
Multi-Omics Analysis Revealed the Accumulation of Flavonoids and Shift of Fungal Community Structure Caused by Tea Grafting (Camellia sinensis L.)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
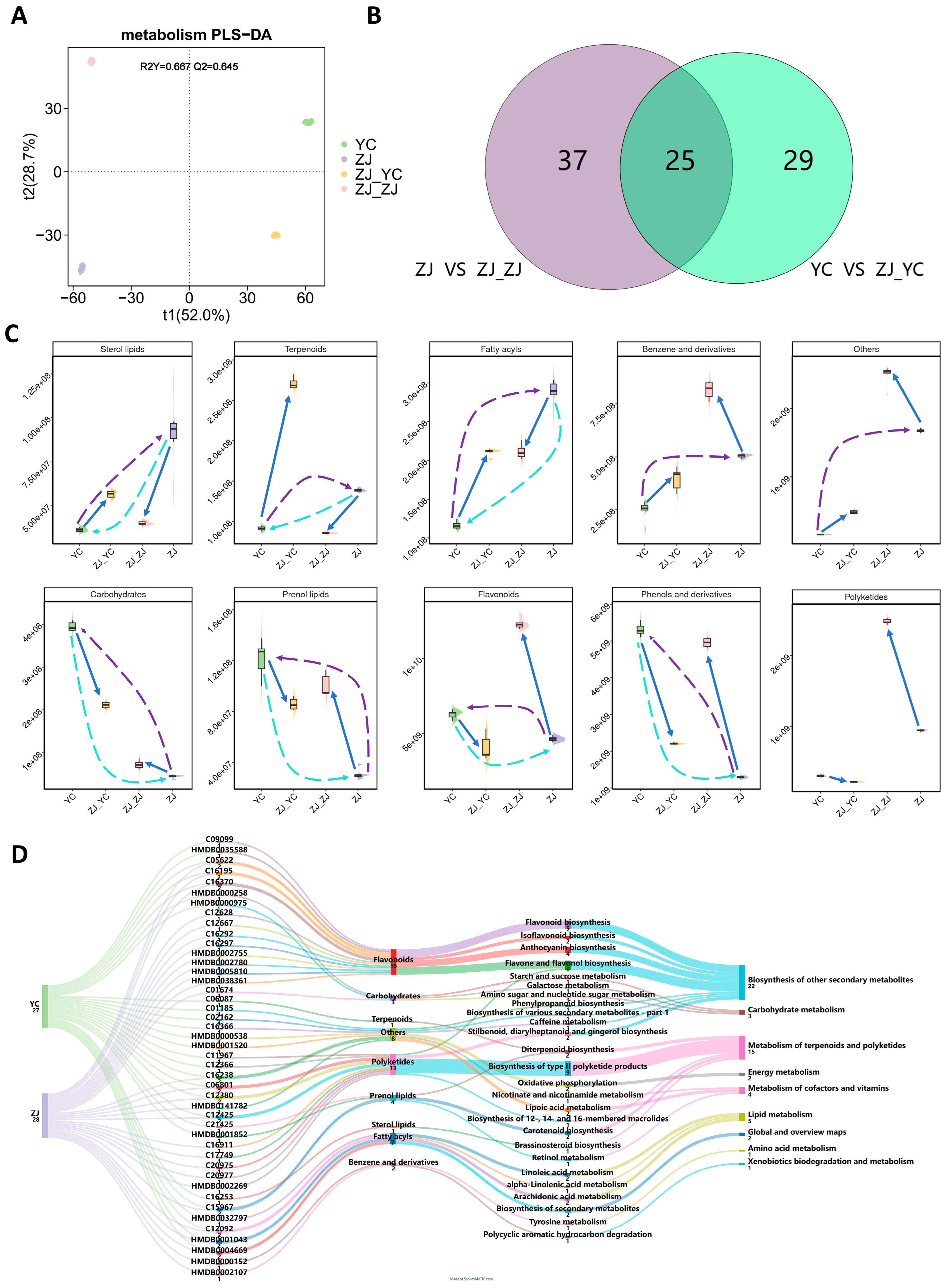
2.1. Metabolic Profile Shift in Tea Leaves After Grafting
2.2. Transcriptional Activity Driven by Grafting
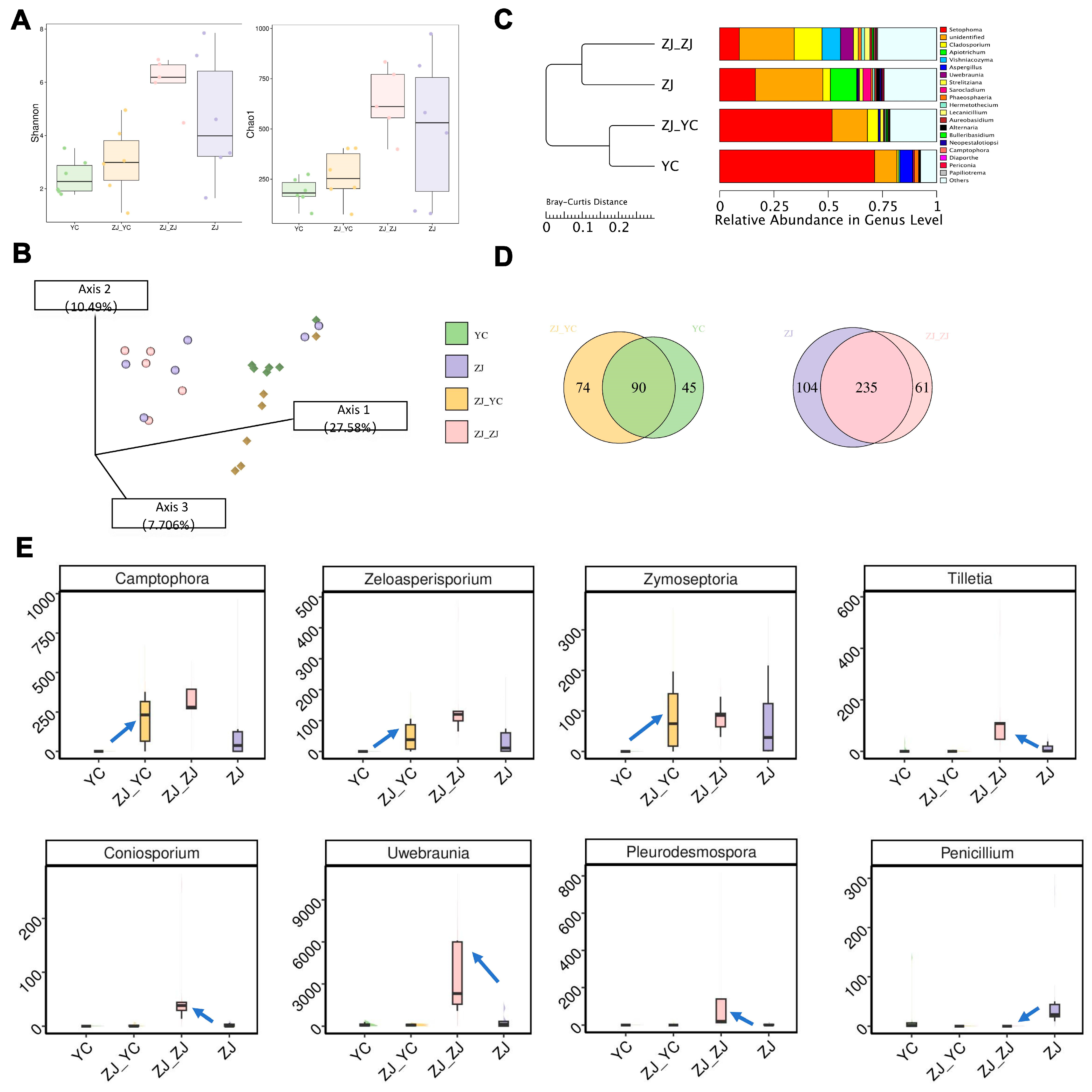
2.3. Shift in Microbial Communities’ Diversity by Grafting
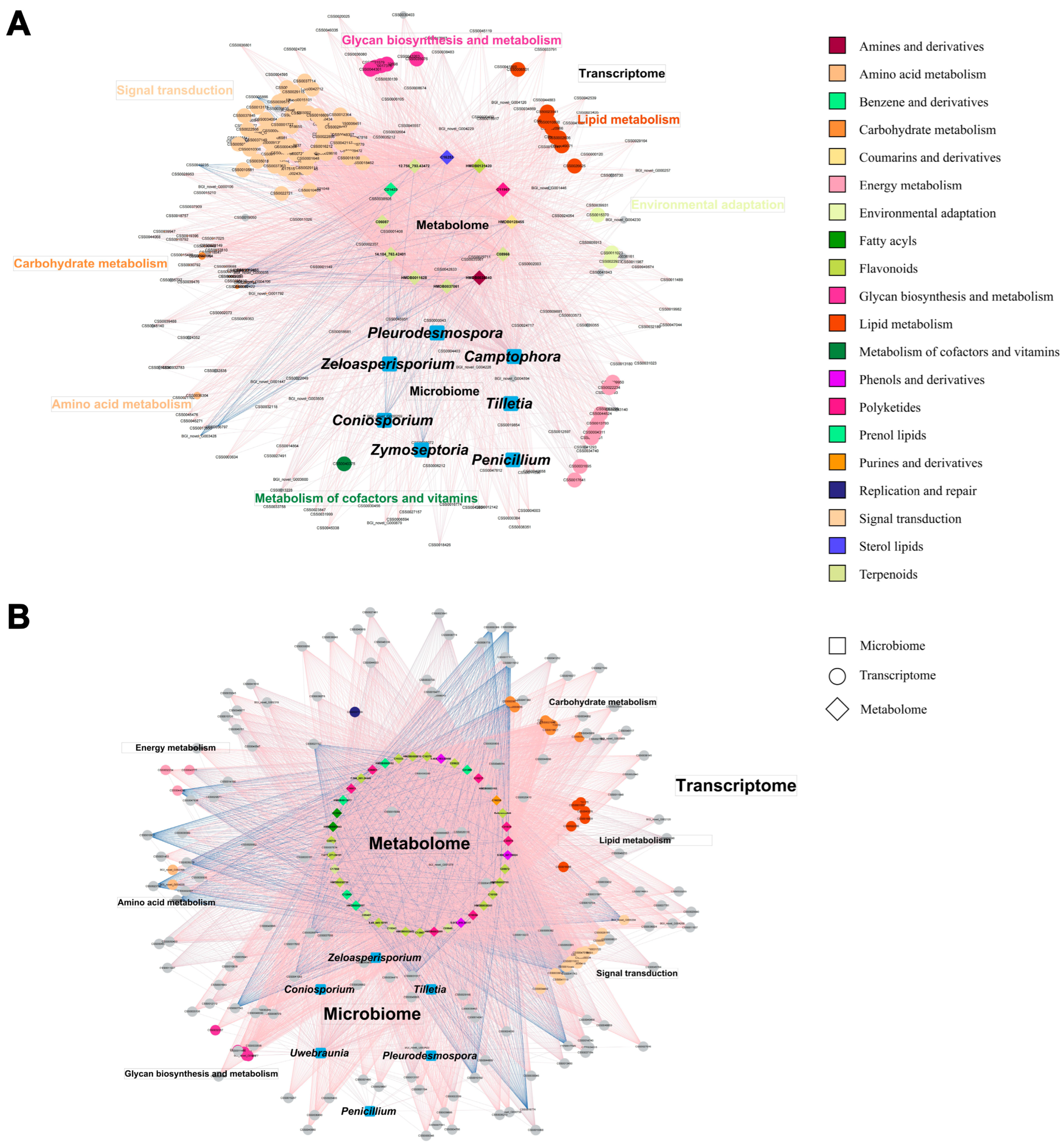
2.4. Responses of Metabolome, Transcriptome, and Microbiome of Tea Plant to Grafting
3. Discussion
4. Materials and Methods
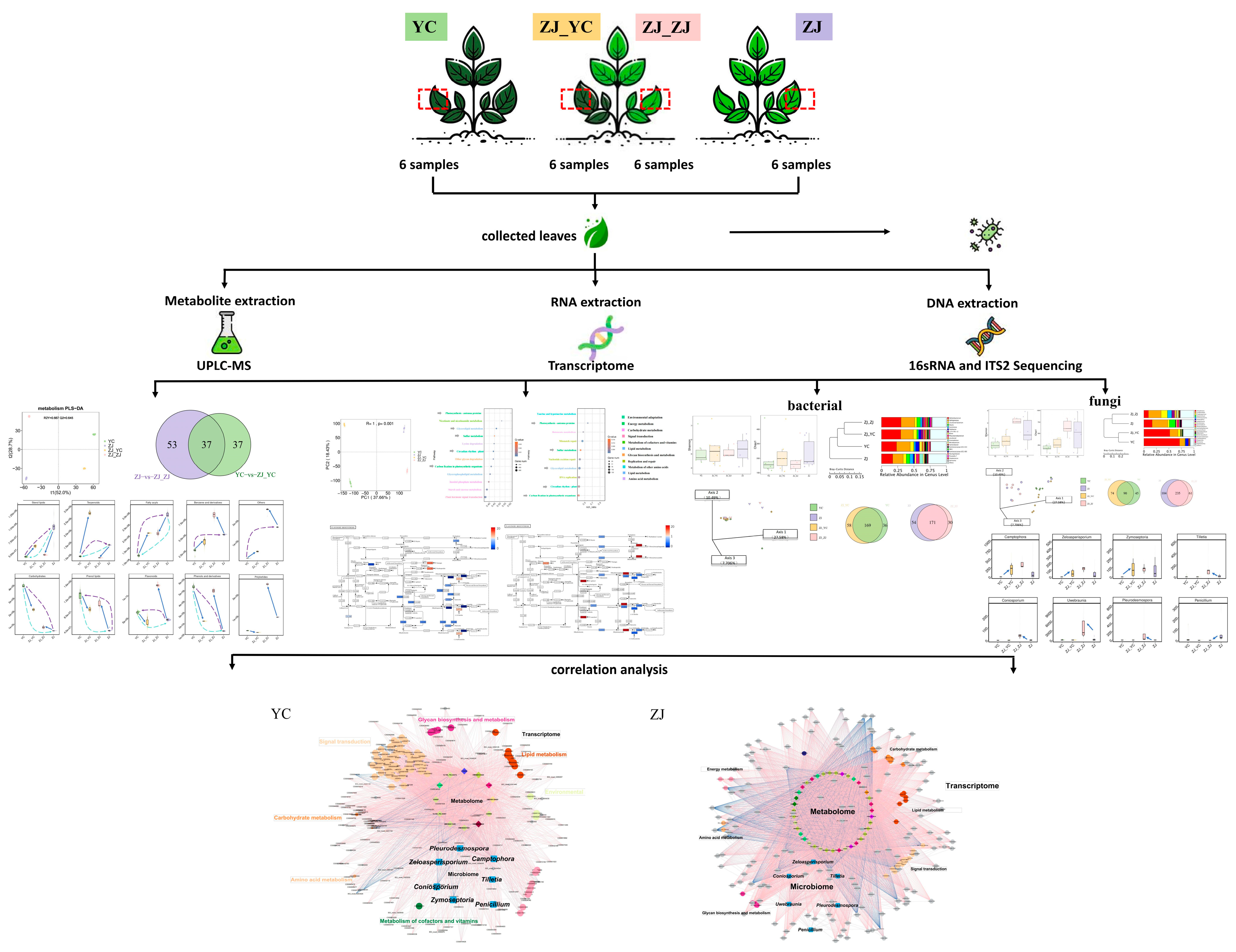
4.1. Experimental Design and Sampling
4.2. DNA Extraction of Fresh Tea Leaf Microbiome and Amplicon Sequencing
4.3. RNA Extraction of Fresh Tea Leaves and Library Construction
4.4. Metabolite Extraction and Identification
4.5. Bioinformatics Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kerio, L.C.; Wachira, F.N.; Wanyoko, J.K.; Rotich, M.K. Characterization of anthocyanins in Kenyan teas: Extraction and identification. Food Chem. 2012, 131, 31–38. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, Q.; Granato, D.; Xu, Y.; Ho, C. Association between chemistry and taste of tea: A review. Trends Food Sci. Technol. 2020, 101, 139–149. [Google Scholar] [CrossRef]
- Zhang, L.; Ho, C.T.; Zhou, J.; Santos, J.S.; Armstrong, L.; Granato, D. Chemistry and biological activities of processed Camellia sinensis teas: A comprehensive review. Compr. Rev. Food Sci. Food Saf. 2019, 18, 1474–1495. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Cheng, H.; Xu, P.; Wang, Y. Regulation of biosynthesis of the main flavor-contributing metabolites in tea plant (Camellia sinensis): A review. Crit. Rev. Food Sci. Nutr. 2023, 63, 10520–10535. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, G.; Li, X.; Shen, Q.; Wu, Q.; Zhuang, J.; Zhang, X.; Xia, E.; Zhang, Z.; Qian, Y.; et al. Comparative analysis of phenolic compound metabolism among tea plants in the section Thea of the genus Camellia. Food Res. Int. 2020, 135, 109276. [Google Scholar] [CrossRef]
- Li, M.; Shen, Y.; Ling, T.; Ho, C.T.; Li, D.; Guo, H.; Xie, Z. Analysis of differentiated chemical components between Zijuan purple tea and Yunkang green tea by UHPLC-Orbitrap-MS/MS combined with chemometrics. Foods 2021, 10, 1070. [Google Scholar] [CrossRef]
- Chen, L.; Shi, X.; Nian, B.; Duan, S.; Jiang, B.; Wang, X.; Lv, C.; Zhang, G.; Ma, Y.; Zhao, M. Alternative splicing regulation of anthocyanin biosynthesis in Camellia sinensis var. assamica unveiled by PacBio Iso-Seq. G3 2020, 10, 2713–2723. [Google Scholar] [CrossRef]
- Chen, W.; Qi, D.; Wang, W.; Miao, A.; Ma, C. GC-MS analysis combined with sensory analysis revealed the various aroma characteristics of black tea resulted from different grafting rootstocks. J. Food Sci. 2021, 86, 813–823. [Google Scholar] [CrossRef]
- Tuwei, G.; Kaptich, F.K.K.; Langat, M.C.; Chomboi, K.C.; Corley, R.H.V. Effects of grafting on tea1. growth, yield and quality. Exp. Agric. 2008, 44, 521–535. [Google Scholar] [CrossRef]
