scholar] [CrossRef]
- Mo, Y.; Bier, R.; Li, X.; Daniels, M.; Smith, A.; Yu, L.; Kan, J. Agricultural Practices Influence Soil Microbiome Assembly and Interactions at Different Depths Identified by Machine Learning. Commun. Biol. 2024, 7, 1349. [Google Scholar] [CrossRef]
- Mondaca, P.; Celis-Diez, J.L.; Díaz-Siefer, P.; Olmos-Moya, N.; Montero-Silva, F.; Molina, S.; Fontúrbel, F.E.; Aponte, H.; Mandakovic, D.; Bastidas, B.; et al. Effects of Sustainable Agricultural Practices on Soil Microbial Diversity, Composition, and Functions. Agric. Ecosyst. Environ. 2024, 370, 109053. [Google Scholar] [CrossRef]
- Ferreira, J.J.; Campa, A.; Pérez-Vega, E.; Rodríguez-Suárez, C.; Giraldez, R. Introgression and Pyramiding into Common Bean Market Class Fabada of Genes Conferring Resistance to Anthracnose and Potyvirus. Theor. Appl. Genet. 2012, 124, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles Instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of RRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A Comprehensive Online Resource for Quality Checked and Aligned Ribosomal RNA Sequence Data Compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- Kõljalg, U.; Larsson, K.H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; et al. UNITE: A Database Providing Web-Based Methods for the Molecular Identification of Ectomycorrhizal Fungi. New Phytol. 2005, 166, 1063–1068. [Google Scholar] [CrossRef]
- Pielou, E.C. The Measurement of Diversity in Different Types of Biological Collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Ripley, B.; Venables, B. MASS: Support Functions and Datasets for Venables and Ripley’s MASS. In CRAN: Contributed Packages; CRAN: Wirtschaftsuniversität Wien, Austria, 2009. [Google Scholar]
- Cribari-Neto, F.; Zeileis, A. Beta Regression in R. J. Stat. Softw. 2010, 34, 1–24. [Google Scholar] [CrossRef]
- Fox, J.; Weisberg, S.; Price, B. Car: Companion to Applied Regression. In CRAN: Contributed Packages; CRAN: Wirtschaftsuniversität Wien, Austria, 2001. [Google Scholar]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package. In CRAN: Contributed Packages; CRAN: Wirtschaftsuniversität Wien, Austria, 2001. [Google Scholar]
- Kuhn, M.; Jackson, S.; Cimentada, J. Corrr: Correlations in R. Available online: https://github.com/tidymodels/corrr (accessed on 31 November 2024).
- Wei, T.; Simko, V.; Levy, M.; Xie, Y.; Jin, Y.; Zemla, J.; Freidank, M.; Cai, J.; Protivinksy, T. Corrplot: Visualization of a Correlation Matrix; CRAN: Wirtschaftsuniversität Wien, Austria, 2024. [Google Scholar]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Peterson, B.G.; Carl, P.; Boudt, K.; Bennett, R.; Ulrich, J.; Zivot, E.; Cornilly, D.; Hung, E.; Lestel, M.; Balkissoon, K.; et al. Econometric Tools for Performance and Risk Analysis; CRAN: Wirtschaftsuniversität Wien, Austria, 2022. [Google Scholar]
