
Complete Chloroplast Genome Analysis of Casearia kurzii: Gene Loss at the IR Boundary and Monophyletic Evolution Within Casearia

 ,
,
Abstract

1. Introduction
2. Results
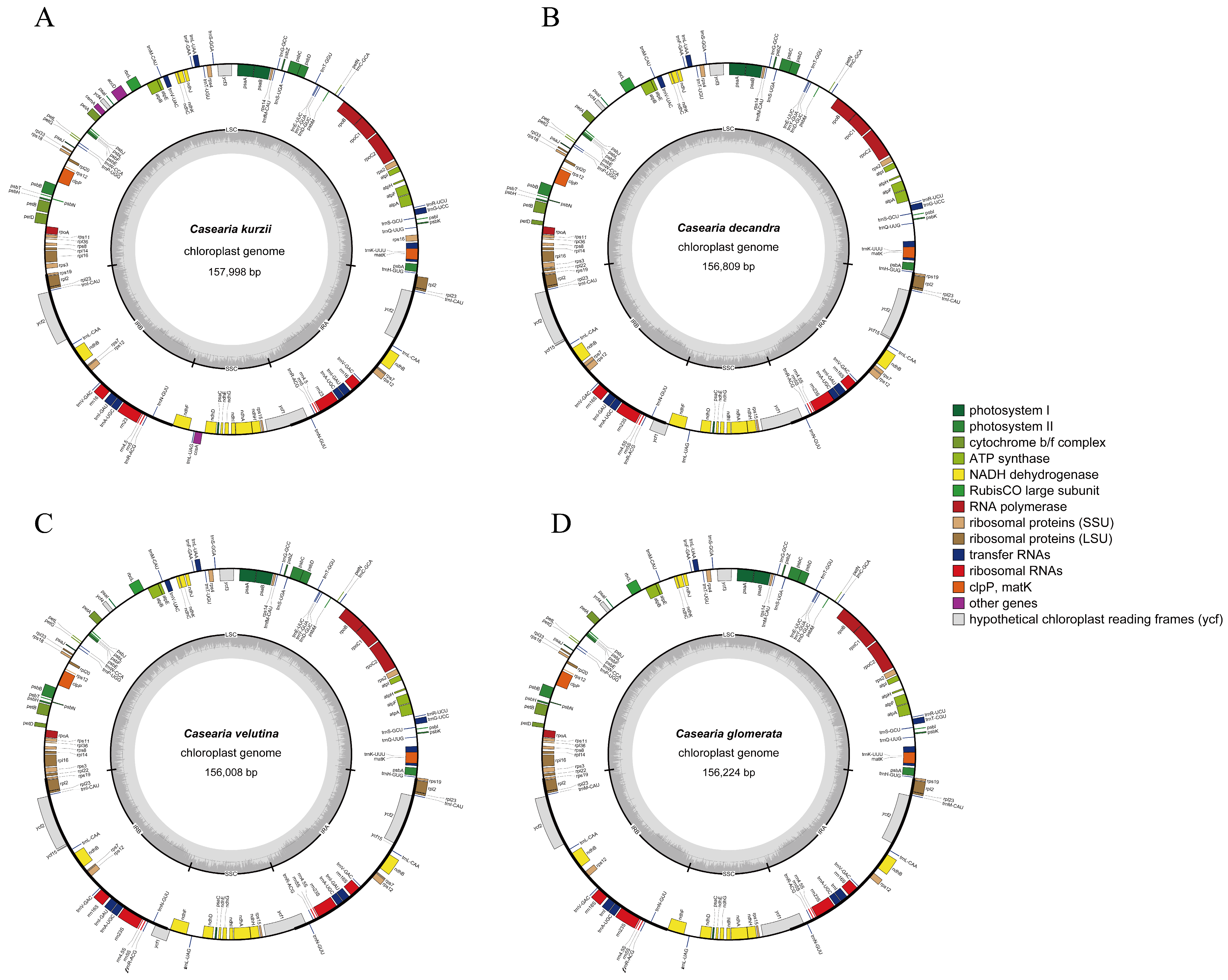
2.1. Main Features of the Chloroplast Genome
2.2. Dispersed Repeats and SSR Analysis
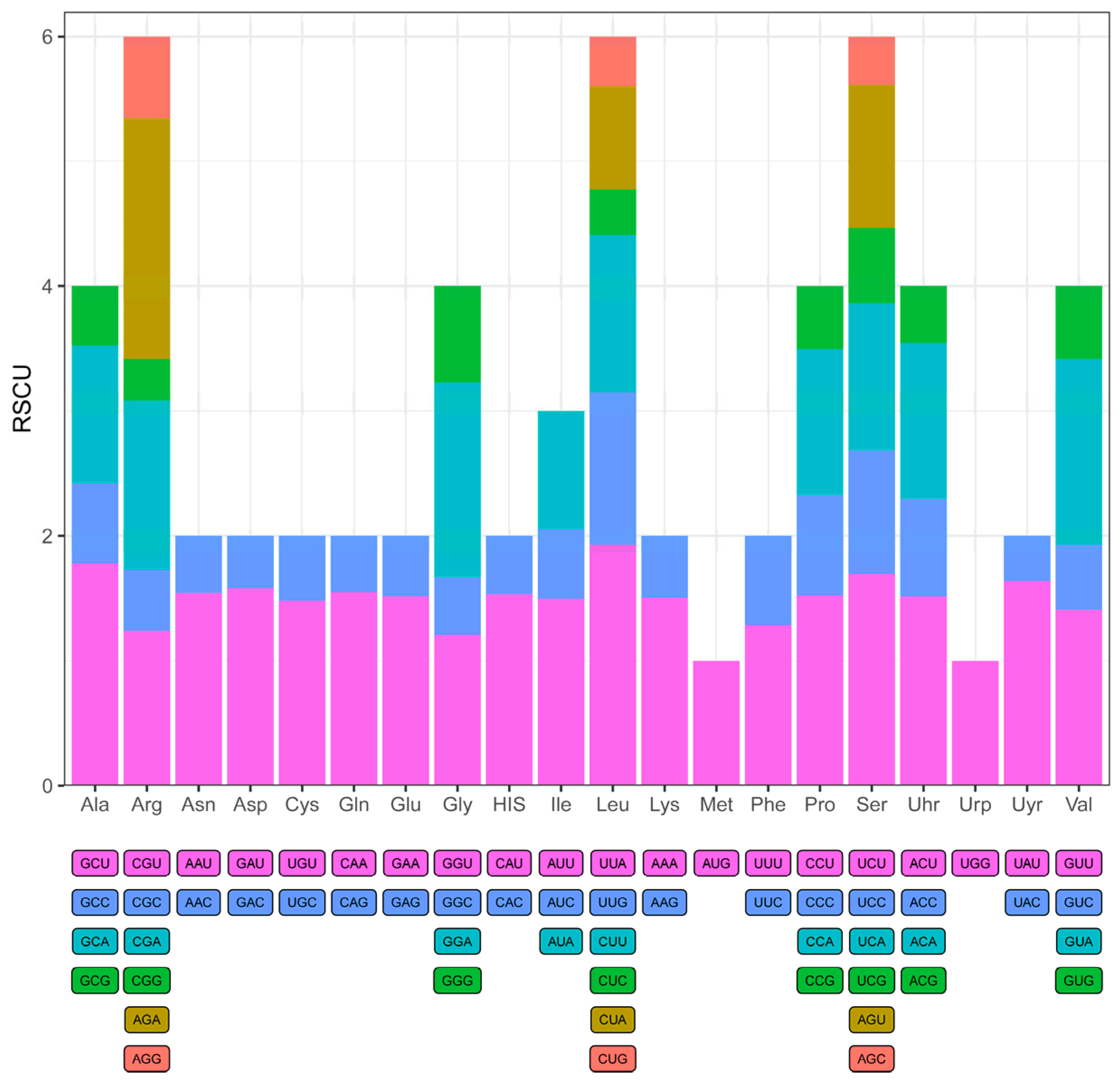
2.3. Codon Usage Analysis
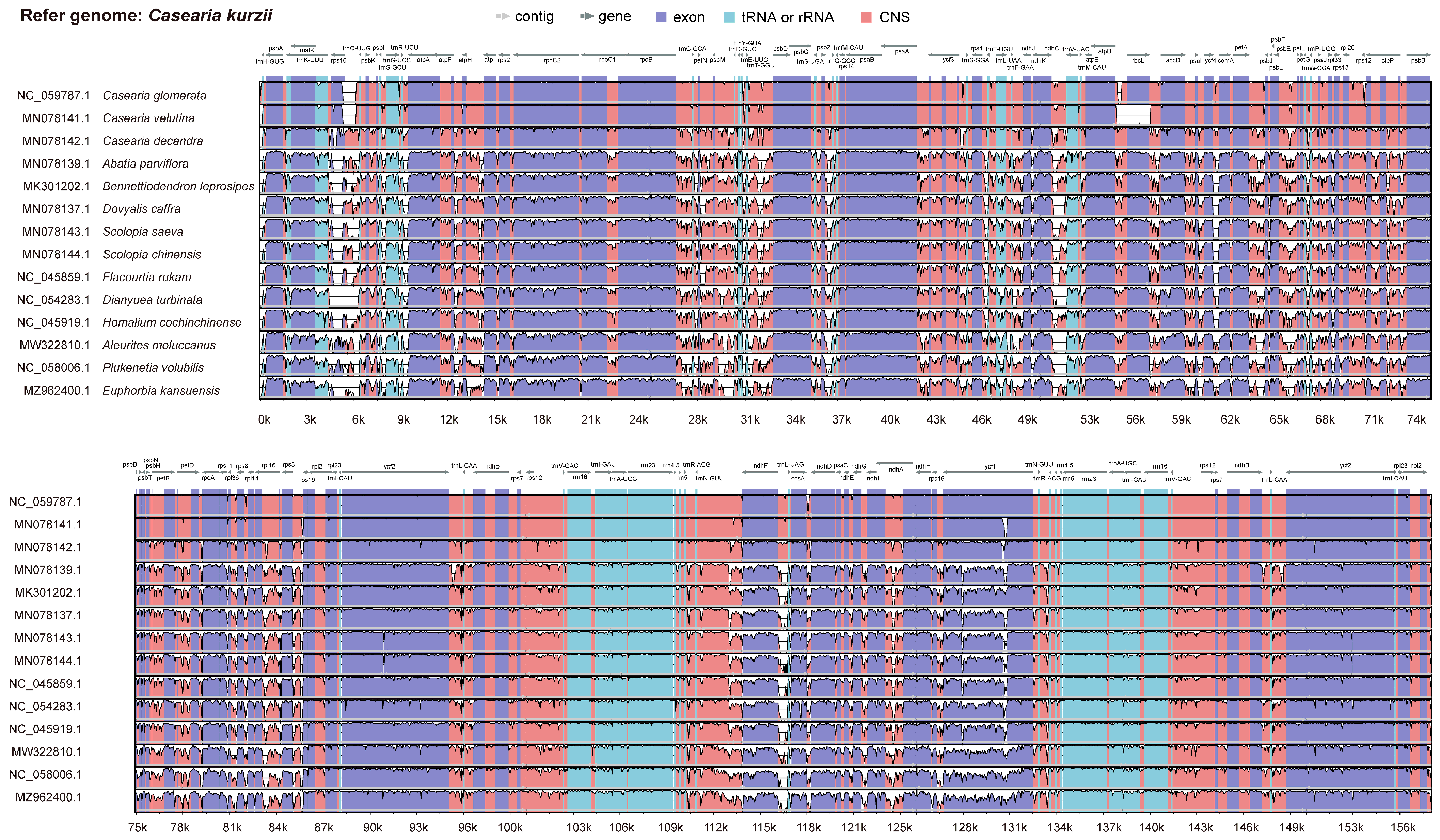
2.4. Sequence Divergence Analysis
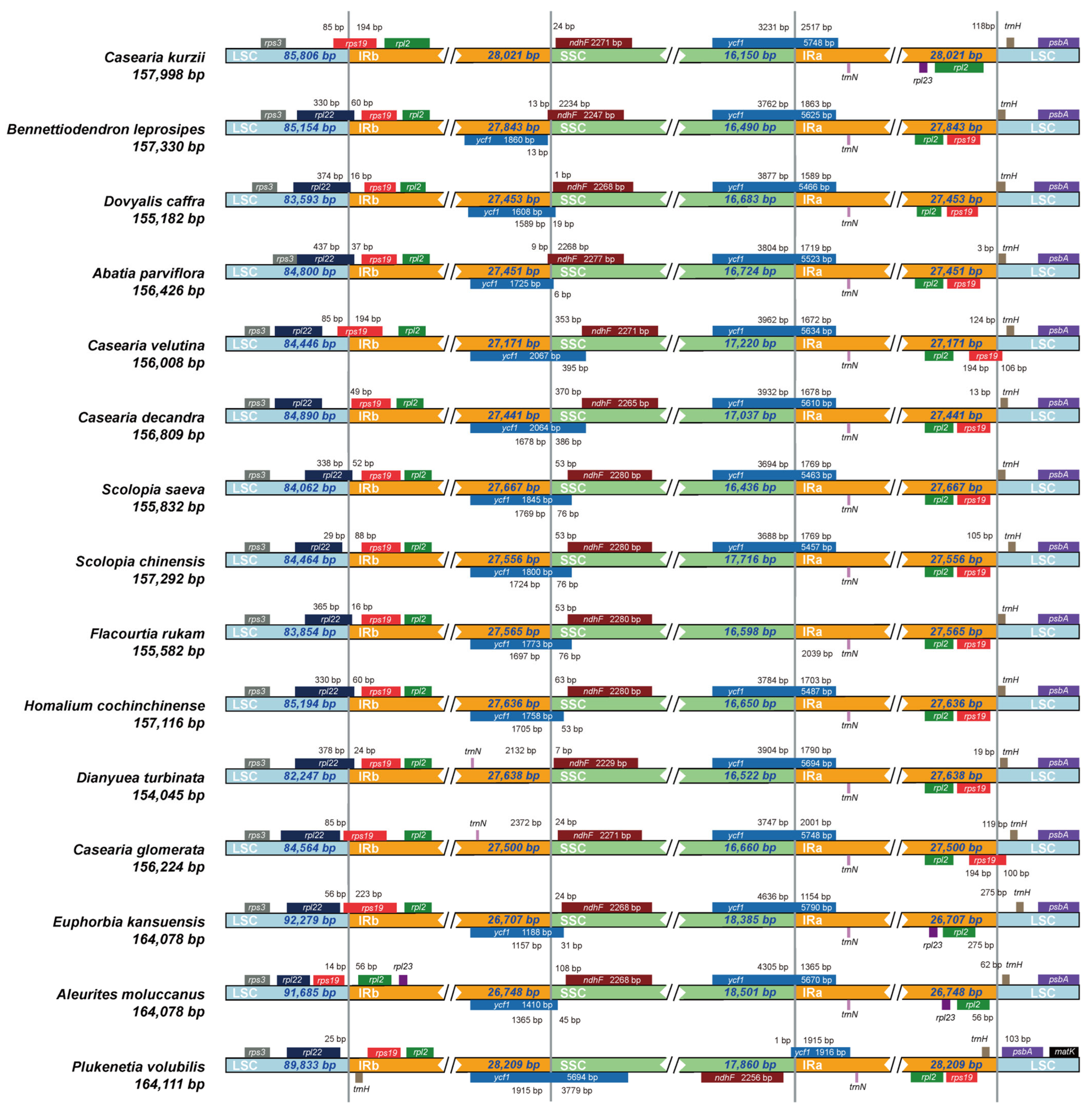
2.5. Expansion and Contraction of IR Boundaries
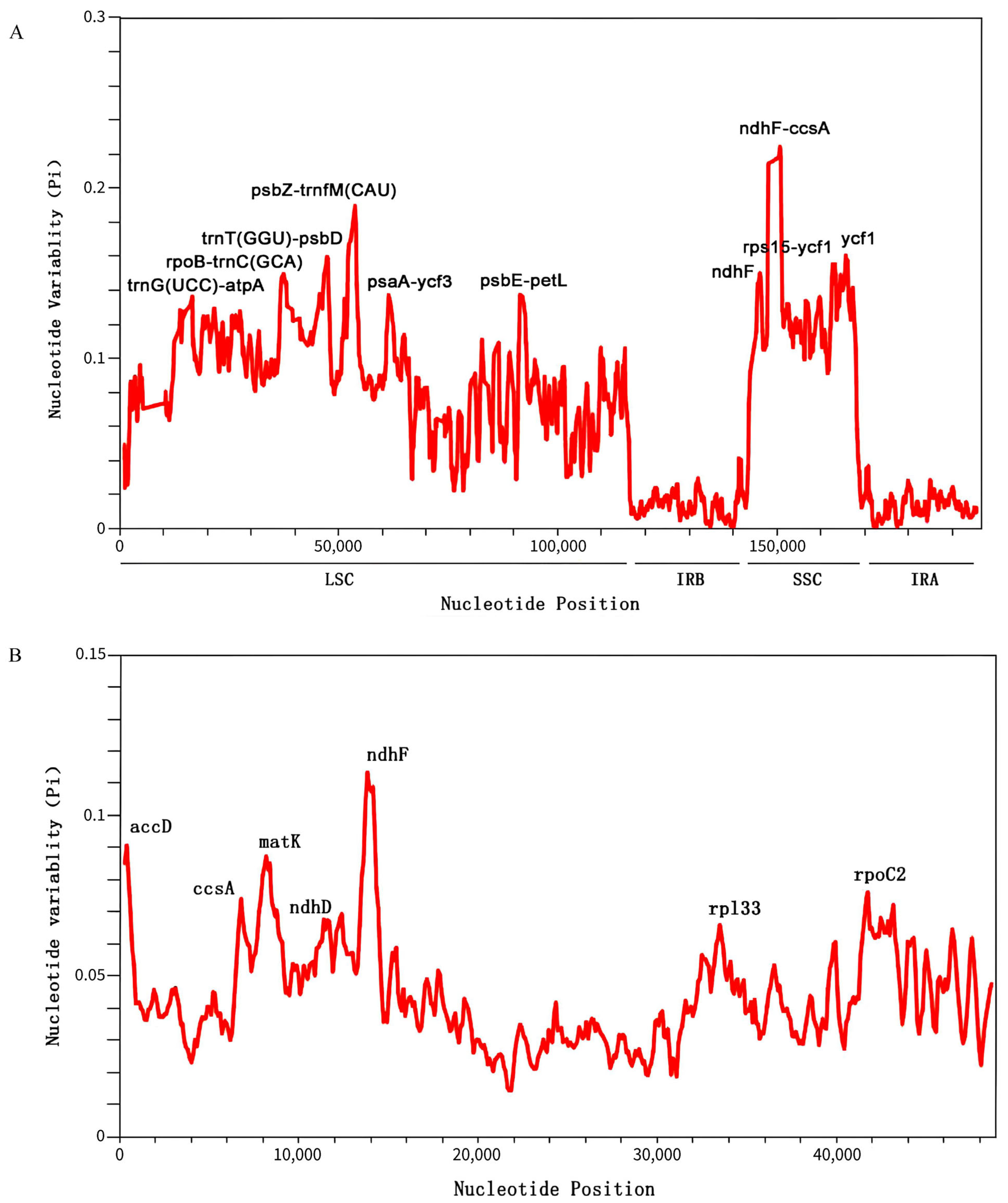
2.6. Nucleotide Polymorphism Analysis
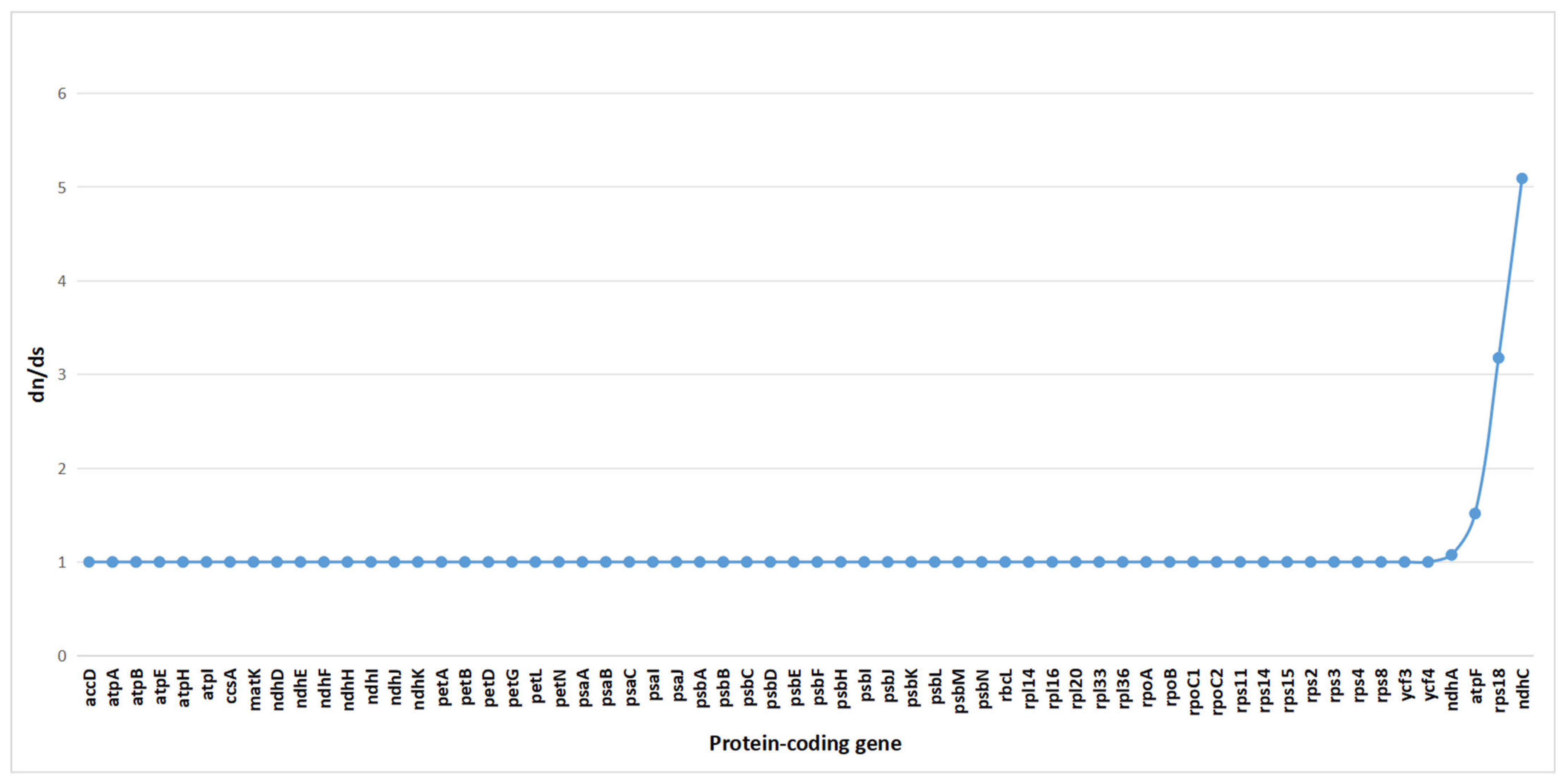
2.7. Selection Pressure Analysis
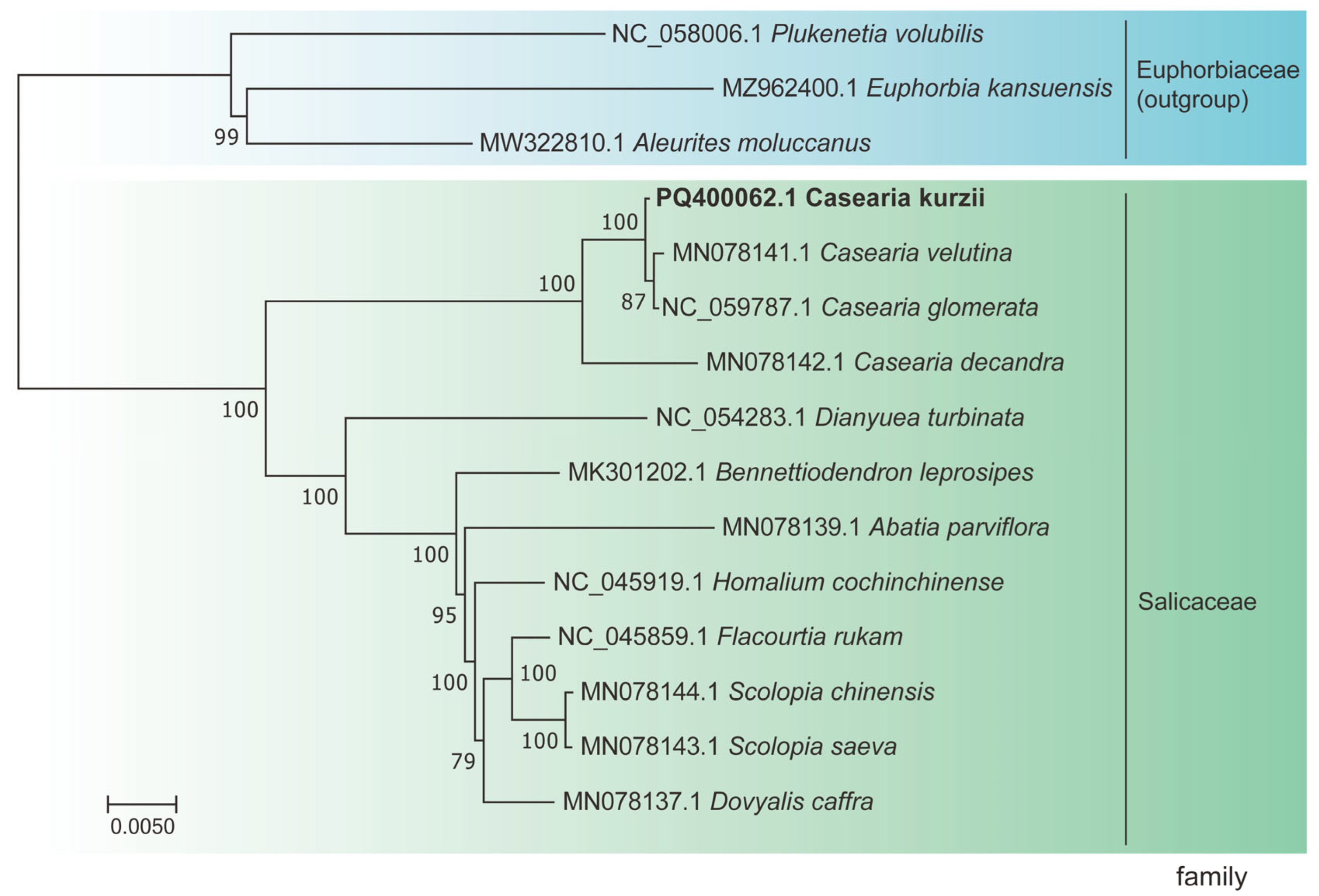