- Qi, D.; Li, J.; Qiao, X.; Lu, M.; Chen, W.; Miao, A.; Guo, W.; Ma, C. Non-targeted metabolomic analysis based on ultra-high-performance liquid chromatography quadrupole time-of-flight tandem mass spectrometry reveals the effects of grafting on non-volatile metabolites in fresh tea leaves (Camellia sinensis L.). J. Agric. Food Chem. 2019, 67, 6672–6682. [Google Scholar] [CrossRef]
- Cheng, Y.; Mao, J.; Cao, L.; Wang, H.; Li, S.; Jin, X.; Zhang, X.; Li, Y. Molecular basis of flavonoid accumulation in tea leaves grafted with Camellia sinensis var. assamica cv. “Yinghong9” as rootstock based on multi-omics analysis. Sci. Hortic. 2023, 321, 112290. [Google Scholar] [CrossRef]
- Yao, S.H.; Zhou, C.; Li, S.J.; Li, Y.H.; Shen, C.W.; Tao, Y.; Li, X. Microbial diversity across tea varieties and ecological niches: Correlating tea polyphenol contents with stress resistance. Front. Microbiol 2024, 15, 1439630. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Xie, H.; Liu, S.; Zhu, J.; Zhao, S.; Wei, C. Metabolites and transcriptional profiling analysis reveal the molecular mechanisms of the anthocyanin metabolism in the “Zijuan” tea plant (Camellia sinensis var. assamica). J. Agric. Food Chem. 2021, 69, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Summary of the National Plant Protection Variety Zijuan and Yuncha No.1 Experimental Demonstration Project. In Ke Ji Tui Guang; House, C.A.J.E.P., Ed.; Chinese Agricultural Science and Technology: Beijing, China, 2007. [Google Scholar]
- Zhang, J.; Tian, Y.; Xu, P.; Zhang, H. Breeding of high quality and disease resistance Yunnan big-leaf tea cultivar “Yuncha1”. J. Tea 2008, 34, 39–41+9. [Google Scholar]
