
Multi-Omics Analysis Reveals Differential Molecular Responses of RNA Polymerase Common Subunit ZmRPABC5b for Seedling Development in Maize
 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
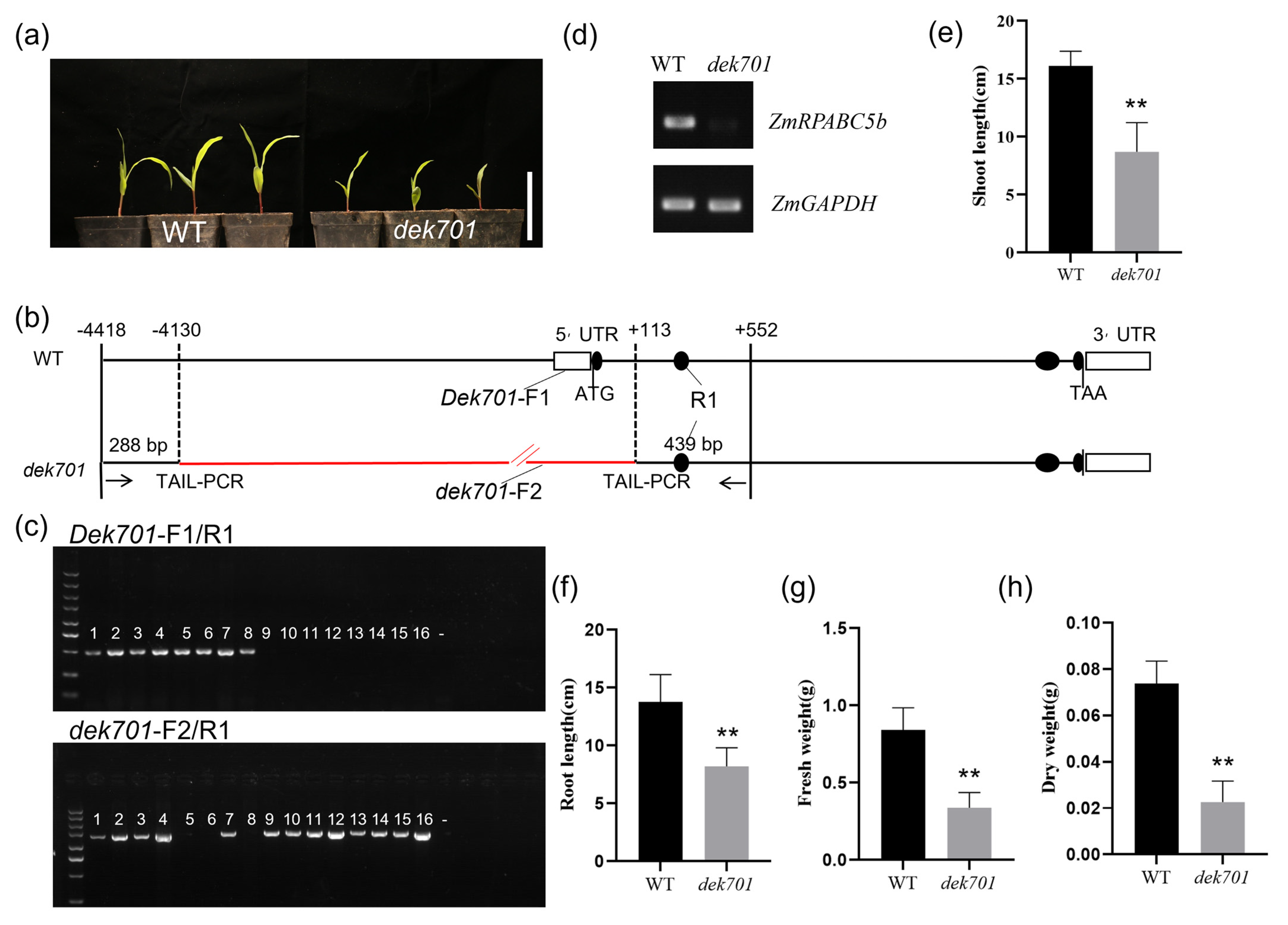
2.1. ZmRPABC5b Mutation Affects Seedlings Growth
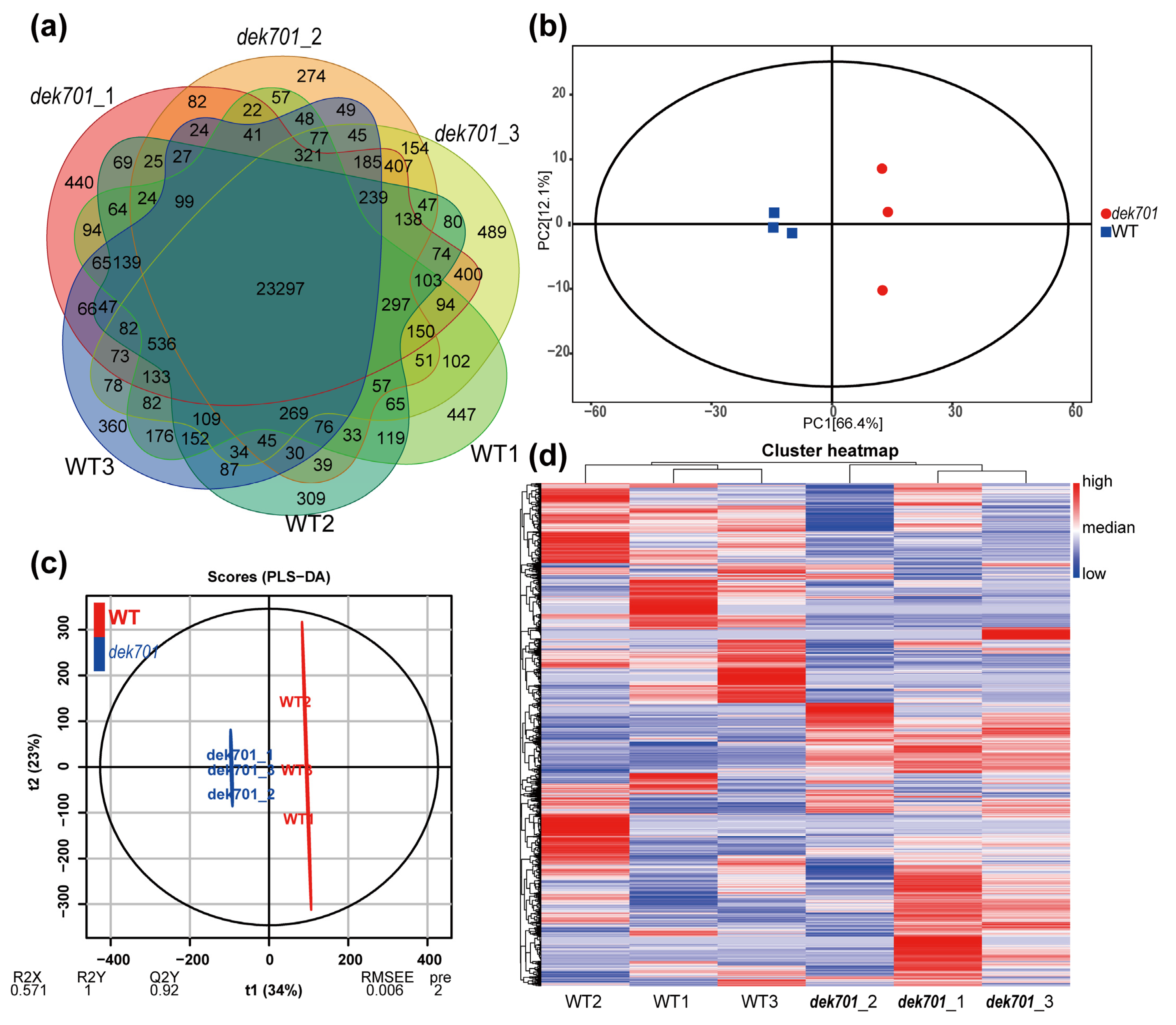
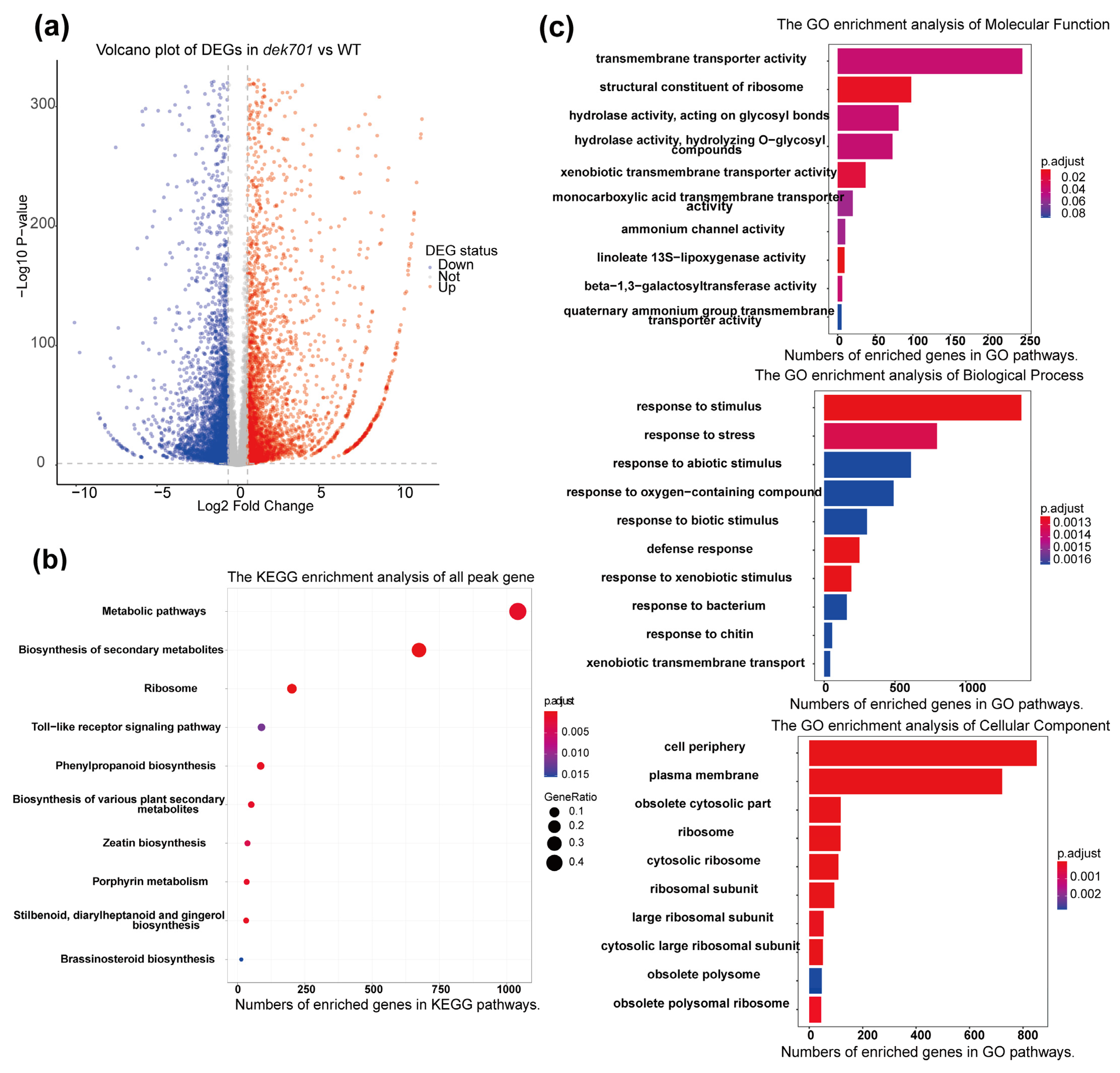
2.2. Transcriptome Analysis of the dek701 Seedlings
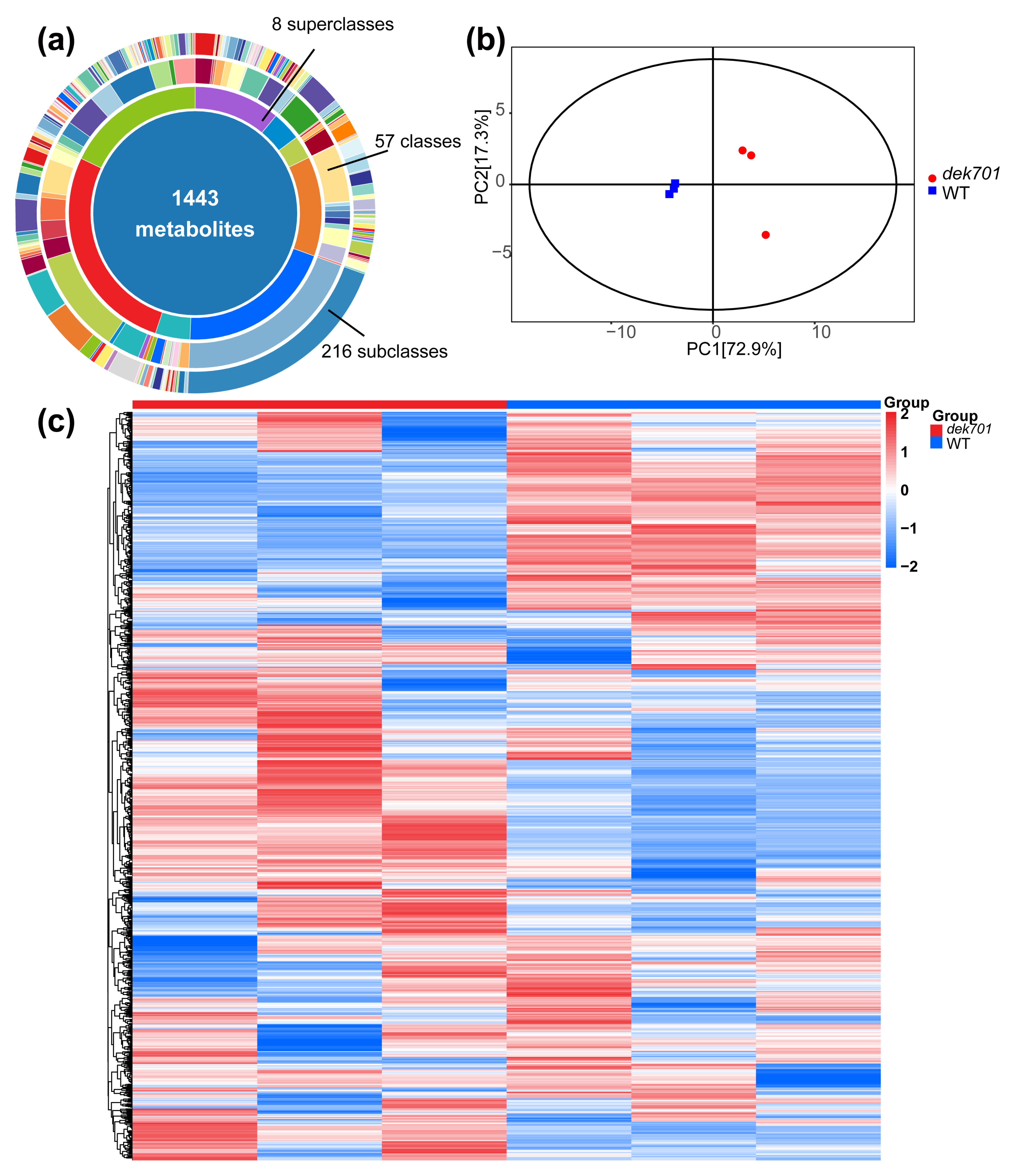
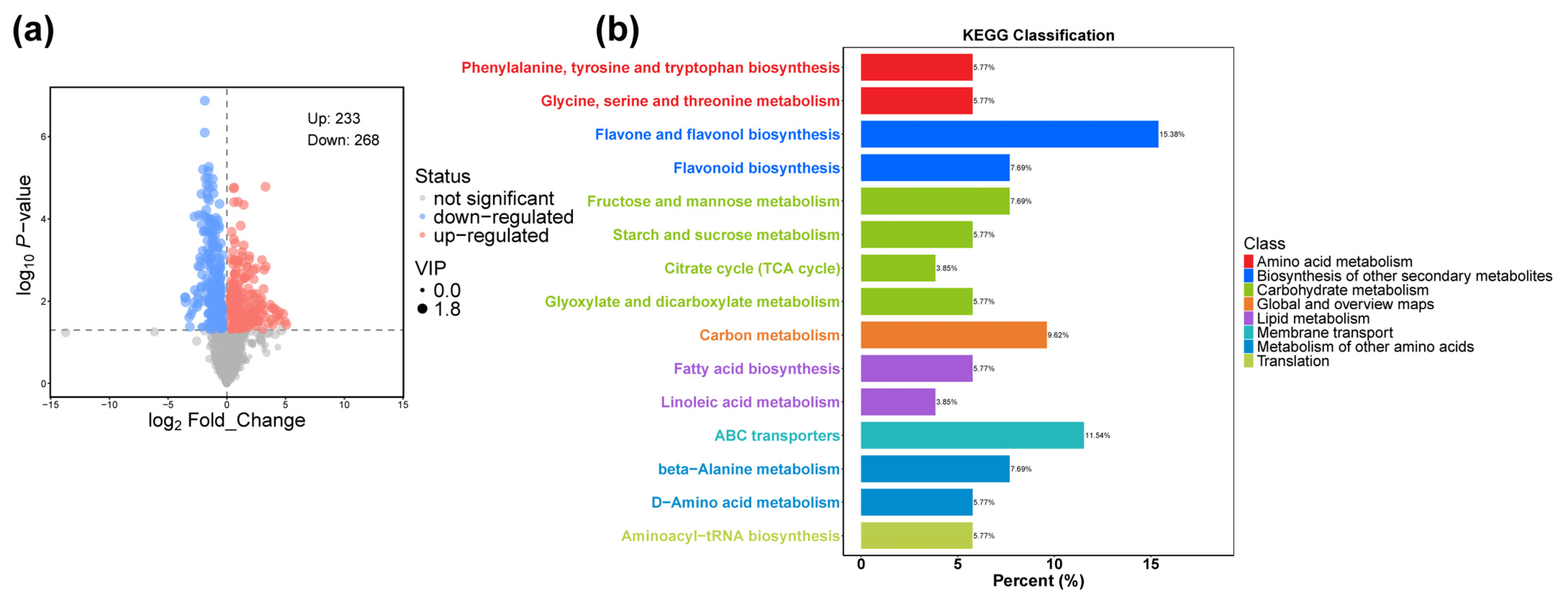
2.3. Metabolomes Analysis of the dek701 Seedlings
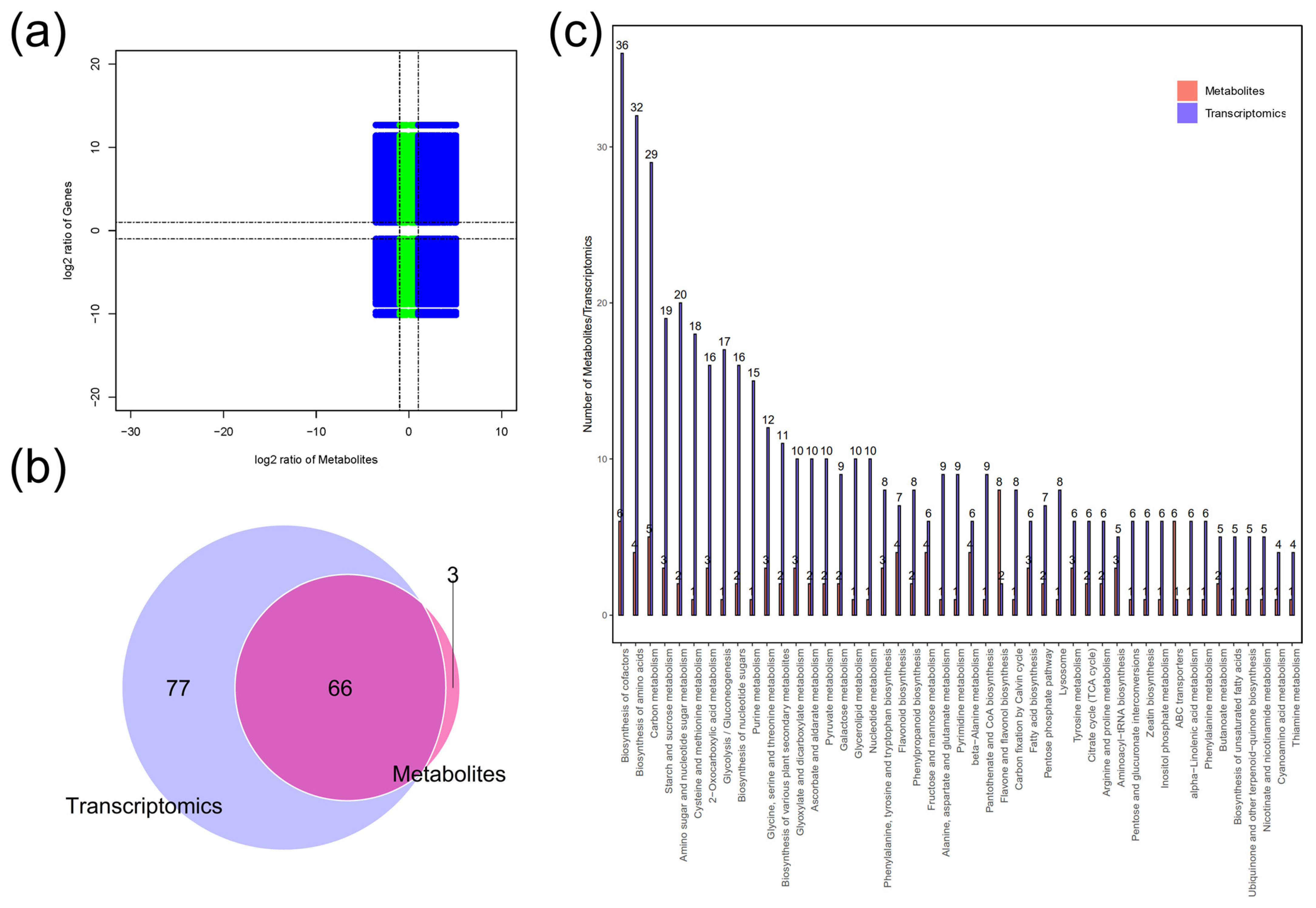
2.4. Combined Transcriptome and Metabolome Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. DNA Extraction, RNA Extraction and Genotype Identification
4.3. Transcriptome Profiling Analysis
4.4. Enrichment Analysis of Differentially Expressed Genes
4.5. Metabolome Profiling Analysis
4.6. Combined Analysis of Transcriptome and Metabolome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- von Hippel, P.H. An Integrated Model of the Transcription Complex in Elongation, Termination, and Editing. Science 1998, 281, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Nudler, E. Transcription Elongation: Structural Basis and Mechanisms. J. Mol. Biol. 1999, 288, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schramm, L.; Hernandez, N. Recruitment of RNA Polymerase III to Its Target Promoters. Genes Dev. 2002, 16, 2593–2620. [Google Scholar] [CrossRef] [PubMed]
