
The Involvement of Amino Acid Metabolism in the Mechanisms of Salt Tolerance Adaptation in Medicago sativa and Medicago truncatula

 ,
,  ,
,
Abstract
1. Introduction
2. Results
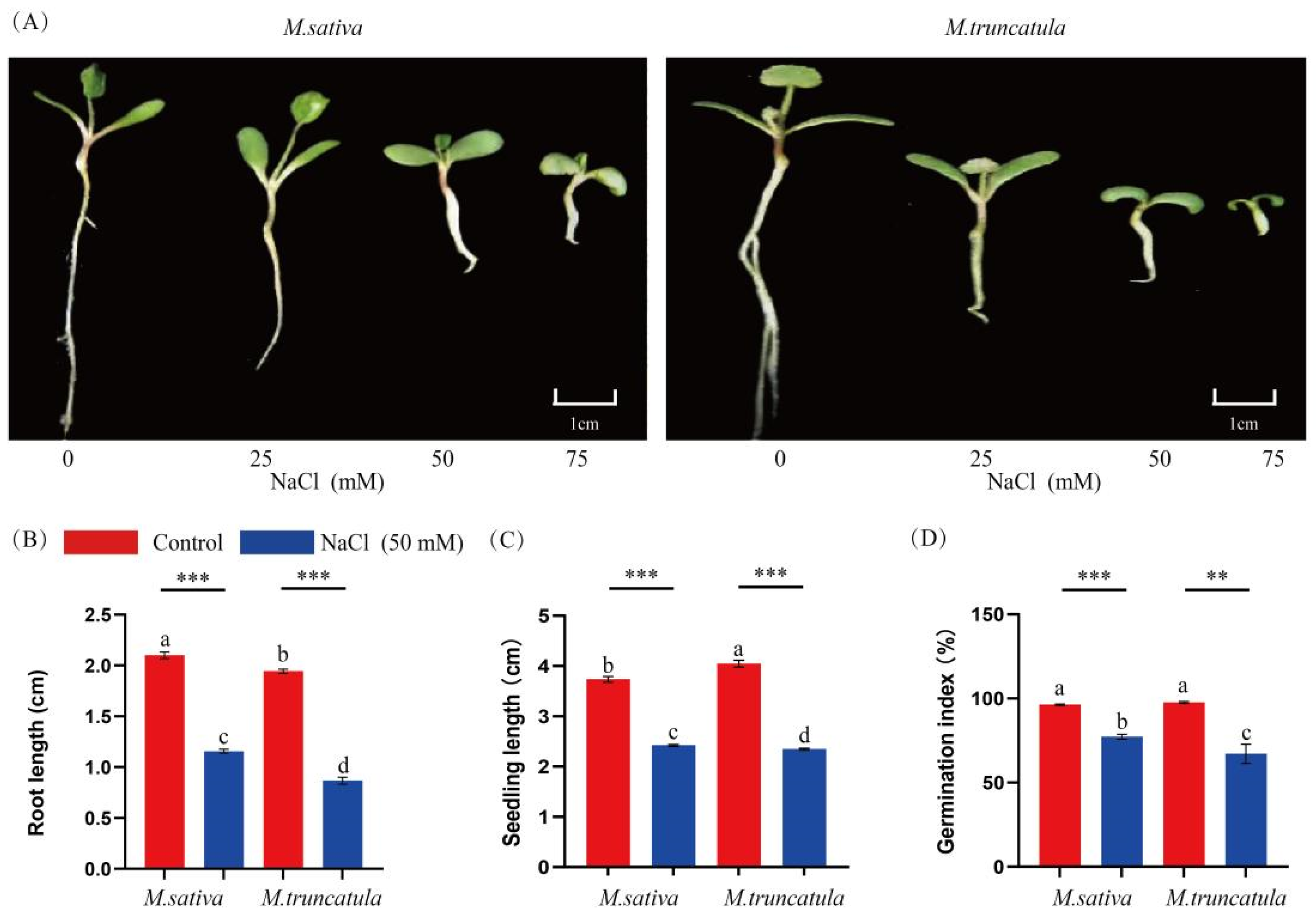
2.1. Effects of NaCl Stress on the Germination of M. sativa (Medicago sativa L.) and M. truncatula (Medicago truncatula L.) Seeds
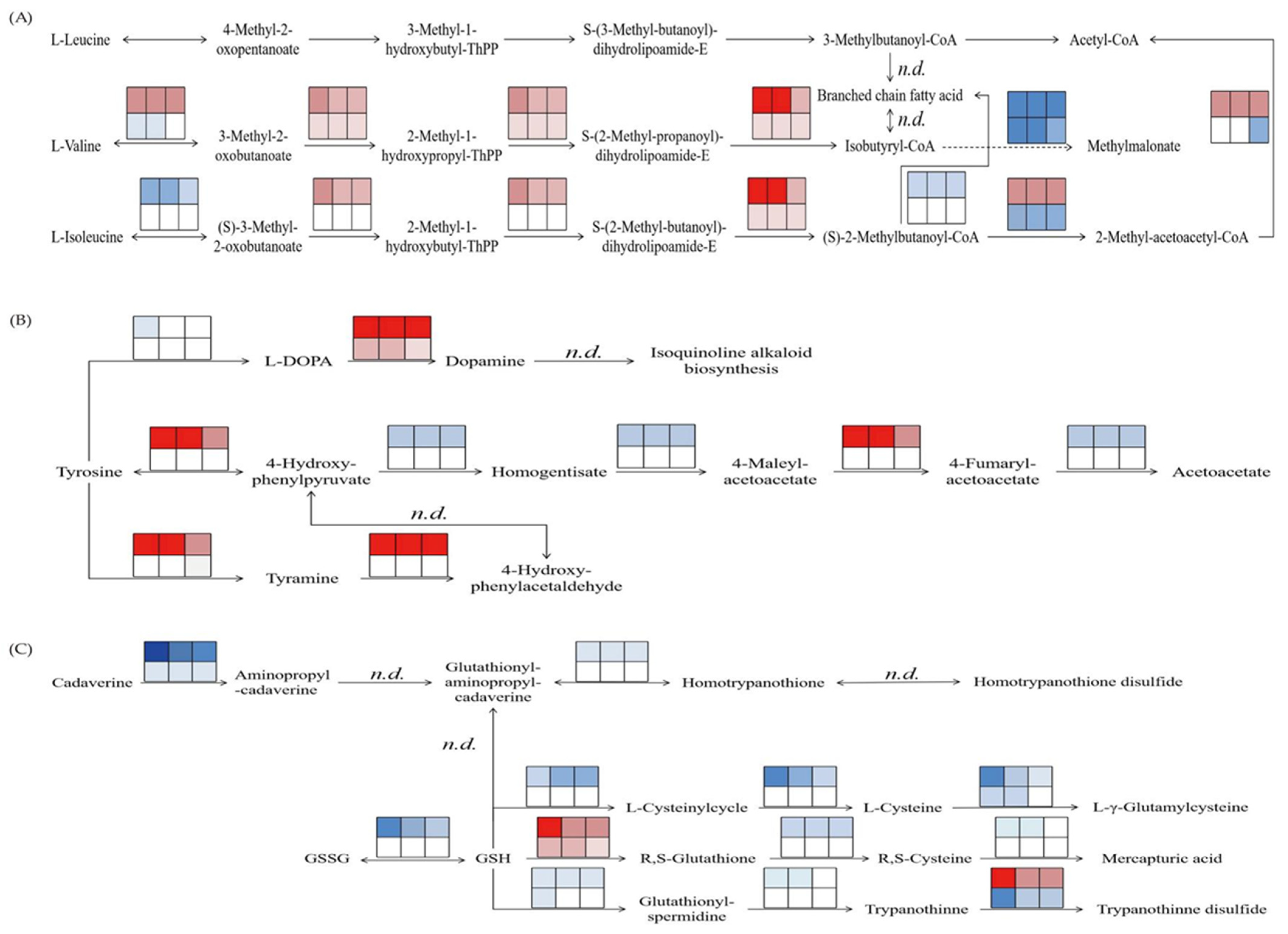
2.2. Effects of NaCl Stress on Amino Acid Metabolism in M. sativa and M. truncatula Seedlings
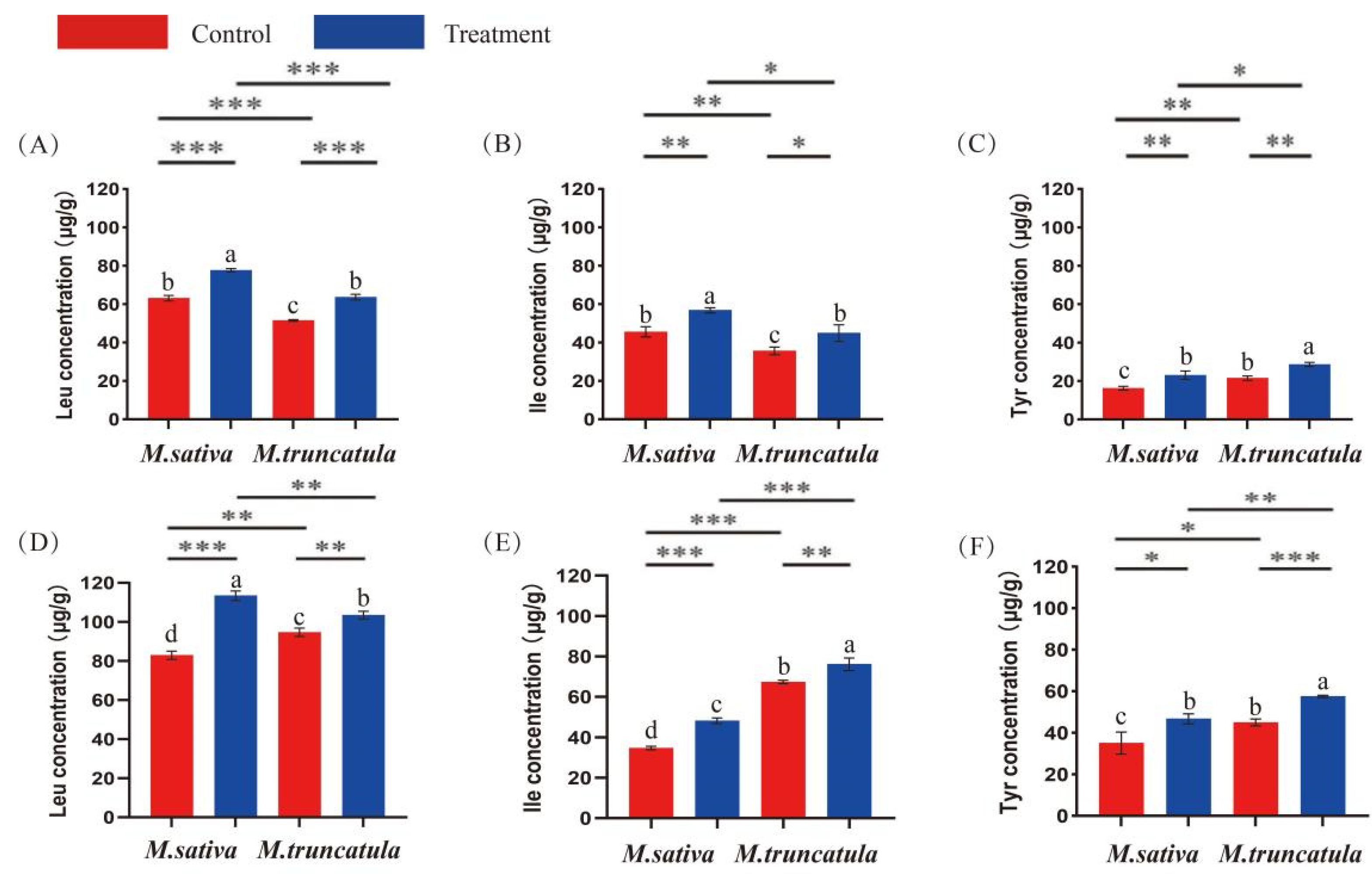
2.3. Quantitative Analysis of Amino Acid Concentrations
2.4. Exogenous Application of Leu, Ile, and Tyr
3. Discussion
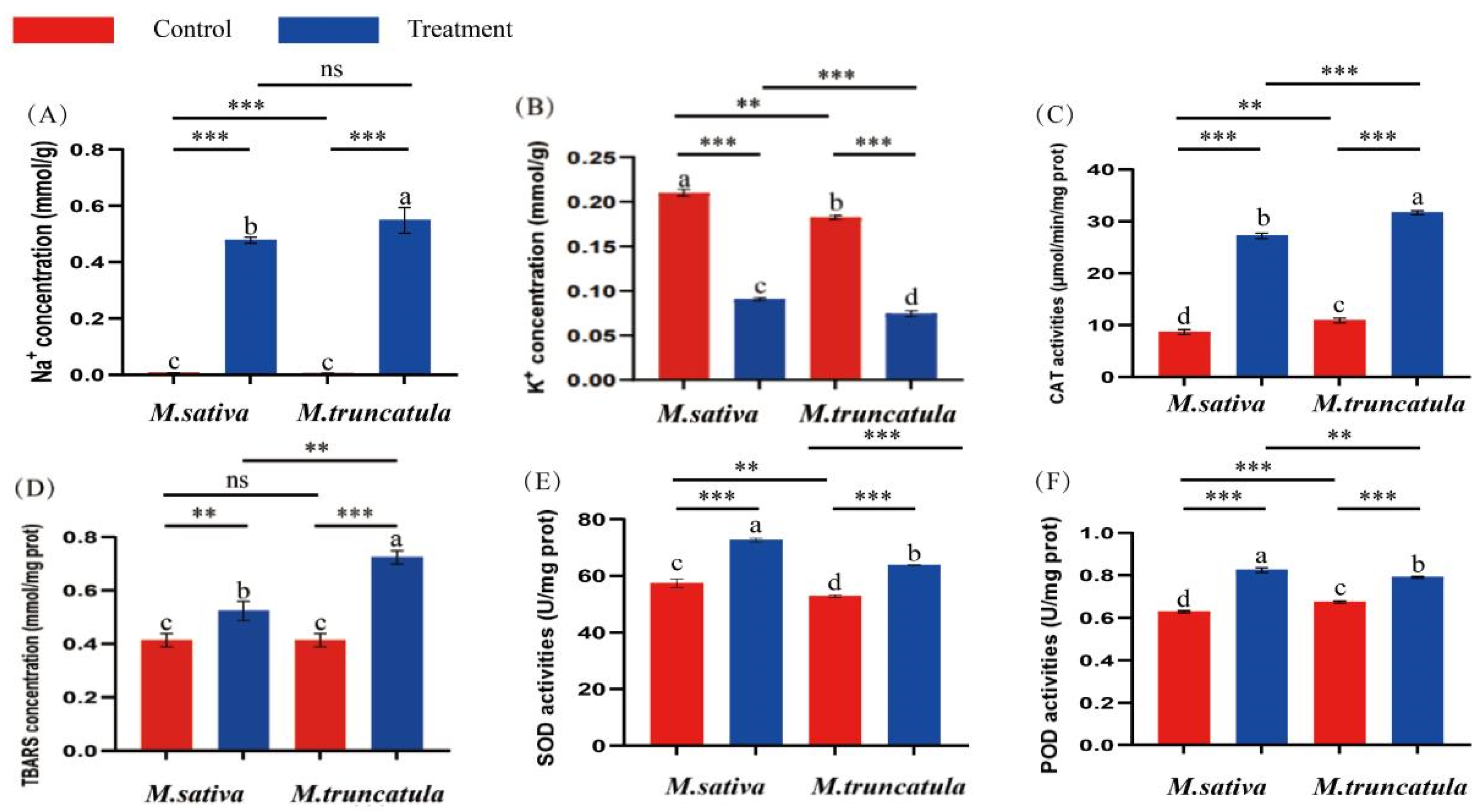
3.1. Changes in the Levels of Reactive Oxygen Species (ROS) in M. sativa (Medicago sativa L.) and M. truncatula (Medicago truncatula L.) Seedlings Under Salt Stress
3.2. Relationship Between the Amino Acid Metabolism and Salt Tolerance Mechanisms in M. sativa and M. truncatula
3.3. Roles of Amino Acids in M. sativa and M. truncatula Under Salt Stress
4. Materials and Methods
4.1. Plant Growth Conditions and Stress Treatment
4.2. Transcriptome Sequencing and Data Analysis
4.3. Proteome Profiling and Statistical Analysis
4.4. Determination of Amino Acids
4.5. Integrated Transcriptome and Proteome Analysis
4.6. Assays of the Antioxidant Enzymes and Other Physiological and Biochemical Indices
4.7. Statistical Analysis
5. Conclusions
6. Future Perspectives
