

A Protoplast System for CRISPR-Cas Ribonucleoprotein Delivery in Pinus taeda and Abies fraseri
 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. CRISPR-sgRNA Design for Knockout of the P. taeda PAL Gene
2.2. Initiation of Pinus taeda Somatic Embryogenic Tissue Cultures
2.3. Overnight Cell Wall Digestion Improves Transfection Efficiency of P. taeda Protoplasts
2.4. Transfection Buffer Conditions Affect Protoplast DNA-Uptake Efficiency
2.5. W5 Buffer Facilitates Post-Transfection Recovery of Protoplasts
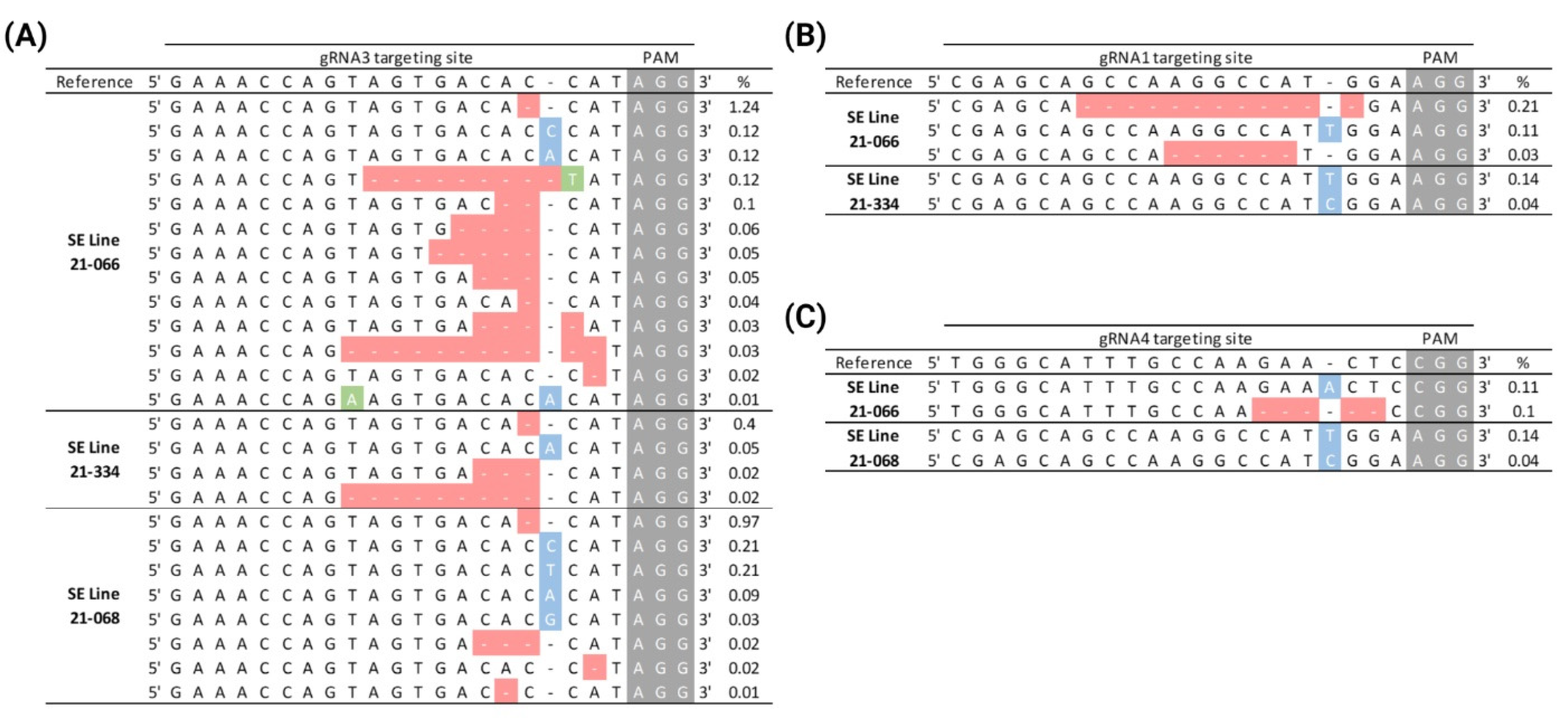
2.6. Genome Editing of PtPAL Through CRISPR-RNP Protoplast Transfection in P. taeda
2.7. CRISPR-sgRNA Design for Knockout of the A. fraseri PDS Gene
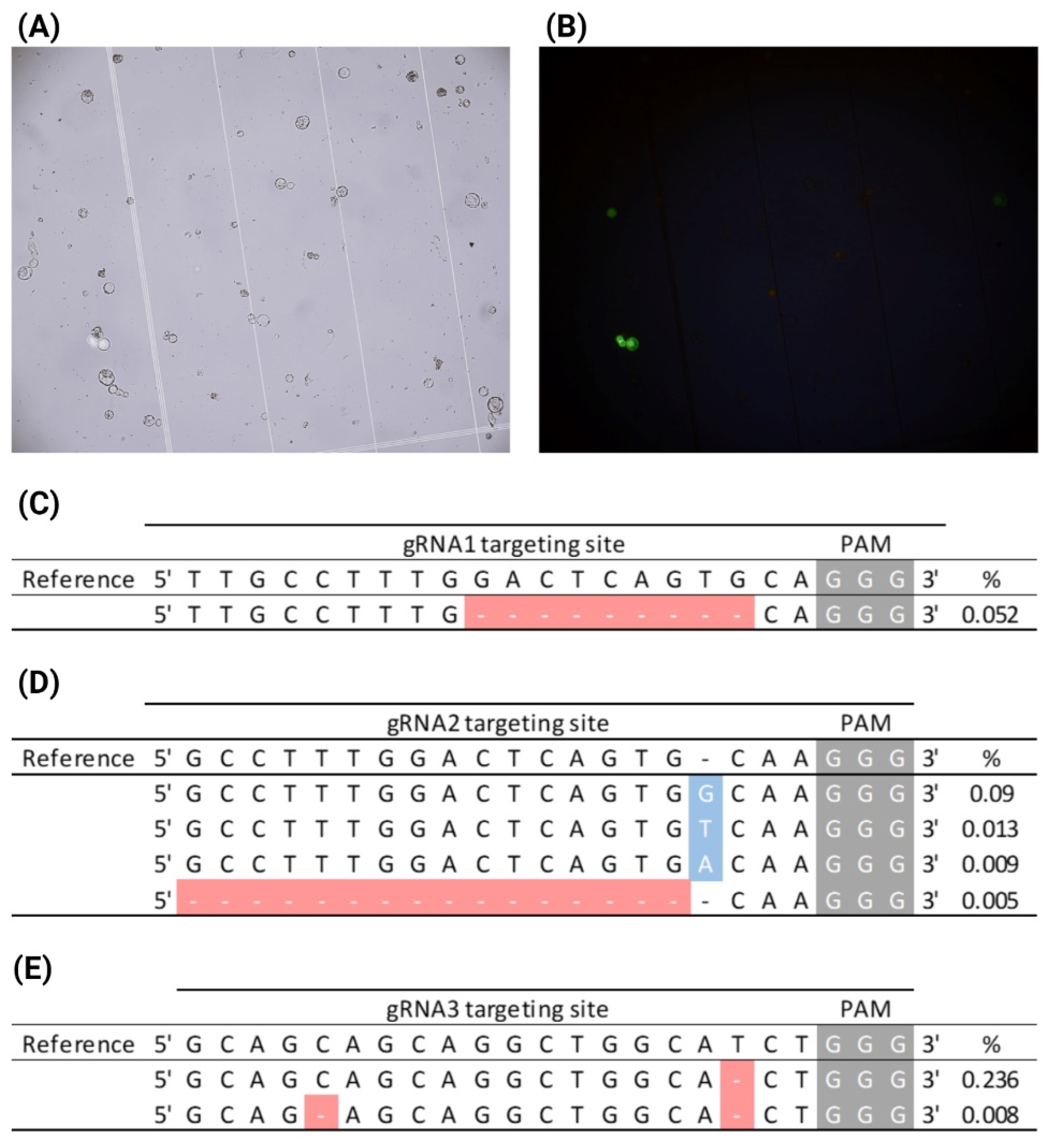
2.8. AfPDS Editing in CRISPR-RNP-Transfected A. fraseri Protoplasts
3. Discussion
4. Methods
4.1. Pinus taeda Somatic Embryogenic Tissue Initiation and Maintenance
4.2. Abies fraseri Somatic Embryogenic Tissue Initiation and Maintenance
4.3. Gene Amplification and Primer Design
4.3.1. Pinus taeda
4.3.2. Abies fraseri
4.4. Recombinant SpCas9 Expression and Purification
4.5. CRISPR sgRNA Design and CRISPR In Vitro Cleavage Assay
4.6. Protoplast Isolation
4.7. CRISPR-RNP Transfection
4.8. CRISPR-Driven Gene Editing Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Frenne, P.; Lenoir, J.; Luoto, M.; Scheffers, B.R.; Zellweger, F.; Aalto, J.; Ashcroft, M.B.; Christiansen, D.M.; Decocq, G.; De Pauw, K. Forest microclimates and climate change: Importance, drivers and future research agenda. Glob. Change Biol. 2021, 27, 2279–2297. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization of the United Nations. Global Forest Resources Assessment. 2020. Available online: https://www.fao.org/interactive/forest-resources-assessment/2020/en/ (accessed on 14 February 2025).