- Woychik, N.A.; Hampsey, M. The RNA Polymerase II Machinery: Structure Illuminates Function. Cell 2002, 108, 453–463. [Google Scholar] [CrossRef]
- Grummt, I. Life on a Planet of Its Own: Regulation of RNA Polymerase I Transcription in the Nucleolus. Genes Dev. 2003, 17, 1691–1702. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, W.; Chen, K.; Wu, Z.; Yang, H.; Xu, Y. Structure of the Human RNA Polymerase I Elongation Complex. Cell Discov. 2021, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Vassylyev, D.G.; Sekine, S.; Laptenko, O.; Lee, J.; Vassylyeva, M.N.; Borukhov, S.; Yokoyama, S. Crystal Structure of a Bacterial RNA Polymerase Holoenzyme at 2.6 Å Resolution. Nature 2002, 417, 712–719. [Google Scholar] [CrossRef]
- Hirata, A.; Klein, B.J.; Murakami, K.S. The X-Ray Crystal Structure of RNA Polymerase from Archaea. Nature 2008, 451, 851–854. [Google Scholar] [CrossRef]
- Ebright, R.H. RNA Polymerase: Structural Similarities Between Bacterial RNA Polymerase and Eukaryotic RNA Polymerase II. J. Mol. Biol. 2000, 304, 687–698. [Google Scholar] [CrossRef]
- Wild, T.; Cramer, P. Biogenesis of Multisubunit RNA Polymerases. Trends Biochem. Sci. 2012, 37, 99–105. [Google Scholar] [CrossRef]
- Cramer, P.; Armache, K.-J.; Baumli, S.; Benkert, S.; Brueckner, F.; Buchen, C.; Damsma, G.E.; Dengl, S.; Geiger, S.R.; Jasiak, A.J.; et al. Structure of Eukaryotic RNA Polymerases. Annu. Rev. Biophys. 2008, 37, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Mann, C.; Micouin, J.Y.; Chiannilkulchai, N.; Treich, I.; Buhler, J.M.; Sentenac, A. RPC53 Encodes a Subunit of Saccharomyces Cerevisiae RNA Polymerase C (III) Whose Inactivation Leads to a Predominantly G1 Arrest. Mol. Cell. Biol. 1992, 12, 4314–4326. [Google Scholar] [CrossRef]