7. Limitations
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAT | Catalase |
| DEG | Differentially expressed genes |
| DEP | Differentially expressed proteins |
| DMSO | Dimethyl sulfoxide |
| DTT | Dithiothreitol |
| FDR | False discovery rate |
| FPKM | Fragments per kilobase of exon model per million mapped fragments |
| Glu | Glutamic acid |
| GSEA | Gene set enrichment analysis |
| GO | Gene Ontology |
| Ile | Isoleucine |
| KEGG | Kyoto encyclopedia of genes and genomes |
| Leu | Leucine |
| Na+ | Sodium ions |
| NES | Normalised enrichment score |
| POD | Peroxidase |
| PSM | Peptide spectrum match |
| SOD | Superoxide dismutase |
| TBARS | tiobarbituric acid-reactive substances |
| TCA | tricarboxylic acid |
| TEAB | Tetraethylammonium bromide |
| TMT | Tandem mass tag |
| TOR | target of rapamycin |
| Tyr | Tyrosine |
| UV | Ultraviolet |
References
- Chen, J.; Zhang, H.Q.; Zhang, X.L.; Tang, M. Arbuscular Mycorrhizal Symbiosis Alleviates Salt Stress in Black Locust through Improved Photosynthesis, Water Status, and K+/Na+ Homeostasis. Front. Plant. Sci. 2017, 8, 1739. [Google Scholar] [CrossRef]
- Ruan, C.J.; da Silva, J.A.T.; Mopper, S.; Qin, P.; Lutts, S. Halophyte improvement for a salinized world. Crit. Rev. Plant Sci. 2010, 6, 329–359. [Google Scholar] [CrossRef]
- Kristin, M.R.; Noah, F.; Daniel, V.M.; Johannes, R. Linking bacterial community composition to soil salinity along environmental gradients. ISME J. 2019, 13, 836–846. [Google Scholar]