- Wei, J.; Yang, Y.; Peng, Y.; Wang, S.; Zhang, J.; Liu, X.; Liu, J.; Wen, B.; Li, M. Biosynthesis and the transcriptional regulation of terpenoids in tea plants (Camellia sinensis). Int. J. Mol. Sci. 2023, 24, 6937. [Google Scholar] [CrossRef]
- Dong, D.; Shi, Y.N.; Mou, Z.M.; Chen, S.Y.; Zhao, D.K. Grafting: A potential method to reveal the differential accumulation mechanism of secondary metabolites. Hortic. Res. 2022, 9, uhac050. [Google Scholar] [CrossRef]
- Liu, Z.W.; Shi, X.Y.; Duan, S.M.; Nian, B.; Chen, L.J.; Zhang, G.H.; Lv, C.Y.; Ma, Y.; Zhao, M. Multiomics analysis of the mechanisms behind flavonoid differences between purple and green tender shoots of Camellia sinensis var. assamica. G3 2023, 13, jkac297. [Google Scholar] [CrossRef]
- Song, S.; Tao, Y.; Gao, L.; Liang, H.; Tang, D.; Lin, J.; Wang, Y.; Gmitter, F.G., Jr.; Li, C. An integrated metabolome and transcriptome analysis reveal the regulation mechanisms of flavonoid biosynthesis in a purple tea plant cultivar. Front. Plant. Sci. 2022, 13, 880227. [Google Scholar] [CrossRef]
- Ruan, Y.; Wang, T.; Guo, S.; Ling, N.; Shen, Q. Plant grafting shapes complexity and co-occurrence of rhizobacterial assemblages. Microb. Ecol. 2020, 80, 643–655. [Google Scholar] [CrossRef]
- Ramirez-Villacis, D.X.; Erazo-Garcia, P.; Quijia-Pillajo, J.; Llerena-Llerena, S.; Barriga-Medina, N.; Jones, C.D.; Leon-Reyes, A. Influence of grafting on rootstock rhizosphere microbiome assembly in Rosa sp. ’Natal Brier’. Biology 2023, 12, 663. [Google Scholar] [CrossRef]