- Yee, N.S.; Gong, W.; Huang, Y.; Lorent, K.; Dolan, A.C.; Maraia, R.J.; Pack, M. Mutation of RNA Pol III Subunit Rpc2/Polr3b Leads to Deficiency of Subunit Rpc11 and Disrupts Zebrafish Digestive Development. PLoS Biol. 2007, 5, e312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-Q.; Li, Y.; Fu, Z.-Y.; Liu, X.-B.; Yuan, K.; Fang, Y.; Liu, Y.; Li, G.; Zhang, X.-S.; Chong, K.; et al. Intact Arabidopsis RPB1 Functions in Stem Cell Niches Maintenance and Cell Cycling Control. Plant J. 2018, 95, 150–167. [Google Scholar] [CrossRef]
- Chen, L.; Guan, L.; Qian, P.; Xu, F.; Wu, Z.; Wu, Y.; He, K.; Gou, X.; Li, J.; Hou, S. NRPB3, the Third Largest Subunit of RNA Polymerase II, Is Essential for Stomatal Patterning and Differentiation in Arabidopsis. Development 2016, 143, 1600–1611. [Google Scholar] [CrossRef]
- Wang, A.; Hou, Q.; Si, L.; Huang, X.; Luo, J.; Lu, D.; Zhu, J.; Shangguan, Y.; Miao, J.; Xie, Y.; et al. The PLATZ Transcription Factor GL6 Affects Grain Length and Number in Rice. Plant Physiol. 2019, 180, 2077–2090. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, J.; Ye, J.; Zheng, X.; Xiang, X.; Li, C.; Fu, M.; Wang, Q.; Zhang, Z.; Wu, Y. The Maize Imprinted Gene Floury3 Encodes a PLATZ Protein Required for tRNA and 5S rRNA Transcription through Interaction with RNA Polymerase III. Plant Cell 2017, 29, 2661–2675. [Google Scholar] [CrossRef]