- Xiao, S.; Chen, C.; Xia, Q.; Liu, Y.; Yao, Y.; Chen, Q.; Hartsfield, M.; Brozena, A.; Tu, K.; Eichhorn, S.J. Lightweight, strong, moldable wood via cell wall engineering as a sustainable structural material. Science 2021, 374, 465–471. [Google Scholar] [PubMed]
- U.S. Environmental Protection Agency. Greenhouse Gas Reporting Program (GHGRP). 2021. Available online: https://www.epa.gov/ghgreporting/ghgrp-pulp-and-paper#map-facilities (accessed on 10 March 2025).
- Vanholme, R.; Cesarino, I.; Rataj, K.; Xiao, Y.; Sundin, L.; Goeminne, G.; Kim, H.; Cross, J.; Morreel, K.; Araujo, P. Caffeoyl shikimate esterase (CSE) is an enzyme in the lignin biosynthetic pathway in Arabidopsis. Science 2013, 341, 1103–1106. [Google Scholar] [PubMed]
- Sulis, D.B.; Jiang, X.; Yang, C.; Marques, B.M.; Matthews, M.L.; Miller, Z.; Lan, K.; Cofre-Vega, C.; Liu, B.; Sun, R. Multiplex CRISPR editing of wood for sustainable fiber production. Science 2023, 381, 216–221. [Google Scholar]
- Sulis, D.B.; Wang, J.P. Regulation of lignin biosynthesis by post-translational protein modifications. Front. Plant Sci. 2020, 11, 544521. [Google Scholar]
- Susaeta, A.; Carter, D.R.; Adams, D.C. Impacts of climate change on economics of forestry and adaptation strategies in the southern United States. J. Agric. Appl. Econ. 2014, 46, 257–272. [Google Scholar]
- Pureswaran, D.S.; Roques, A.; Battisti, A. Forest insects and climate change. Curr. For. Rep. 2018, 4, 35–50. [Google Scholar] [CrossRef]
- United States Department of Agriculture, National Agricultural Statistics Service. Census of Agriculture. 2017. Available online: www.nass.usda.gov/AgCensus (accessed on 14 February 2025).