2.8. Phylogenetic Tree Reconstruction
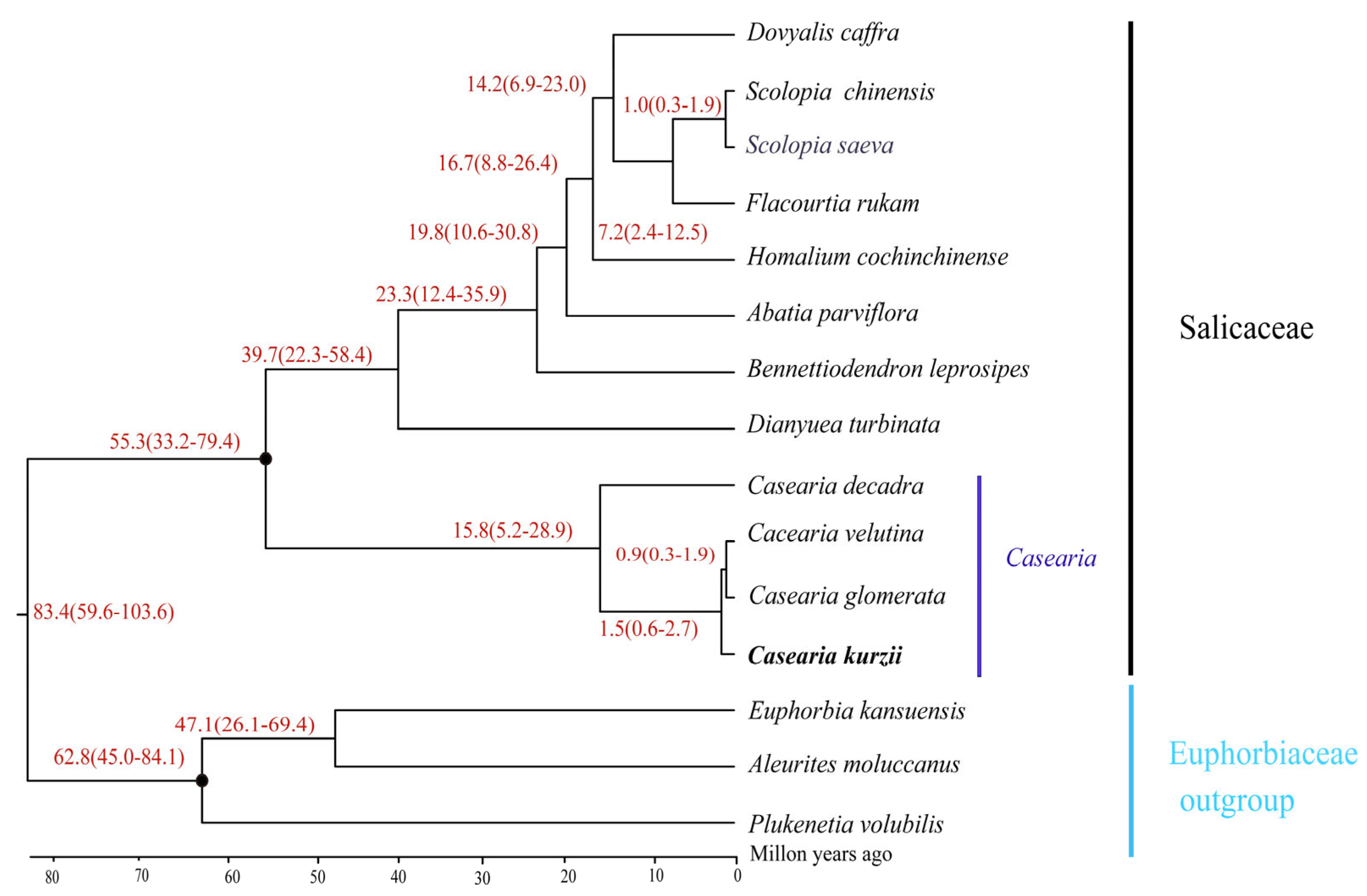
2.9. Divergence Time Tree
3. Materials and Methods
3.1. Plant Materials and DNA Extraction
3.2. DNA Sequencing, Assembly, and Annotation
3.3. Repeat Sequence Analysis
3.4. Codon Usage Bias Analysis
3.5. Comparative Genomic Analysis
3.6. Phylogenetic Analysis
3.7. Divergence Time Estimation
4. Discussion
4.1. Chloroplast Genome Structure and Basic Characteristics
4.2. Codon Usage and Selection Pressure
4.3. Repetitive Sequences and Identification of Highly Variable Regions
4.4. Phylogenetic and Evolutionary Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| LSC | large single-copy region |
| SSC | small single-copy region |