- R Core Team, R. A Language and Environment for Statistical Computing; CRAN: Wirtschaftsuniversität Wien, Austria, 2024. [Google Scholar]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling Function and Taxonomy in the Global Ocean Microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An Open Annotation Tool for Parsing Fungal Community Datasets by Ecological Guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Camacho-Sanchez, M.; Herencia, J.F.; Arroyo, F.T.; Capote, N. Soil Microbial Community Responses to Different Management Strategies in Almond Crop. J. Fungi 2023, 9, 95. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil | Year | Seq. | Input | Input Passed Filter (%) | Denoised | Merged Input (%) | Non-Chimeric Input (%) | Observed OTUs |
|---|---|---|---|---|---|---|---|---|
| CS | 2022 | 16S | 120,179 ± 27,066 | 77.86 | 70,955 ± 16,364 | 34.04 | 33.64 | 634 ± 117 |
| ITS | 160,773 ± 69,498 | 74.43 | 117,357 ± 45,972 | 68.57 | 67.09 | 167 ± 32 | ||
| 2023 | 16S | 91,369 ± 68,881 | 59.10 | 41,285 ± 32,190 | 27.62 | 27.08 | 453 ± 53 | |
| ITS | 48,594 ± 20,817 | 50.10 | 22,807 ± 12,277 | 41.15 | 40.51 | 147 ± 44 | ||
| OS | 2022 | 16S | 86,807 ± 11,692 | 77.89 | 52,710 ± 7877 | 31.69 | 31.35 | 489 ± 78 |
| ITS | 126,259 ± 34,298 | 74.43 | 93,871 ± 28,359 | 66.13 | 65.28 | 260 ± 76 | ||
| 2023 | 16S | 93,863 ± 55,541 | 54.53 | 39,946 ± 29,127 | 22.65 | 22.05 | 352 ± 7 | |
| ITS | 41,308 ± 10,520 | 54.66 | 20,731 ± 7278 | 43.98 | 43.44 | 217 ± 78 |
| Diversity | Statistic Model | Organism | Coefficient | Z Value | F Value | Pr (>|z|) |
|---|---|---|---|---|---|---|
| Alpha diversity: Richness | Binomial negative | 16S | Farming | 2.78 | - | 5.50 × 10−3 |
| Year | 3.38 | - | 7.31 × 10−3 | |||
| Farming:Year | 0.06 | - | 0.95 | |||
| ITS | Farming | 2.89 | - | 3.84 × 10−3 | ||
| Year | 0.53 | - | 0.60 | |||
| Farming:Year | 0.33 | - | 0.74 | |||
| Alpha diversity: Evenness | Beta regresion | 16S | Farming | 1.54 | - | 0.12 |
| Year | 0.46 | - | 0.64 | |||
| Farming:Year | 2.47 | - | 0.01 | |||
| ITS | Farming | 5.34 | - | 9.34 × 10−8 | ||
| Year | 1.80 | - | 0.07 | |||
| Farming:Year | 1.78 | - | 0.08 | |||
| Alpha diversity: Shannon Index | ANOVA | 16S | Farming | - | 0.60 | 0.45 |
| Year | - | 2.40 | 0.14 | |||
| Farming:Year | - | 0.14 | 0.71 | |||
| ITS | Farming | - | 27.31 | 8.33 × 10−5 | ||
| Year | - | 0 | 0.99 | |||
| Farming:Year | - | 2.10 | 0.17 | |||
| Beta diversity | Permanova of Jaccard distances | 16S | Farming | - | 1.16 | 1.00 × 10−3 |
| Year | - | 1.15 | 1.00 × 10−3 | |||
| Farming:Year | - | 1.09 | 2.00 × 10−3 | |||
| ITS | Farming | - | 3.67 | 1.00 × 10−3 | ||
| Year | - | 2.20 | 1.00 × 10−3 | |||
| Farming:Year | - | 1.96 | 3.00 × 10−3 |
| 2022 | 2023 | |||||
|---|---|---|---|---|---|---|
| CS | OS | CS | OS | |||
| Taxonomy | Mean ± SD | Mean ± SD | p-Value | Mean ± SD | Mean ± SD | p-Value |
| Uncultured verrucomicrobia (g) | 2.49 ± 6.91 | 0 | 2.00 × 10−3 | 2.73 ± 8.79 | 0 | 1.24 × 10−2 |
| Uncultured planctomycete (g) | 4.33 ± 13.6 | 0.38 ± 2.24 | 2.59 × 10−2 | 2.00 ± 7.28 | 0 | 4.33 × 10−2 |