- Potter, K.M.; Hargrove, W.W.; Koch, F.H. Predicting climate change extirpation risk for central and southern Appalachian forest tree species. In Proceedings of the Conference on the Ecology and Management of High-Elevation Forests in the Central and Southern Appalachian Mountains, Snowshoe, WV, USA, 14–15 May 2010. [Google Scholar]
- Kaylor, S.D.; Hughes, M.J.; Franklin, J.A. Recovery trends and predictions of Fraser fir (Abies fraseri) dynamics in the Southern Appalachian Mountains. Can. J. For. Res. 2017, 47, 125–133. [Google Scholar]
- Mckeever, K.M.; Chastagner, G.A.; Pathology, P.; State, W. A Survey of Phytophthora spp. Associated with Abies in U.S. Christmas Tree Farms. Plant Disease 2016, 100, 1161–1169. [Google Scholar]
- Prunier, J.; Verta, J.P.; MacKay, J.J. Conifer genomics and adaptation: At the crossroads of genetic diversity and genome function. New Phytol. 2016, 209, 44–62. [Google Scholar]
- Kovach, A.; Wegrzyn, J.L.; Parra, G.; Holt, C.; Bruening, G.E.; Loopstra, C.A.; Hartigan, J.; Yandell, M.; Langley, C.H.; Korf, I. The Pinus taeda genome is characterized by diverse and highly diverged repetitive sequences. BMC Genom. 2010, 11, 420. [Google Scholar]
- Liu, B.; Liu, J.; Yu, J.; Wang, Z.; Sun, Y.; Li, S.; Lin, Y.-C.J.; Chiang, V.L.; Li, W.; Wang, J.P. Transcriptional reprogramming of xylem cell wall biosynthesis in tension wood. Plant Physiol. 2021, 186, 250–269. [Google Scholar] [PubMed]
- Chen, H.; Wang, J.P.; Liu, H.; Li, H.; Lin, Y.-C.J.; Shi, R.; Yang, C.; Gao, J.; Zhou, C.; Li, Q. Hierarchical transcription factor and chromatin binding network for wood formation in Populus trichocarpa. Plant Cell 2019, 31, 602–626. [Google Scholar] [PubMed]
- Wang, Z.; Mao, Y.; Guo, Y.; Gao, J.; Liu, X.; Li, S.; Lin, Y.-C.J.; Chen, H.; Wang, J.P.; Chiang, V.L. MYB transcription factor161 mediates feedback regulation of secondary wall-associated NAC-Domain1 family genes for wood formation. Plant Physiol. 2020, 184, 1389–1406. [Google Scholar]
- González, M.N.; Massa, G.A.; Andersson, M.; Turesson, H.; Olsson, N.; Fält, A.-S.; Storani, L.; Décima Oneto, C.A.; Hofvander, P.; Feingold, S.E. Reduced enzymatic browning in potato tubers by specific editing of a polyphenol oxidase gene via ribonucleoprotein complexes delivery of the CRISPR/Cas9 system. Front. Plant Sci. 2020, 10, 1649. [Google Scholar]
- Woo, J.W.; Kim, J.; Kwon, S.I.; Corvalán, C.; Cho, S.W.; Kim, H.; Kim, S.-G.; Kim, S.-T.; Choe, S.; Kim, J.-S. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat. Biotechnol. 2015, 33, 1162–1164. [Google Scholar]
- Fan, Y.; Xin, S.; Dai, X.; Yang, X.; Huang, H.; Hua, Y. Efficient genome editing of rubber tree (Hevea brasiliensis) protoplasts using CRISPR/Cas9 ribonucleoproteins. Ind. Crops Prod. 2020, 146, 112146. [Google Scholar]
- Pavese, V.; Moglia, A.; Abbà, S.; Milani, A.M.; Torello Marinoni, D.; Corredoira, E.; Martínez, M.T.; Botta, R. First Report on Genome Editing via Ribonucleoprotein (RNP) in Castanea sativa Mill. Int. J. Mol. Sci. 2022, 23, 5762. [Google Scholar] [CrossRef]
- Qin, G.; Gu, H.; Ma, L.; Peng, Y.; Deng, X.W.; Chen, Z.; Qu, L.-J. Disruption of phytoene desaturase gene results in albino and dwarf phenotypes in Arabidopsis by impairing chlorophyll, carotenoid, and gibberellin biosynthesis. Cell Res. 2007, 17, 471–482. [Google Scholar]