- Zhang, P.; Duo, T.Q.; Wang, F.D.; Zhang, X.Z.; Yang, Z.Z.; Hu, G.F. De novo transcriptome in roots of switchgrass (Panicum virgatum L.) reveals gene expression dynamic and act network under alkaline salt stress. BMC Genom. 2021, 22, 1197–1210. [Google Scholar] [CrossRef]
- Cheng, T.L.; Chen, J.H.; Zhang, J.B.; Shi, S.Q.; Zhou, Y.W.; Lu, L.; Wang, P.K.; Jiang, Z.P.; Yang, J.C.; Zhang, S.G.; et al. Physiological and proteomic analyses of leaves from the halophyte Tangut Nitraria reveals diverse response pathways critical for high salinity tolerance. Front. Plant Sci. 2015, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Huang, X.; Lan, H.X.; Zhang, H.S.; Huang, J. Rearrangement of nitrogen metabolism in rice (Oryza sativa L.) under salt stress. Plant Signal. Behav. 2016, 11, 4–7. [Google Scholar] [CrossRef]
- Sui, D.Z.; Wang, B.S. Comparative Proteomics on Leaves of Triadica sebifera Clones under Salt Stress. Bull. Bot. Res. 2023, 43, 679–689. [Google Scholar]
- Jiao, Y.; Bai, Z.Z.; Xu, J.Y.; Zhao, M.L.; Khan, Y.; Hu, Y.J.; Shi, L.X. Metabolomics and its physiological regulation process reveal the salt-tolerant mechanism in Glycine soja seedling roots. Plant Physiol. Biochem. 2018, 126, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.M. Synthesis versus degradation: Directions of amino acid metabolism during Arabidopsis abiotic stress response. Plant Mol. Biol. 2018, 98, 121–135. [Google Scholar] [CrossRef]