| IRs | inverted repeat regions |
| GC | guanine-cytosine |
| SSRs | simple sequence repeats |
| RSCU | relative synonymous codon usage |
| CNSs | conserved noncoding sequences |
| Pi | nucleotide diversity |
| ML | maximum likelihood |
| HPD | highest posterior density |
References
- Jensen, P.E.; Leister, D. Chloroplast Evolution, Structure and Functions. F1000Prime Rep. 2014, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Müller, K.F.; Quandt, D. The Evolution of the Plastid Chromosome in Land Plants: Gene Content, Gene Order, Gene Function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.R.; Nunes, R.; Sobreiro, M.B.; Dias, R.O.; Corvalán, L.C.J.; Braga-Ferreira, R.S.; Targueta, C.P.; Telles, M.P.C. The Complete Chloroplast Genome Sequence of Eugenia Klotzschiana O. Berg Unveils the Evolutionary Dynamics in Plastomes of Myrteae DC. Tribe (Myrtaceae). Gene 2023, 876, 147488. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.; Yu, M.; Chang, W. Chloroplast Genomes: Diversity, Evolution, and Applications in Genetic Engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, K.; Dong, W.; Dong, S.; Wang, Y.; Xu, C.; Li, E.; Sun, J. Chloroplast Genome Evolution of Hamamelidaceae at Subfamily Level. Ecol. Evol. 2025, 15, e71141. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, H.; Wu, M.; Zhang, X.; Baasanmukh, S.; Li, H.; Sun, H.; Chen, S. Comparative Chloroplast Genomes of Incarvillea Species (Bignoniaceae) Unveiled Genomic Diversity and Shed Light on Phylogenetic Relationships. BMC Plant Biol. 2025, 25, 399. [Google Scholar] [CrossRef]
- Nguyen, P.A.T.; Nguyen, H.A.; Do, H.D.K.; Tan Khang, D. The Complete Chloroplast Genome of Baccaurea Ramiflora Lour. Cultivar Ha Chau (Phyllanthaceae, Malpighiales). Ecol. Evol. 2025, 15, e71145. [Google Scholar] [CrossRef]
- Ma, J.; Yang, X.; Zhang, Q.; Zhang, X.; Xie, C.; Tuerhong, M.; Zhang, J.; Jin, D.; Lee, D.; Xu, J.; et al. Cytotoxic Clerodane Diterpenoids from the Leaves of Casearia Kurzii. Bioorg. Chem. 2019, 85, 558–567. [Google Scholar] [CrossRef]
- iPlant. Flora of China. Available online: http://www.iplant.cn/foc (accessed on 10 March 2025).