- Murovec, J.; Guček, K.; Bohanec, B.; Avbelj, M.; Jerala, R. DNA-free genome editing of Brassica oleracea and B. rapa protoplasts using CRISPR-Cas9 ribonucleoprotein complexes. Front. Plant Sci. 2018, 9, 1594. [Google Scholar] [CrossRef]
- Banakar, R.; Schubert, M.; Collingwood, M.; Vakulskas, C.; Eggenberger, A.L.; Wang, K. Comparison of CRISPR-Cas9/Cas12a ribonucleoprotein complexes for genome editing efficiency in the rice phytoene desaturase (OsPDS) gene. Rice 2020, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, H.; Liu, J.; Yang, Q.; Shao, X.; Bi, F.; Hu, C.; Huo, H.; Chen, K.; Yi, G. Establishment of a PEG-mediated protoplast transformation system based on DNA and CRISPR/Cas9 ribonucleoprotein complexes for banana. BMC Plant Biol. 2020, 20, 425. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Zhang, C.; Yu, L.; Yang, J.; Li, C. CRISPR/Cas9 ribonucleoprotein mediated DNA-free genome editing in larch. For. Res. 2024, 4, e036. [Google Scholar] [CrossRef] [PubMed]
- Poovaiah, C.; Phillips, L.; Geddes, B.; Reeves, C.; Sorieul, M.; Thorlby, G. Genome editing with CRISPR/Cas9 in Pinus radiata (D. Don). BMC Plant Biol. 2021, 21, 363. [Google Scholar] [CrossRef]
- Cao, H.X.; Vu, G.T.H.; Gailing, O. From Genome Sequencing to CRISPR-Based Genome Editing for Climate-Resilient Forest Trees. Int. J. Mol. Sci. 2022, 23, 966. [Google Scholar] [CrossRef]
- Chang, S.; Mahon, E.L.; MacKay, H.A.; Rottmann, W.H.; Strauss, S.H.; Pijut, P.M.; Powell, W.A.; Coffey, V.; Lu, H.; Mansfield, S.D. Genetic engineering of trees: Progress and new horizons. Vitr. Cell. Dev. Biol.-Plant 2018, 54, 341–376. [Google Scholar] [CrossRef]
- Wang, J.P.; Matthews, M.L.; Williams, C.M.; Shi, R.; Yang, C.; Tunlaya-Anukit, S.; Chen, H.-C.; Li, Q.; Liu, J.; Lin, C.-Y. Improving wood properties for wood utilization through multi-omics integration in lignin biosynthesis. Nat. Commun. 2018, 9, 1579. [Google Scholar] [CrossRef]
- Wang, J.P.; Liu, B.; Sun, Y.; Chiang, V.L.; Sederoff, R.R. Enzyme-enzyme interactions in monolignol biosynthesis. Front. Plant Sci. 2019, 9, 1942. [Google Scholar] [CrossRef]
- Matthews, M.L.; Wang, J.P.; Sederoff, R.; Chiang, V.L.; Williams, C.M. Modeling cross-regulatory influences on monolignol transcripts and proteins under single and combinatorial gene knockdowns in Populus trichocarpa. PLoS Comput. Biol. 2020, 16, e1007197. [Google Scholar] [CrossRef]
- Matthews, M.L.; Wang, J.P.; Sederoff, R.; Chiang, V.L.; Williams, C.M. A multiscale model of lignin biosynthesis for predicting bioenergy traits in Populus trichocarpa. Comput. Struct. Biotechnol. J. 2021, 19, 168–182. [Google Scholar] [CrossRef]
- Whetten, R.; Sederoff, R. Lignin biosynthesis. Plant Cell 1995, 7, 1001. [Google Scholar] [PubMed]
- Shi, R.; Shuford, C.M.; Wang, J.P.; Sun, Y.-H.; Yang, Z.; Chen, H.-C.; Tunlaya-Anukit, S.; Li, Q.; Liu, J.; Muddiman, D.C. Regulation of phenylalanine ammonia-lyase (PAL) gene family in wood forming tissue of Populus trichocarpa. Planta 2013, 238, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, C.-J. Multifaceted regulations of gateway enzyme phenylalanine ammonia-lyase in the biosynthesis of phenylpropanoids. Mol. Plant 2015, 8, 17–27. [Google Scholar] [PubMed]