- Lailheugue, V.; Darriaut, R.; Tran, J.; Morel, M.; Marguerit, E.; Lauvergeat, V. Both the scion and rootstock of grafted grapevines influence the rhizosphere and root endophyte microbiomes, but rootstocks have a greater impact. Environ. Microbiome 2024, 19, 24. [Google Scholar] [CrossRef]
- Aboody, M.S.A.; Mickymaray, S. Anti-fungal efficacy and mechanisms of flavonoids. Antibiotics 2020, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Mutha, R.E.; Tatiya, A.U.; Surana, S.J. Flavonoids as natural phenolic compounds and their role in therapeutics: An overview. Futur. J. Pharm. Sci. 2021, 7, 25. [Google Scholar] [CrossRef]
- Simonetti, G.; Brasili, E.; Pasqua, G. Antifungal activity of phenolic and polyphenolic compounds from different matrices of Vitis vinifera L. against human pathogens. Molecules 2020, 25, 3748. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, G.R.; Ellepola, K.; Seneviratne, C.J.; Koga-Ito, C.Y. Potential use of phenolic acids as anti-Candida agents: A Review. Front. Microbiol. 2015, 6, 1420. [Google Scholar] [CrossRef]
- Zhu, X.; Xia, L.; Chen, L.; Sun, Y.; Tian, Y.; Song, W.; Jiang, H. Full-length transcriptome analysis of protected cultivation ‘Yuncha 1’ (Camellia sinensis Var assamica). J. Tea Sci. 2018, 38, 193–201. [Google Scholar] [CrossRef]
- Sun, X.; Hu, Y.H.; Wang, J.; Fang, C.; Li, J.; Han, M.; Wei, X.; Zheng, H.; Luo, X.; Jia, Y.; et al. Efficient and stable metabarcoding sequencing data using a DNBSEQ-G400 sequencer validated by comprehensive community analyses. GigaByte 2021, 2021, gigabyte16. [Google Scholar] [CrossRef]