- An, L.; Ma, J.; Yang, X.; Liang, Y.; Wang, H.; Tuerhong, M.; Lall, N.; Abudukeremu, M.; Zhang, Y.; Lee, D.; et al. Caseahomopene A, a Ring-Expanded Homotriterpenoid from Casearia Kurzii Showing Anti-Inflammatory Activities in Vitro and in Vivo. Bioorg. Chem. 2020, 98, 103758. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, Q.; Yang, X.; Li, Y.; Zhang, X.; Li, Y.; Du, Q.; Jin, D.; Cui, J.; Lall, N.; et al. Diterpenoids from the Leaves of Casearia Kurzii Showing Cytotoxic Activities. Bioorg. Chem. 2020, 98, 103741. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Wang, T.; Zhang, J.; Huang, Z.; Shen, T.; Lou, H.; Ren, D.; Wang, X. Dolabellane and Clerodane Diterpenoids from the Twigs and Leaves of Casearia Kurzii. J. Nat. Prod. 2020, 83, 2817–2830. [Google Scholar] [CrossRef] [PubMed]
- Shuo, Y.; Zhang, C.; Yang, X.; Liu, F.; Zhang, Q.; Li, A.; Ma, J.; Lee, D.; Ohizumi, Y.; Guo, Y. Clerodane Diterpenoids from Casearia Kurzii and Their Cytotoxic Activities. J. Nat. Med. 2019, 73, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Tulasne, L. Florae Madagascariensis fragmenta. Fragmentum tertium. Violarieae, Sauvagesieae, Turneraceae, Samydeae, et Bixaceae. Ann. Sci. Nat. Sér 1868, 5, 298–344. [Google Scholar]
- Perrier de la Bâthie, H. Révision des Flacourtiacées de Madagascar et des Comores. Mém. Mus. Natl. Hist. Nat. 1940, 13, 261–301. [Google Scholar]
- Perrier de la Bâthie, H. Flacourtiacées. In Flore de Madagascar et des Comores; Muséum National d’Histoire Naturelle: Paris, France, 1946; Volume 140. [Google Scholar]
- Sleumer, H. Le genre Casearia Jacq. (Flacourtiaceae) en Afrique, à Madagascar et aux Mascareignes. Bull. Jard. Bot. Nat. Belg. 1971, 41, 397–426. [Google Scholar] [CrossRef]
- Sleumer, H. Flacourtiaceae. Flora Neotropica; New York Botanical Garden Press: New York, NY, USA, 1980; Volume 22, pp. 1–499. [Google Scholar]
- Alford, M.H. Systematic Studies in Flacourtiaceae. Ph.D. Thesis, Cornell University, Ithaca, NY, USA, 2005. [Google Scholar]
- Samarakoon, T.; Alford, M.H. New Names and Combinations in Neotropical Samydaceae. Novon 2019, 27, 65–71. [Google Scholar] [CrossRef]
- de Mestier, A.; Lücking, R.; Gutierrez, J.; Brokamp, G.; Celis, M.; Borsch, T. Nested Singletons in Molecular Trees: Utility of Adding Morphological and Geographical Data from Digitized Herbarium Specimens to Test Taxon Concepts at Species Level in the Case of Casearia (Salicaceae). Ecol. Evol. 2023, 13, e9736. [Google Scholar] [CrossRef]
- Applequist, W.L.; Gates, M.T. Two New Small-Leaved Species of Casearia (Salicaceae) from Madagascar. Novon 2020, 28, 256–262. [Google Scholar] [CrossRef]
- Costa, M.F.; Pereira, A.A.; Pinheiro, J.B.; Zucchi, M.I.; Araújo, A.S.F.; Gomes, R.L.F.; Valente, S.E.S.; Oliveira, M.E.A.; Lopes, A.C.A. Chloroplast Diversity of Casearia Grandiflora in the Cerrado of Piauí State. Genet. Mol. Res. 2017, 16, gmr16019572. [Google Scholar] [CrossRef]
- de Mestier, A.; Brokamp, G.; Celis, M.; Falcón-Hidalgo, B.; Gutiérrez, J.; Borsch, T. Character Evolution and Biogeography of Casearia (Salicaceae): Evidence for the South American Origin of a Pantropical Genus and for Multiple Migrations to the Caribbean Islands. TAXON 2022, 71, 321–347. [Google Scholar] [CrossRef]
- Huang, H.; Xie, Y.; Luo, S.; Zhang, L.; Deng, C.; Chen, Z. The Complete Chloroplast Genome Sequence of Casearia Glomerata Roxb. (Malpighiales; Salicaceae) from Fujian, China. Mitochondrial DNA B Resour. 2021, 6, 1932–1933. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-M.; Wang, D.-Y.; Zhang, L.; Kang, M.; Lu, Z.; Zhu, R.; Mao, X.; Xi, Z.; Ma, T. Intergeneric Relationships within the Family Salicaceae s.l. Based on Plastid Phylogenomics. Int. J. Mol. Sci. 2019, 20, 3788. [Google Scholar] [CrossRef]
- Ma, H.; Liu, Z.; Lan, W.; Yang, M.; Mo, Q.; Huang, X.; Wu, P.; Huang, H.; Huang, M. Complete Chloroplast Genomes of 9 Impatiens Species: Genome Structure, Comparative Analysis, and Phylogenetic Relationships. Int. J. Mol. Sci. 2025, 26, 536. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Wang, Y.; Song, H.; Dong, W.; Dong, N. Insights into Phylogeny, Taxonomy, Origins and Evolution of Crataegus and Mespilus, Based on Comparative Chloroplast Genome Analysis. Genes 2025, 16, 204. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 September 2024).
- Martin, M. CUTADAPT Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet.J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Jin, J.; Yu, W.; Yang, J.; Song, Y.; dePamphilis, C.; Yi, T.; Li, D. GetOrganelle: A Fast and Versatile Toolkit for Accurate de Novo Assembly of Organelle Genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive Visualization of de Novo Genome Assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) Version 1.3.1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-Web: A Web Server for Microsatellite Prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The Manifold Applications of Repeat Analysis on a Genomic Scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An Integrated and Scalable Desktop Platform for Streamlined Molecular Sequence Data Management and Evolutionary Phylogenetics Studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.; Tian, C.; Yang, Y.; Liu, Q.; Liu, L.; Tao, Q.; Li, Z.; Wu, Z. Complete Chloroplast Genome of Corethrodendron Fruticosum (Papilionoideae: Fabaceae): Comparative and Phylogenetic Analysis. Genes 2023, 14, 1289. [Google Scholar] [CrossRef]
- Jia, X.; Wei, J.; Chen, Y.; Zeng, C.; Deng, C.; Zeng, P.; Tang, Y.; Zhou, Q.; Huang, Y.; Zhu, Q. Codon Usage Patterns and Genomic Variation Analysis of Chloroplast Genomes Provides New Insights into the Evolution of Aroideae. Sci. Rep. 2025, 15, 4333. [Google Scholar] [CrossRef]
- Group, T.A.P. An Update of the Angiosperm Phylogeny Group Classification for the Orders and Families of Flowering Plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef]
- Xi, Z.; Ruhfel, B.R.; Schaefer, H.; Amorim, A.M.; Sugumaran, M.; Wurdack, K.J.; Endress, P.K.; Matthews, M.L.; Stevens, P.F.; Mathews, S.; et al. Phylogenomics and a Posteriori Data Partitioning Resolve the Cretaceous Angiosperm Radiation Malpighiales. Proc. Natl. Acad. Sci. USA 2012, 109, 17519–17524. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An Online Program to Visualize the Junction Sites of Chloroplast Genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational Tools for Comparative Genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. Available online: http://www.timetree.org/ (accessed on 28 October 2024). [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarisation in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree, v1.4.4. Tree Figure Drawing Tool. Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2009.