- Vejnar, C.E.; Moreno-Mateos, M.A.; Cifuentes, D.; Bazzini, A.A.; Giraldez, A.J. Optimized CRISPR–Cas9 system for genome editing in zebrafish. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot086850. [Google Scholar] [CrossRef]
- Pullman, G.S. Embryogenic tissue initiation in loblolly pine (Pinus taeda L.). In Step Wise Protocols for Somatic Embryogenesis of Important Woody Plants; Springer: Berlin/Heidelberg, Germany, 2018; pp. 13–31. [Google Scholar]
- Cairney, J.; Pullman, G.S. The cellular and molecular biology of conifer embryogenesis. New Phytol. 2007, 176, 511–536. [Google Scholar]
- Lin, Y.-C.; Li, W.; Chen, H.; Li, Q.; Sun, Y.-H.; Shi, R.; Lin, C.-Y.; Wang, J.P.; Chen, H.-C.; Chuang, L. A simple improved-throughput xylem protoplast system for studying wood formation. Nat. Protoc. 2014, 9, 2194–2205. [Google Scholar]
- Hsu, C.-T.; Lee, W.-C.; Cheng, Y.-J.; Yuan, Y.-H.; Wu, F.-H.; Lin, C.-S. Genome editing and protoplast regeneration to study plant–pathogen interactions in the model plant Nicotiana benthamiana. Front. Genome Ed. 2021, 2, 39. [Google Scholar]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Chou, E.Y. Understanding the Patterned Deposition of Lignin in Secondary Cell Walls; University of British Columbia: Vancouver, BC, Canada, 2016. [Google Scholar]
- Masani, M.Y.A.; Noll, G.A.; Parveez, G.K.A.; Sambanthamurthi, R.; Prüfer, D. Efficient transformation of oil palm protoplasts by PEG-mediated transfection and DNA microinjection. PLoS ONE 2014, 9, e96831. [Google Scholar]
- Malnoy, M.; Viola, R.; Jung, M.-H.; Koo, O.-J.; Kim, S.; Kim, J.-S.; Velasco, R.; Nagamangala Kanchiswamy, C. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Front. Plant Sci. 2016, 7, 1904. [Google Scholar] [CrossRef]
- Carlsen, F.M.; Johansen, I.E.; Yang, Z.; Liu, Y.; Westberg, I.N.; Kieu, N.P.; Jørgensen, B.; Lenman, M.; Andreasson, E.; Nielsen, K.L. Strategies for efficient gene editing in protoplasts of Solanum tuberosum theme: Determining gRNA efficiency design by utilizing protoplast. Front. Genome Ed. 2022, 3, 795644. [Google Scholar]
- Zhang, Y.; Ren, Q.; Tang, X.; Liu, S.; Malzahn, A.A.; Zhou, J.; Wang, J.; Yin, D.; Pan, C.; Yuan, M. Expanding the scope of plant genome engineering with Cas12a orthologs and highly multiplexable editing systems. Nat. Commun. 2021, 12, 1944. [Google Scholar] [PubMed]
- Tang, W.; Newton, R. Genetic transformation of conifers and its application in forest biotechnology. Plant Cell Rep. 2003, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Klimaszewska, K.; Hargreaves, C.; Lelu-Walter, M.-A.; Trontin, J.-F. Advances in conifer somatic embryogenesis since year 2000. Vitr. Embryog. High. Plants 2016, 1359, 131–166. [Google Scholar]
- Trontin, J.-F.; Walter, C.; Klimaszewska, K.; Park, Y.-S.; Lelu-Walter, M.-A. Recent progress in genetic transformation of four Pinus spp. Transgenic Plant J. 2007, 1, 314–329. [Google Scholar]
- Bewg, W.P.; Ci, D.; Tsai, C.-J. Genome editing in trees: From multiple repair pathways to long-term stability. Front. Plant Sci. 2018, 9, 1732. [Google Scholar]
- Tsai, C.-J.; Xue, L.-J. CRISPRing into the woods. GM Crops Food 2015, 6, 206–215. [Google Scholar] [CrossRef]