- Zhao, H.; Qin, Y.; Xiao, Z.; Li, Q.; Yang, N.; Pan, Z.; Gong, D.; Sun, Q.; Yang, F.; Zhang, Z.; et al. Loss of Function of an RNA Polymerase III Subunit Leads to Impaired Maize Kernel Development. Plant Physiol. 2020, 184, 359–373. [Google Scholar] [CrossRef]
- Woychik, N.A.; Liao, S.M.; Kolodziej, P.A.; Young, R.A. Subunits Shared by Eukaryotic Nuclear RNA Polymerases. Genes Dev. 1990, 4, 313–323. [Google Scholar] [CrossRef]
- Woychik, N.A.; Young, R.A. RNA Polymerase II Subunit RPB10 Is Essential for Yeast Cell Viability. J. Biol. Chem. 1993, 268, 12230. [Google Scholar] [CrossRef]
- Chen, Q.; Guo, Y.; Zhang, J.; Zheng, N.; Wang, J.; Liu, Y.; Lu, J.; Zhen, S.; Du, X.; Li, L.; et al. RNA Polymerase Common Subunit ZmRPABC5b Is Transcriptionally Activated by Opaque2 and Essential for Endosperm Development in Maize. Nucleic Acids Res. 2023, 51, 7832–7850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, T.; Gao, M.; Ye, M.; Lin, M.; Wu, D.; Guo, J.; Guan, W.; Wang, J.; Yang, K.; et al. Transcriptome and Metabolome Profiling Reveal the Resistance Mechanisms of Rice against Brown Planthopper. Int. J. Mol. Sci. 2022, 23, 4083. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, J.; Wang, J.; Xie, Y.; Cui, Y.; Du, X.; Li, L.; Fu, J.; Liu, Y.; Wang, J.; et al. Small Kernel 501 (Smk501) Encodes the RUBylation Activating Enzyme E1 Subunit ECR1 (E1 C-TERMINAL RELATED 1) and Is Essential for Multiple Aspects of Cellular Events during Kernel Development in Maize. New Phytol. 