- Pires, M.V.; Pereira Júnior, A.A.; Medeiros, D.B.; Daloso, D.M.; Pham, P.A.; Barros, K.A.; Fernie, A.R. The influence of alternative pathways of respiration that utilize branched-chain amino acids following water shortage in Arabidopsis. Plant Cell Environ. 2016, 39, 1304–1319. [Google Scholar] [CrossRef]
- Kopriva, S.; Malagoli, M.; Takahashi, H. Sulfur nutrition: Impacts on plant development, metabolism, and stress responses. J. Exp. Bot. 2019, 70, 4069–4073. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, T.; Whittaker, A.; Bochicchio, A.; Vazzana, C.; Suzuki, A.; Masclaux-Daubresse, C. Amino acid pattern and glutamate metabolism during dehydration stress in the ‘resurrection’ plant Sporobolus stapfianus: A comparison between desiccation-sensitive and desiccation-tolerant leaves. J. Exp. Bot. 2007, 58, 3037–3046. [Google Scholar] [CrossRef] [PubMed]
- Ferreira Júnior, D.C.; Gaion, L.A.; Sousa Júnior, G.S.; Santos, D.M.M.; Carvalho, R.F. Drought-induced proline synthesis depends on root-to-shoot communication mediated by light perception. Acta Physiol. Plant. 2018, 40, 363. [Google Scholar] [CrossRef]
- Forde, B.G.; Lea, P.J. Glutamate in plants: Metabolism, regulation, and signalling. J. Exp. Bot. 2007, 58, 2339–2358. [Google Scholar] [CrossRef]
- Wang, L.X.; Pan, D.Z.; Lv, X.J.; Cheng, C.L.; Li, J.; Liang, W.Y.; Xing, J.H.; Chen, W. A multilevel investigation to discover why Kandelia candel thrives in high salinity. Plant Cell Environ. 2016, 39, 2486–2497. [Google Scholar] [CrossRef] [PubMed]