- Pullman, G.S.; Olson, K.; Egertsdotter, U.; Bucalo, K. Fraser fir somatic embryogenesis: High frequency initiation, maintenance, embryo development, germination and cryopreservation. New For. 2016, 47, 453–480. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar]
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files, version 1.33; GitHub Inc.: San Francisco, CA, USA, 2014. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar]
- Hart, A.J.; Ginzburg, S.; Xu, M.; Fisher, C.R.; Rahmatpour, N.; Mitton, J.B.; Paul, R.; Wegrzyn, J.L. EnTAP: Bringing faster and smarter functional annotation to non-model eukaryotic transcriptomes. Mol. Ecol. Resour. 2020, 20, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [PubMed]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Efficiency Scores | Outcome Scores | |||||
|---|---|---|---|---|---|---|
| Name | Target Sequence | Position | Doench’16 | Moreno-Mateos | Out-of-Frame | Lindel |
| PtPAL_gRNA1 | CGAGCAGCCAAGGCCATGGA | 106–125 | 55 | 57 | 65 | 84 |
| PtPAL_gRNA2 | CACTTGCGATCTTCGAGCAA | 230–249 | 59 | 58 | 65 | 81 |
| PtPAL_gRNA3 | GAAACCAGTAGTGACACCAT | 341–360 | 66 | 60 | 54 | 69 |
| PtPAL_gRNA4 | TGGGCATTTGCCAAGAACTC | 434–453 | 51 | 66 | 80 | 77 |
| PtPAL_gRNA5 | CCCGGGCTGCCATGCTGGTT | 479–498 | 39 | 72 | 62 | 68 |
| AfPDS_gRNA1 | TTGCCTTTGGACTCAGTGCA | 61–80 | 55 | 50 | 53 | 91 |
| AfPDS_gRNA2 | TGCCTTTGGACTCAGTGCAA | 62–81 | 59 | 59 | 60 | 77 |
| AfPDS_gRNA3 | GCAGCAGCAGGCTGGCATCT | 110–129 | 45 | 49 | 63 | 86 |
| AfPDS_gRNA4 | TCTGGGAGGAGTGAATATTT | 127–146 | 39 | 57 | 66 | 75 |
| Media and Components (mg/L) | |||
|---|---|---|---|
| PTIM | PTMM | AFMM | |
| NH4NO3 | 200 | 200 | ˉ |
| KNO3 | 909.9 | 900 | ˉ |
| KH2PO4 | 136.1 | 130 | 340 |
| Ca(NO3)2·4H2O | 236.2 | 230 | ˉ |
| MgSO4·7H2O | 246.5 | 250 | 394 |
| Mg(NO3)2·6H2O | 256.5 | 260 | ˉ |
| MgCl2·6H2O | 50 | 100 | ˉ |
| CaSO4·2H2O | ˉ | ˉ | 37.8 |
| H3PO4 | ˉ | ˉ | 373 |
| KI | 4.15 | 4.15 | 0.083 |
| H3BO3 | 15.5 | 15.5 | 2.48 |
| MnSO4·H2O | 10.5 | 10.5 | 18.6 |
| ZnSO4·7H2O | 14.668 | 14.4 | 5.76 |
| NaMoO4·2H2O | 0.125 | 0.125 | 0.103 |
| CuSO4·5H2O | 0.1725 | 0.125 | 3.75 |
| COCl2·6H2O | 0.125 | 0.125 | 0.012 |
| NiCl2·6H2O | ˉ | ˉ | 1.188 |
| AgNO3 | 3.398 | ˉ | ˉ |
| FeSO4·7H2O | 13.9 | 13.9 | ˉ |
| Na2EDTA | 18.65 | 18.65 | ˉ |
| Maltose | 15,000 | ˉ | ˉ |
| Sucrose | ˉ | 15,000 | 10,000 |
| Myo-inositol | 100 | 500 | 1000 |
| C12H10Mg3O14·9H2O | ˉ | ˉ | 266 |
| Casamino acids | 500 | ˉ | ˉ |
| L-glutamine | 450 | 2500 | 2000 |
| Thiamine HCl | 1 | 1 | 1 |
| Pyridoxine HCl | 0.5 | 0.5 | 0.5 |
| Nicotinic Acid | 0.5 | 0.5 | 0.5 |
| Glycine | 2 | 2 | ˉ |
| D-xylose | 100 | ˉ | ˉ |
| MES | 250 | ˉ | ˉ |
| Biotin | 0.05 | ˉ | ˉ |
| Folic acid | 0.5 | ˉ | ˉ |
| Vitamin B12 | 0.1 | ˉ | ˉ |
| Vitamin E | 0.1 | ˉ | ˉ |
| α-ketoglutaric acid | 100 | ˉ | ˉ |
| Sodium thiosulfate | 1 mM | ˉ | ˉ |
| NAA | 2 | ˉ | ˉ |
| 2,4-D | ˉ | 2 | ˉ |
| BAP | 0.63 | 0.333 | 1.1 |
| Kinetin | 0.61 | ˉ | ˉ |
| Abscisic acid | ˉ | 5 | ˉ |
| 24-epibrassinolide | 2.1 μM | ˉ | ˉ |
| Gellan Gum | 5000 | 5000 | 3000 |
| pH | 5.7 | 5.7 | 5.7 |
| Name | Sequence | Purpose |
|---|---|---|
| AfPDS_gRNA1 | TAATACGACTCACTATAGTTGCCTTTGGACTCAGTGCAGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGG | In vitro transcription |
| AfPDS_gRNA2 | TAATACGACTCACTATAGTGCCTTTGGACTCAGTGCAAGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGG | In vitro transcription |