2021, 230, 2337–2354. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Zhang, W.; Yang, Y.; Ma, Y.; Li, X.; Meng, D.; Luo, H.; Xue, W.; Lv, X.; et al. BSA-Seq and Transcriptomic Analysis Provide Candidate Genes Associated with Inflorescence Architecture and Kernel Orientation by Phytohormone Homeostasis in Maize. Int. J. Mol. Sci. 2023, 24, 10728. [Google Scholar] [CrossRef]
- Werner, F. Structure and Function of Archaeal RNA Polymerases. Mol. Microbiol. 2007, 65, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Wimberly, B.T.; Brodersen, D.E.; Clemons, W.M.; Morgan-Warren, R.J.; Carter, A.P.; Vonrhein, C.; Hartsch, T.; Ramakrishnan, V. Structure of the 30S Ribosomal Subunit. Nature 2000, 407, 327–339. [Google Scholar] [CrossRef]
- Nomura, M.; Erdmann, V.A. Reconstitution of 50S Ribosomal Subunits from Dissociated Molecular Components. Nature 1970, 228, 744–748. [Google Scholar] [CrossRef]
- Lafontaine, D.L.J.; Tollervey, D. The Function and Synthesis of Ribosomes. Nat. Rev. Mol. Cell Biol. 2001, 2, 514–520. [Google Scholar] [CrossRef]
- Dahlberg, A.E. The Functional Role of Ribosomal RNA in Protein Synthesis. Cell 1989, 57, 525–529. [Google Scholar] [CrossRef]
- Ramakrishnan, V. Ribosome Structure and the Mechanism of Translation. Cell 2002, 108, 557–572. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The Mechanism of Eukaryotic Translation Initiation and Principles of Its Regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Schmeing, T.M.; Ramakrishnan, V. What Recent Ribosome Structures Have Revealed about the Mechanism of Translation. Nature 2009, 461, 1234–1242. [Google Scholar] [CrossRef]