| Uncultured chthoniobacter (g) | 0 | 2.31 ± 7.52 | 1.13 × 10−3 | 0 | 1.92 ± 6.95 | 2.34 × 10−2 |
| Uncultured bacteroidetes (g) | 0.13 ± 1.12 | 4.06 ± 13.56 | 8.61 × 10−3 | 0.45 ± 2.54 | 3.61 ± 10.19 | 4.10 × 10−2 |
| Uncultured UTBCD1 (g) | 0.85 ± 2.68 | 0 | 4.28 × 10−2 | 2.91 ± 10.15 | 0 | 4.21 × 10−2 |
| Uncultured adlerbacteria (g) | 1.05 ± 3.50 | 0.14 ± 0.95 | 2.77 × 10−2 | 1.42 ± 4.65 | 0 | 6.87 × 10−3 |
| Uncultured A4b (g) | 1.23 ± 4.71 | 0.24 ± 1.31 | 2.00 × 10−2 | 0.89 ± 3.96 | 0.12 ± 0.99 | 3.13 × 10−2 |
| Uncultured bacteriap25 (g) | 1.43 ± 5.02 | 0.03 ± 0.26 | 7.79 × 10−3 | 1.40 ± 4.44 | 0 | 2.30 × 10−2 |
| Candidatus uhrbacteria (g) | 1.2 ± 3.88 | 0 | 2.28 × 10−2 | 0.69 ± 2.01 | 0 | 4.25 × 10−2 |
| Uncultured desulfuromonas (g) | 0 | 4.55 ± 13.57 | 2.25 × 10−2 | 0 | 2.84 ± 8.78 | 4.21 × 10−2 |
| Uncultured vicinamibacteraceae (g) | 0.08 ± 0.67 | 3.08 ± 8.76 | 4.57 × 10−3 | 0 | 3.11 ± 10.15 | 6.87 × 10−3 |
| Uncultured planctomycete (g) | 0.00 ± 0.00 | 1.48 ± 5.77 | 7.17 × 10−3 | 0.44 ± 2.56 | 2.99 ± 8.70 | 4.29 × 10−2 |
| Ferruginibacter (g) | 0.00 ± 0.00 | 2.49 ± 7.39 | 3.72 × 10−3 | 0.00 ± 0.00 | 3.40 ± 10.82 | 2.31 × 10−2 |
| Rokubacteriales (g) | 3.13 ± 11.44 | 1.75 ± 9.98 | 3.87 × 10−2 | 3.30 ± 12.57 | 1.43 ± 9.32 | 6.48 × 10−3 |
| SJA-28 (g) | 0 | 2.24 ± 6.46 | 2.28 × 10−2 | 0 | 2.94 ± 7.59 | 2.22 × 10−2 |
| Uncultured simkaniaceae (g) | 1.65 ± 5.92 | 0.07 ± 0.54 | 4.87 × 10−2 | 1.18 ± 4.49 | 0 | 2.27 × 10−2 |
| Uncultured sutterellaceae (f) | 0 | 6.23 ± 15.06 | 4.95 × 10−5 | 0 | 4.63 ± 16.63 | 2.34 × 10−2 |
| Roseiflexaceae (f) | 2.34 ± 5.79 | 0.10 ± 1.16 | 1.46 × 10−6 | 0.84 ± 3.46 | 0 | 7.31 × 10−3 |
| Gemmatimonadaceae (f) | 2.96 ± 9.12 | 0 | 3.39 × 10−4 | 2.83 ± 8.88 | 0 | 6.87 × 10−3 |
| 2022 | 2023 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| CS | OS | CS | OS | ||||||
| Taxonomy | Mean ± SD | Mean ± SD | p-Value | Mean ± SD | Mean ± SD | p-Value | Guild | Confidence Ranking | Source |
| Botryotrichum spirotrichum (s) | 55.00 ± 34.66 | 0 | 7.49 × 10−3 | 149.60 ± 95.87 | 2.00 ± 4.47 | 9.70 × 10−3 | Saprotroph | P | [11,23] |
| Claroideoglomus claroideum (s) | 0.28 ± 1.44 | 7.78 ± 21.93 | 4.11 × 10−5 | 0.03 ± 0.39 | 2.32 ± 8.35 | 5.53 × 10−5 | Arbuscular Mycorrhizal | HP | [11,23] |
| Emmonsiellopsis coralliformis (s) | 2.20 ± 1.79 | 0 | 2.48 × 10−2 | 38.20 ± 21.78 | 0 | 7.49 × 10−3 | Saprotroph | P | [23] |
| Exophiala equina (s) | 0 | 4.90 ± 9.15 | 1.49 × 10−2 | 0 | 10.00 ± 14.66 | 1.49 × 10−2 | Animal Parasite | P | [11,23,24] |
| Funneliformis mosseae (s) | 0.13 ± 0.91 | 3.23 ± 10.92 | 3.30 × 10−7 | 0.03 ± 0.32 | 1.80 ± 10.46 | 1.77 × 10−2 | Arbuscular Mycorrhizal | HP | [23,25] |
| Pyrenochaetopsis leptospora (s) | 0.04 ± 0.20 | 5.60 ± 15.62 | 1.77 × 10−2 | 0 | 7.52 ± 22.13 | 2.49 × 10−3 | Endophyte | Ps | [11,23,26] |
| Rhizophagus fasciculatus (s) | 0 | 5.90 ± 21.33 | 4.28 × 10−2 | 0.40 ± 1.81 | 7.48 ± 18.33 | 4.09 × 10−2 | Arbuscular Mycorrhizal | HP | [23,25] |