| AfPDS_gRNA3 | TAATACGACTCACTATAGGCAGCAGCAGGCTGGCATCTGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGG | In vitro transcription |
| AfPDS_gRNA4 | TAATACGACTCACTATAGTCTGGGAGGAGTGAATATTTGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGG | In vitro transcription |
| AfPDS_Forward | GCGTTTCAAGGGTGGATTC | AfPDS amplification |
| AfPDS_Reverse | GTCCTTTGCAGGTTACATGC | AfPDS amplification |
| AfPDS_Forward_seq | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGCGTTTCAAGGGTGGATTC | AfPDS NGS sequencing |
| AfPDS_Reverse_seq | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTGCATGTAACCTGCAAAGGAC | AfPDS NGS sequencing |
| PtPAL_gRNA1 | TAATACGACTCACTATAGCGAGCAGCCAAGGCCATGGAGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGG | In vitro transcription |
| PtPAL_gRNA2 | TAATACGACTCACTATAGCACTTGCGATCTTCGAGCAAGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGG | In vitro transcription |
| PtPAL_gRNA3 | TAATACGACTCACTATAGGAAACCAGTAGTGACACCATGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGG | In vitro transcription |
| PtPAL_gRNA4 | TAATACGACTCACTATAGTGGGCATTTGCCAAGAACTCGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGG | In vitro transcription |
| PtPAL_gRNA5 | TAATACGACTCACTATAGCCCGGGCTGCCATGCTGGTTGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGG | In vitro transcription |
| PtPAL_Forward | CAGCAGCAGAAATAACGCA | PtPAL amplification |
| PtPAL_Reverse | CTGTTGAATGCGTGGCTGA | PtPAL amplification |
| PtPAL_Forward_seq | ACACTCTTTCCCTACACGACGCTCTTCCGATCTCAGCAGCAGAAATAACGCA | PtPAL NGS sequencing |
| PtPAL_Reverse_seq | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTGTTGAATGCGTGGCTGA | PtPAL NGS sequencing |
| BS6 | AAAAAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTAAACTTGCTATGCTGTTTCCAGC | In vitro transcription |
| BS7 | AAAAAAAGCACCGACTCGGTGC | In vitro transcription |
| T25-long | GAAATTAATACGACTCACTATAG | In vitro transcription |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, B.M.; Sulis, D.B.; Suarez, B.; Yang, C.; Cofre-Vega, C.; Thomas, R.D.; Whitehill, J.G.A.; Whetten, R.W.; Barrangou, R.; Wang, J.P. A Protoplast System for CRISPR-Cas Ribonucleoprotein Delivery in Pinus taeda and Abies fraseri. Plants 2025, 14, 996. https://doi.org/10.3390/plants14070996
Marques BM, Sulis DB, Suarez B, Yang C, Cofre-Vega C, Thomas RD, Whitehill JGA, Whetten RW, Barrangou R, Wang JP. A Protoplast System for CRISPR-Cas Ribonucleoprotein Delivery in Pinus taeda and Abies fraseri. Plants. 2025; 14(7):996. https://doi.org/10.3390/plants14070996
Chicago/Turabian StyleMarques, Barbara M., Daniel B. Sulis, Bethany Suarez, Chenmin Yang, Carlos Cofre-Vega, Robert D. Thomas, Justin G. A. Whitehill, Ross W. Whetten, Rodolphe Barrangou, and Jack P. Wang. 2025. "A Protoplast System for CRISPR-Cas Ribonucleoprotein Delivery in Pinus taeda and Abies fraseri" Plants 14, no. 7: 996. https://doi.org/10.3390/plants14070996
APA StyleMarques, B. M., Sulis, D. B., Suarez, B., Yang, C., Cofre-Vega, C., Thomas, R. D., Whitehill, J. G. A., Whetten, R. W., Barrangou, R., & Wang, J. P. (2025). A Protoplast System for CRISPR-Cas Ribonucleoprotein Delivery in Pinus taeda and Abies fraseri. Plants, 14(7), 996. https://doi.org/10.3390/plants14070996