- Warpeha, K.M.; Montgomery, B.L. Light and Hormone Interactions in the Seed-to-Seedling Transition. Environ. Exp. Bot. 2016, 121, 56–65. [Google Scholar] [CrossRef]
- Santner, A.; Estelle, M. Recent Advances and Emerging Trends in Plant Hormone Signalling. Nature 2009, 459, 1071–1078. [Google Scholar] [CrossRef]
- Finkelstein, R.R. The Role of Hormones during Seed Development and Germination. In Plant Hormones: Biosynthesis, Signal Transduction, Action! Davies, P.J., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 549–573. ISBN 978-1-4020-2686-7. [Google Scholar]
- Yuan, K.; Wysocka-Diller, J. Phytohormone Signalling Pathways Interact with Sugars during Seed Germination and Seedling Development. J. Exp. Bot. 2006, 57, 3359–3367. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.-B.; Li, Y.-H.; Shu, X.-C.; Pu, Y.-T.; Wang, X.-J.; Wang, T.; Wang, Z. The Classification, Molecular Structure and Biological Biosynthesis of Flavonoids, and Their Roles in Biotic and Abiotic Stresses. Molecules 2023, 28, 3599. [Google Scholar] [CrossRef]
- Rich, A.; RajBhandary, U.L. Transfer RNA: Molecular Structure, Sequence, and Properties. Annu. Rev. Biochem. 1976, 45, 805–860. [Google Scholar] [CrossRef]