- Cano, E.A.; Perez, F.; Moreno, V.; Bolarin, M.C. Responses to NaCl stress of cultivated and wild tomato species and their hybrids in callus cultures. Plant Cell Rep. 1996, 15, 791–794. [Google Scholar] [CrossRef]
- Lu, Y.Y.; Su, W.L.; Bao, Y.; Wang, S.; He, F.; Wang, D.L.; Yu, X.Q.; Yin, W.L.; Liu, C.; Xia, X.L. Poplar PdPTP1 Gene Negatively Regulates Salt Tolerance by Affecting Ion and ROS Homeostasis in Populus. Int. J. Mol. Sci. 2020, 21, 1065. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef]
- Liu, Y.; Duan, X.; Zhao, X.; Ding, W.; Wang, Y.; Xiong, Y. Diverse nitrogen signals activate convergent ROP2-TOR signaling in Arabidopsis. Dev. Cell 2021, 9, 1283–1301. [Google Scholar] [CrossRef]
- O’Leary, B.M.; Oh, G.G.K.; Lee, C.P.; Millar, A.H. Metabolite regulatory interactions control plant respiratory metabolism via Target of Rapamycin (TOR) kinase activation. Plant Cell 2020, 32, 666–682. [Google Scholar] [CrossRef]
- Dong, Q.; Chen, M.J.; Zhang, Y.R.; Song, P.P.; Yang, H.L.; Zhao, Y.; Yu, C.X.; Zha, L. Integrated physiologic and proteomic analysis of Stropharia rugosoannulata mycelia in response to Cd stress. J. Hazard. Mater. 2023, 441, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Kazem, G.G.; Samira, S.A. Exogenous Cytokinin and Salicylic Acid Improve Amino Acid Content and Composition of Faba Bean Seeds Under Salt Stress. Gesunde Pflanz. 2022, 74, 935–945. [Google Scholar]