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic Analysis of 83 Plastid Genes Further Resolves the Early Diversification of Eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Y.; Tu, X.; Li, C.; Zhu, Y.; Zhong, H.; Liu, Z.; Wu, S.; Zhai, J. Comparative Analysis of Plastomes in Oxalidaceae: Phylogenetic Relationships and Potential Molecular Markers. Plant Divers. 2021, 43, 281–291. [Google Scholar] [CrossRef]
- Yao, J.; Zheng, Z.; Xu, T.; Wang, D.; Pu, J.; Zhang, Y.; Zha, L. Chloroplast Genome Sequencing and Comparative Analysis of Six Medicinal Plants of Polygonatum. Ecol. Evol. 2025, 15, e70831. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; He, L.; Li, Z.; Yuan, B.; Fang, F.; Wang, M.; Li, A.; Liu, C.; He, M.; et al. Comparative Chloroplast Genomics and Codon Usage Bias Analysis in Hevea Genus. Genes 2025, 16, 201. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, M. Analysis of the Codon Usage Bias Pattern in the Chloroplast Genomes of Chloranthus Species (Chloranthaceae). Genes 2025, 16, 186. [Google Scholar] [CrossRef]
- Yan, K.; Lu, X.; Li, W.; Sun, C.; Zhou, X.; Wang, Y. Chloroplast Genome Diversity and Molecular Evolution in Hypericaceae: New Insights from Three Hypericum Species. Int. J. Mol. Sci. 2025, 26, 323. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, S.; Wang, J.; Shen, H.; Ai, B.; Gao, W.; Zhang, C.; Fei, Q.; Yuan, D.; Wu, Z.; et al. Chloroplast Genomes in Populus (Salicaceae): Comparisons from an Intensively Sampled Genus Reveal Dynamic Patterns of Evolution. Sci. Rep. 2021, 11, 9471. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Huang, J.; Wan, X.; Shi, J.; Chen, Z.; Zeng, W. The Population Chloroplast Genomes of Populus Reveal the Phylogenetic Relationship between Three New Taxa of Sect. Leucoides and Their Parents. BMC Genom. 2025, 26, 156. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Ran, J.; Bao, S.; Li, Y.; Islam, R.; Zhang, N.; Zhao, W.; Ma, Y.; Sun, C. The Complete Chloroplast Genome Sequence of Eupatorium Fortunei: Genome Organization and Comparison with Related Species. Genes 2023, 14, 64. [Google Scholar] [CrossRef]
- Zhong, Y.; Bai, B.; Sun, Y.; Wen, K.; Qiao, Y.; Guo, L.; Deng, H.; Ye, Y.; Feng, L.; Feng, X. Comparative Genomics and Phylogenetic Analysis of Six Malvaceae Species Based on Chloroplast Genomes. BMC Plant Biol. 2024, 24, 1245. [Google Scholar] [CrossRef]
- Li, L.; Dong, T.; Wu, D.; Shu, Z. The Chloroplast Genome of Cephalanthera Nanchuanica (Orchidaceae): Comparative and Phylogenetic Analysis with Other Neottieae Species. BMC Genom. 2024, 25, 1090. [Google Scholar] [CrossRef]
- Kim, S.; Park, B.K.; Kim, H. Comparison of the Complete Chloroplast Genomes of Astilbe: Two Korean Endemic Plant Species. Genes 2024, 15, 1410. [Google Scholar] [CrossRef]
- Almeida-Silva, M.A.; Braga-Ferreira, R.S.; Targueta, C.P.; Corvalán, L.C.J.; Silva-Neto, C.M.; Franceschinelli, E.V.; Sobreiro, M.B.; Nunes, R.; Telles, M.P.C. Chloroplast Genomes of Simarouba Aubl., Molecular Evolution and Comparative Analyses within Sapindales. Sci. Rep. 2024, 14, 21358. [Google Scholar] [CrossRef]
- Zeng, C.; Hollingsworth, P.M.; Yang, J.; He, Z.; Zhang, Z.; Li, D.; Yang, J. Genome Skimming Herbarium Specimens for DNA Barcoding and Phylogenomics. Plant Methods 2018, 14, 43. [Google Scholar] [CrossRef]
- Bi, Y.; Zhang, M.; Xue, J.; Dong, R.; Du, Y.; Zhang, X. Chloroplast Genomic Resources for Phylogeny and DNA Barcoding: A Case Study on Fritillaria. Sci. Rep. 2018, 8, 1184. [Google Scholar] [CrossRef]
- Yu, J.; Han, Y.; Xu, H.; Han, S.; Li, X.; Niu, Y.; Chen, S.; Zhang, F. Structural Divergence and Phylogenetic Relationships of Ajania (Asteraceae) from Plastomes and ETS. BMC Genom. 2023, 24, 602. [Google Scholar] [CrossRef]
- Xiao-Ming, Z.; Junrui, W.; Li, F.; Sha, L.; Hongbo, P.; Lan, Q.; Jing, L.; Yan, S.; Weihua, Q.; Lifang, Z.; et al. Inferring the Evolutionary Mechanism of the Chloroplast Genome Size by Comparing Whole-Chloroplast Genome Sequences in Seed Plants. Sci. Rep. 2017, 7, 1555. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yang, J.; Bai, M.; Zhang, G.; Liu, Z. The Chloroplast Genome Evolution of Venus Slipper (Paphiopedilum): IR Expansion, SSC Contraction, and Highly Rearranged SSC Regions. BMC Plant Biol. 2021, 21, 248. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.; et al. The Complete Nucleotide Sequence of the Tobacco Chloroplast Genome: Its Gene Organization and Expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef]
- Gantt, J.; Baldauf, S.; Calie, P.; Weeden, N.; Palmer, J. Transfer of Rpl22 to the Nucleus Greatly Preceded Its Loss from the Chloroplast and Involved the Gain of an Intron. EMBO J. 1991, 10, 3073–3078. [Google Scholar] [CrossRef]
- Nakai, M. YCF1: A Green TIC: Response to the de Vries et al. Commentary. Plant Cell 2015, 27, 1834–1838. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. Ycf1, the Most Promising Plastid DNA Barcode of Land Plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef]
- Souza, M.; Kawakita, K.; Slusarski, S.; Pereira, G. Vascular Flora of the Upper Paraná River Floodplain. Braz. J. Biol. 2009, 69, 735–745. [Google Scholar] [CrossRef]
- Cavallari, M.; Billot, C.; Bouvet, J.; Favreau, B.; Zucchi, M.I.; Palmieri, D.; Gimenes, M. Isolation and Characterization of Microsatellite Markers for Casearia Sylvestris Sw. (Salicaceae), a Neotropical Medicinal Tree. Mol. Ecol. Resour. 2008, 8, 802–804. [Google Scholar] [CrossRef]
- Ogutcen, E.; de Lima Ferreira, P.; Wagner, N.; Marinček, P.; Vir Leong, J.; Aubona, G.; Cavender-Bares, J.; Michálek, J.; Schroeder, L.; Sedio, B.; et al. Phylogenetic Insights into the Salicaceae: The Evolution of Willows and Beyond. Mol. Phylogenet Evol. 2024, 199, 108161. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Wu, P.; Cheng, T.; Yu, J.; Zhou, S.; Hong, D. Resolving the Systematic Positions of Enigmatic Taxa: Manipulating the Chloroplast Genome Data of Saxifragales. Mol. Phylogenet Evol. 2018, 126, 321–330. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author (Year) | Taxonomic Treatment or Viewpoint on Casearia | Representative Conclusions or Changes | Reference |
|---|---|---|---|
| Tulasne (1868) | First conducted a systematic study of Malagasy Casearia, describing C. lucida, C. nigrescens, C. amplissima, C. parvifolia, C. elliptica, etc. | Established several new species based on morphological traits (e.g., leaf size and shape), laying the foundation for subsequent taxonomic research on this genus in Madagascar. | Tulasne, 1868 [14] |