- Kirchner, S.; Ignatova, Z. Emerging Roles of tRNA in Adaptive Translation, Signalling Dynamics and Disease. Nat. Rev. Genet. 2015, 16, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Frelin, O.; Dervinis, C.; Wegrzyn, J.L.; Davis, J.M.; Hanson, A.D. Drought Stress in Pinus Taeda L. Induces Coordinated Transcript Accumulation of Genes Involved in the Homogentisate Pathway. Tree Genet. Genomes 2017, 13, 27. [Google Scholar] [CrossRef]
- Le, X.H.; Lee, C.-P.; Millar, A.H. The Mitochondrial Pyruvate Carrier (MPC) Complex Mediates One of Three Pyruvate-Supplying Pathways That Sustain Arabidopsis Respiratory Metabolism. Plant Cell 2021, 33, 2776–2793. [Google Scholar] [CrossRef]
- Allen, G.C.; Flores-Vergara, M.A.; Krasynanski, S.; Kumar, S.; Thompson, W.F. A Modified Protocol for Rapid DNA Isolation from Plant Tissues Using Cetyltrimethylammonium Bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef] [PubMed]
- El-Ashram, S.; Al Nasr, I.; Suo, X. Nucleic Acid Protocols: Extraction and Optimization. Biotechnol. Rep. 2016, 12, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. Clusterprofiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, J.; Wang, W.; Wang, Y.; Xu, J.; Li, Z.; Zhao, X.; Fu, B. Integrated Analysis of the Transcriptome and Metabolome Revealed the Molecular Mechanisms Underlying the Enhanced Salt Tolerance of Rice Due to the Application of Exogenous Melatonin. Front. Plant Sci. 2021, 11, 618680. [Google Scholar] [CrossRef]
- Yin, D.-J.; Ye, S.-J.; Sun, X.-Y.; Chen, Q.-Y.; Min, T.; Wang, H.-X.; Wang, L.-M. Integrative Analysis of the Transcriptome and Metabolome Reveals Genes Involved in Phenylpropanoid and Flavonoid Biosynthesis in the Trapa Bispinosa Roxb. Front. Plant Sci. 2022, 13, 913265. [Google Scholar] [CrossRef]