| Acrocalymma (g) | 127.60 ± 119.94 | 7.80 ± 11.37 | 1.12 × 10−2 | 41.80 ± 24.72 | 154.60 ± 75.45 | 2.16 × 10−2 | - | - | - |
| Ciliophora (g) | 0.31 ± 1.62 | 0.69 ± 2.60 | 1.73 × 10−2 | 0.81 ± 3.11 | 0.28 ± 2.34 | 2.79 × 10−3 | - | - | - |
| Cylindrocarpon (g) | 0.25 ± 1.12 | 5.25 ± 5.75 | 9.97 × 10−5 | 0.05 ± 0.22 | 4.80 ± 7.90 | 3.20 × 10−2 | Plant Pathogen | P | [11,23] |
| Diversispora (g) | 0 | 1.83 ± 5.33 | 3.65 × 10−3 | 0 | 0.92 ± 3.39 | 2.34 × 10−2 | Arbuscular Mycorrhizal | HP | [23,25] |
| Dominikia (g) | 0 | 2.88 ± 8.70 | 2.09 × 10−2 | 0 | 1.72 ± 4.06 | 4.12 × 10−2 | Arbuscular Mycorrhizal | HP | [25] |
| Pyrenochaeta (g) | 1.10 ± 3.82 | 4.85 ± 7.38 | 3.01 × 10−2 | 0.30 ± 1.34 | 7.25 ± 12.61 | 1.64 × 10−2 | Saprotroph | HP | [11,23] |
| Ramicandelaber (g) | 26.48 ± 69.44 | 0.60 ± 1.68 | 3.37 × 10−2 | 22.44 ± 46.03 | 0.16 ± 0.80 | 3.66 × 10−2 | Saprotroph | P | [11,23] |
| Rhizophagus (g) | 0.07 ± 0.54 | 2.76 ± 9.07 | 3.97 × 10−4 | 0.00 | 0.69 ± 2.96 | 1.32 × 10−2 | Arbuscular Mycorrhizal | HP | [25] |
| Talaromyces (g) | 5.78 ± 14.64 | 2.80 ± 15.89 | 8.30 × 10−3 | 16.67 ± 52.81 | 3.09 ± 11.29 | 7.16 × 10−3 | Saprotroph | Ps | [27] |
| Tetracladium (g) | 0 | 4.05 ± 7.25 | 4.53 × 10−3 | 0 | 4.65 ± 9.48 | 1.98 × 10−2 | Saprotroph | P | [11,23] |
| Agaricales (o) | 0 | 2.73 ± 8.33 | 4.19 × 10−2 | 0 | 0.63 ± 2.04 | 4.19 × 10−2 | Saprotroph | Ps | [28] |
| Helotiales (o) | 0 | 1.13 ± 2.19 | 4.17 × 10−4 | 0 | 4.70 ± 13.79 | 6.17 × 10−3 | - | - | - |
| Rhizophydiales (o) | 0 | 8.77 ± 23.04 | 3.26 × 10−9 | 0.01 ± 0.09 | 0.42 ± 2.15 | 3.01 × 10−2 | - | - | - |
| Didymellaceae (f) | 0.08 ± 0.28 | 4.36 ± 8.64 | 4.56 × 10−2 | 0.32 ± 1.11 | 5.72 ± 8.66 | 4.94 × 10−3 | - | - | - |
| Glomeraceae (f) | 0 | 1.33 ± 4.60 | 1.91 × 10−4 | 0.14 ± 1.43 | 2.97 ± 7.89 | 8.04 × 10−6 | Arbuscular Mycorrhizal | HP | [25] |
| Nectriaceae (f) | 0.67 ± 1.91 | 9.67 ± 11.49 | 1.01 × 10−2 | 2.47 ± 5.46 | 38.73 ± 66.26 | 3.67 × 10−2 | Saprotroph | Ps | [27] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suarez-Fernandez, M.; Ferreira, J.J.; Campa, A. Impact of Farming System on Soil Microbial Communities Associated with Common Bean in a Region of Northern Spain. Plants 2025, 14, 1359. https://doi.org/10.3390/plants14091359
Suarez-Fernandez M, Ferreira JJ, Campa A. Impact of Farming System on Soil Microbial Communities Associated with Common Bean in a Region of Northern Spain. Plants. 2025; 14(9):1359. https://doi.org/10.3390/plants14091359
Chicago/Turabian StyleSuarez-Fernandez, Marta, Juan Jose Ferreira, and Ana Campa. 2025. "Impact of Farming System on Soil Microbial Communities Associated with Common Bean in a Region of Northern Spain" Plants 14, no. 9: 1359. https://doi.org/10.3390/plants14091359
APA StyleSuarez-Fernandez, M., Ferreira, J. J., & Campa, A. (2025). Impact of Farming System on Soil Microbial Communities Associated with Common Bean in a Region of Northern Spain. Plants, 14(9), 1359. https://doi.org/10.3390/plants14091359