| Perrier de la Bâthie (1940, 1946) | In his treatment of the Flore de Madagascar, continued to subdivide Malagasy Casearia, mainly adopting and revising Tulasne’s classification; added several new varieties (var. onivensis, var. ovata, var. subtrinervia, etc.) and new combinations based on C. nigrescens and others. | Continued to treat multiple “small-leaf” taxa as varieties under C. nigrescens, and revised species such as C. amplissima and C. parvifolia (= C. tulasneana). Provided substantial morphological evidence for classifying this genus in Madagascar at that time. | Perrier de la Bâthie, 1940, 1946 [15,16] |
| Sleumer (1971, 1980) | In revising Casearia from the Africa–Madagascar–Mascarene region, tended toward extensive lumping: most previously described Malagasy species (including those named by Perrier) were merged into C. nigrescens (further divided into var. nigrescens and var. lucida). | Emphasized the high degree of floral similarity among Malagasy species, thereby merging most morphologically distinct species into C. nigrescens, resulting in extreme nomenclatural “lumping.” However, many specimens did not fully match Sleumer’s classification morphologically, prompting future reexamination. | Sleumer, 1971, 1980 [17,18] |
| Alford (2005) | Based on molecular phylogenetics, proposed separating Samydaceae (or including it in a broadly circumscribed Salicaceae) from the “old,” polyphyletic Flacourtiaceae. Within this framework, Casearia shares lineage features with related genera (Laetia, Zuelania, Samyda, etc.). | Molecular data support a close relationship between Casearia and several smaller genera (Laetia, Samyda, Zuelania, etc.), but sampling of most Malagasy species was not detailed at the time. Also noted that some species groups and boundary issues remain unresolved in this genus. | Alford, 2005 [19] |
| Samarakoon and Alford (2019) | Integrated molecular and morphological evidence to merge Samyda, Laetia, Zuelania, etc. into Casearia; clarified that Casearia spans multiple lineages between the Old World and New World, although sampling of Malagasy and African species remains limited. | Molecular phylogenetic analyses revealed that Samyda, Laetia, Zuelania, etc., previously separated in traditional classifications, are nested within Casearia, thus supporting a broader “Casearia sensu lato.” Nonetheless, further evolutionary and taxonomic reexaminations are needed for various African–Malagasy species. | Samarakoon and Alford, 2019 [20] |
| Applequist and Gates (2020) | Emphasized that Malagasy Casearia urgently requires further revision; described two new Malagasy species (C. anosyensis and C. montigena). Also reaffirmed that Sleumer’s broad lumping of numerous small-leaf taxa into C. nigrescens does not adequately reflect actual diversity. | Stressed a comprehensive approach integrating morphology and taxonomy, confirming at least two new “small-leaf” species distinct from known taxa (e.g., C. tulasneana). Called for further molecular and micromorphological studies to refine classification of Malagasy and African Casearia; also conducted IUCN assessments (e.g., C. anosyensis was deemed Endangered). | Applequist and Gates, 2020 [22] |
| Species Names | C. kurzii | C. velutina | C. decandra | C. glomerata |
|---|---|---|---|---|
| Total length (bp) | 157,998 | 156,008 | 156,809 | 156,224 |
| LSC length (bp) | 85,806 | 84,446 | 84,890 | 84,564 |
| IRB length (bp) | 28,021 | 27,171 | 27,441 | 27,500 |
| SSC length (bp) | 16,150 | 17,220 | 17,037 | 16,660 |
| IRA length (bp) | 28,021 | 27,171 | 27,441 | 27,500 |
| Coding length (bp) | 78,334 | 79,662 | 80,322 | 77,730 |
| Non-coding length (bp) | 79,664 | 76,346 | 76,487 | 78,494 |
| Total number of genes | 127 | 130 | 130 | 124 |
| Protein-coding genes (duplicated) | 82 (6) | 85 (9) | 85 (9) | 80 (6) |
| tRNA genes (duplicated) | 37 (7) | 37 (7) | 37 (7) | 34 (7) |
| rRNA genes (duplicated) | 8 (4) | 8 (4) | 8 (4) | 8 (4) |
| Pseudo genes | 0 | 1 | 1 | 0 |
| GC content of genome (%) | 36.72 | 36.83 | 36.8 | 36.81 |
| GC content of LSC (%) | 34.46 | 34.61 | 34.57 | 34.59 |
| GC content of IRB (%) | 42.05 | 42.38 | 42.27 | 42.19 |
| GC content of SSC (%) | 30.21 | 30.19 | 30.29 | 30.33 |
| GC content of IRA (%) | 42.05 | 42.38 | 42.27 | 42.19 |
| Gene Function | Gene Category | Name of Genes | Total Number |
|---|---|---|---|
| Self-replication | Large subunit of ribosome | rpl20, rpl23(X2), rpl14, rpl33, rpl16 *, rpl36, rpl2 * (X2) | 9 |
| Small subunit of ribosome | rps11, rps14, rps15, rps16, rps2, rps3, rps18, rps4, rps19, rps7(X2), rps8, rps12 ** (X2) | 14 | |
| DNA dependent RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | 4 | |
| rRNA gene | rrn5(X2), rrn4.5(X2), rrn16(X2), rrn23(X2) | 8 | |
| tRNA gene | trnR-UCU, trnE-UUC, trnT-GGU, trnS-GGA, trnI-CAU(X2), trnV-GAC(X2), trnR-ACG(X2), trnL-UAA *, trnG-GCC, trnD-GUC, trnY-GUA, trnP-UGG, trnM-CAU, trnL-CAA(X2), trnS-GCU, trnW-CCA, trnF-GAA, trnT-UGU, trnS-UGA, trnV-UAC *, trnG-UCC *, trnL-UAG, trnI-GAU * (X2), trnH-GUG, trnfM-CAU, trnQ-UUG, trnN-GUU(X2), trnK-UUU *, trnA-UGC * (X2), trnC-GCA | 37 | |
| Gene for photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ | 5 |
| Subunits of photosystem II | psbL, psbZ, psbM, psbN, psbA, psbB, psbC, psbD, psbE, psbF, psbT, psbH, psbI, psbJ, psbK | 15 | |
| Subunits of NADH-dehydrogenase | ndhG, ndhH, ndhI, ndhJ, ndhK, ndhA *, ndhB * (X2), ndhC, ndhD, ndhE, ndhF | 12 | |
| Subunits of cytochrome b/f complex | petL, petN, petA, petB *, petD *, petG | 6 | |
| Subunit for ATP synthase | atpI, atpA, atpB, atpE, atpF *, atpH | 6 | |
| Large subunit of rubisco | rbcL | 1 | |
| Other genes | Maturase | matK | 1 |
| Protease | clpP ** | 1 | |
| Envelope membrane protein | cemA | 1 | |
| Subunit of Acetyl-carboxylase | accD | 1 | |
| C-type cytochrome synthesis gene | ccsA | 1 | |
| Unknown function | Open reading frames (ORF,ycf) | ycf1, ycf2(X2), ycf3 **, ycf4 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, K.; Li, W.; Sun, C.; Lu, X.; Zhou, X.; Wang, Y.; Tian, Y. Complete Chloroplast Genome Analysis of Casearia kurzii: Gene Loss at the IR Boundary and Monophyletic Evolution Within Casearia. Plants 2025, 14, 1356. https://doi.org/10.3390/plants14091356
Yan K, Li W, Sun C, Lu X, Zhou X, Wang Y, Tian Y. Complete Chloroplast Genome Analysis of Casearia kurzii: Gene Loss at the IR Boundary and Monophyletic Evolution Within Casearia. Plants. 2025; 14(9):1356. https://doi.org/10.3390/plants14091356
Chicago/Turabian StyleYan, Kan, Wandi Li, Chao Sun, Xin Lu, Xueqiong Zhou, Youyou Wang, and Yongqiang Tian. 2025. "Complete Chloroplast Genome Analysis of Casearia kurzii: Gene Loss at the IR Boundary and Monophyletic Evolution Within Casearia" Plants 14, no. 9: 1356. https://doi.org/10.3390/plants14091356
APA StyleYan, K., Li, W., Sun, C., Lu, X., Zhou, X., Wang, Y., & Tian, Y. (2025). Complete Chloroplast Genome Analysis of Casearia kurzii: Gene Loss at the IR Boundary and Monophyletic Evolution Within Casearia. Plants, 14(9), 1356. https://doi.org/10.3390/plants14091356