- Liu, J.; Li, L.; Liu, Y.; Kong, Z.; Zhang, P.; Wang, Q.; Cheng, S.; Qin, P. Combined Transcriptome and Metabolome Analysis of the Resistance Mechanism of Quinoa Seedlings to Spodoptera Exigua. Front. Plant Sci. 2022, 13, 931145. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, Y.; Zhang, J.; Guo, S.; Du, X.; Gu, R.; Wang, J.; Chen, Q. Multi-Omics Analysis Reveals Differential Molecular Responses of RNA Polymerase Common Subunit ZmRPABC5b for Seedling Development in Maize. Plants 2025, 14, 941. https://doi.org/10.3390/plants14060941
Yi Y, Zhang J, Guo S, Du X, Gu R, Wang J, Chen Q. Multi-Omics Analysis Reveals Differential Molecular Responses of RNA Polymerase Common Subunit ZmRPABC5b for Seedling Development in Maize. Plants. 2025; 14(6):941. https://doi.org/10.3390/plants14060941
Chicago/Turabian StyleYi, Yaoran, Jie Zhang, Shuangqi Guo, Xuemei Du, Riliang Gu, Jianhua Wang, and Quanquan Chen. 2025. "Multi-Omics Analysis Reveals Differential Molecular Responses of RNA Polymerase Common Subunit ZmRPABC5b for Seedling Development in Maize" Plants 14, no. 6: 941. https://doi.org/10.3390/plants14060941
APA StyleYi, Y., Zhang, J., Guo, S., Du, X., Gu, R., Wang, J., & Chen, Q. (2025). Multi-Omics Analysis Reveals Differential Molecular Responses of RNA Polymerase Common Subunit ZmRPABC5b for Seedling Development in Maize. Plants, 14(6), 941. https://doi.org/10.3390/plants14060941