- Hiroshi, O.; Ruben, C.A.; David, Y.; Daisuke, T.; Takashi, S.; Bonifacino, J.S. . The Medium Subunits of Adaptor Complexes Recognize Distinct but Overlapping Sets of Tyrosine-based Sorting Signals. J. Biol. Chem. 1998, 40, 25915–25921. [Google Scholar]
- Szabados, L.; Savouré, A. Proline: A multifunctional amino acid. Trends Plant Sci. 2010, 15, 89–97. [Google Scholar] [CrossRef]
- Heinemann, B.; Künzler, P.; Eubel, H.; Braun, H.P.; Hildebr, T.M. Estimating the number of protein molecules in a plant cell: Protein and amino acid homeostasis during drought. Plant Physiol. 2021, 185, 385–404. [Google Scholar] [CrossRef]
- Dennison, K.L.; Spalding, E.P. Glutamate-gated calcium fluxes in Arabidopsis. Plant Physiol. 2001, 124, 1511. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, Q.Y.; Zhang, W.H. Glutamate receptors are involved in mitigating effects of amino acids on seed germination of Arabidopsis thaliana under salt stress. Environ. Exp. Bot. 2016, 130, 68–78. [Google Scholar] [CrossRef]
- Abdelkader, M.; Voronina, L.; Baratova, L.; Shelepova, O.; Zargar, M.; Puchkov, M.; Loktionova, E.; Amantayev, B.; Kipshakbaeva, A.; Arinov, B. Biostimulants-Based Amino Acids Augment Physio-Biochemical Responses and Promote Salinity Tolerance of Lettuce Plants (Lactuca sativa L.). Horticulture 2023, 9, 807. [Google Scholar] [CrossRef]