- Emmanuel, B.; Fagbola, O.; Abaidoo, R.; Osonubi, O.; Oyetunji, O. Abundance and distribution of arbuscular mycorrhizal fungi species in long-term soil fertility management systems in northern Nigeria. J. Plant Nutr. 2010, 33, 1264–1275. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 2018, 7, 1–6. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbo, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Liu, J.; Tian, Y.; Ye, S.; Pang, D.; Chen, L.; Qu, H. Multi-Omics Analysis Revealed the Accumulation of Flavonoids and Shift of Fungal Community Structure Caused by Tea Grafting (Camellia sinensis L.). Plants 2025, 14, 1176. https://doi.org/10.3390/plants14081176
Liu Y, Liu J, Tian Y, Ye S, Pang D, Chen L, Qu H. Multi-Omics Analysis Revealed the Accumulation of Flavonoids and Shift of Fungal Community Structure Caused by Tea Grafting (Camellia sinensis L.). Plants. 2025; 14(8):1176. https://doi.org/10.3390/plants14081176
Chicago/Turabian StyleLiu, Yue, Jun Liu, Yiping Tian, Shuang Ye, Dandan Pang, Linbo Chen, and Hao Qu. 2025. "Multi-Omics Analysis Revealed the Accumulation of Flavonoids and Shift of Fungal Community Structure Caused by Tea Grafting (Camellia sinensis L.)" Plants 14, no. 8: 1176. https://doi.org/10.3390/plants14081176
APA StyleLiu, Y., Liu, J., Tian, Y., Ye, S., Pang, D., Chen, L., & Qu, H. (2025). Multi-Omics Analysis Revealed the Accumulation of Flavonoids and Shift of Fungal Community Structure Caused by Tea Grafting (Camellia sinensis L.). Plants, 14(8), 1176. https://doi.org/10.3390/plants14081176