- Guo, T.; Bai, J.; Wang, J. Research on the present situation and countermeasures of alfalfa grass industry in China. Chin. J. Grassl. 2018, 40, 111–115. [Google Scholar]
- Raiesi, F. The conversion of overgrazed pastures to almond orchards and alfalfa cropping systems may favor microbial indicators of soil quality in central Iran. Agric. Ecosyst. Environ. 2007, 121, 309–318. [Google Scholar] [CrossRef]
- Su, Y. Soil carbon and nitrogen sequestration following the conversion of cropland to alfalfa forage land in northwest China. Soil Tillage Res. 2007, 92, 181–189. [Google Scholar] [CrossRef]
- Tadege, M.; Ratet, P.; Mysore, K.S. Insertional mutagenesis: A Swiss Army knife for functional genomics of Medicago truncatula. Trends Plant Sci. 2005, 10, 229–235. [Google Scholar] [CrossRef]
- Peel, M.D.; Waldron, B.L.; Jensen, K.B.; Chatterton, N.J.; Horton, H.; Dudley, L.M. Screening for salinity tolerance in alfalfa. Crop Sci. 2004, 44, 2049–2053. [Google Scholar] [CrossRef]
- Xiong, J.; Sun, Y.; Yang, Q.; Tian, H.; Zhang, H.; Liu, Y.; Chen, M. Proteomic analysis of early salt stress responsive proteins in alfalfa roots and shoots. Proteome Sci. 2017, 15, 19. [Google Scholar] [CrossRef]
- Mittler, R. ROS are good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Waszczak, C.; Carmody, M.; Kangasjärvi, J. Annual review of plant biology reactive oxygen species in plant signaling. Annu. Rev. Plant Biol. 2018, 69, 209–236. [Google Scholar] [CrossRef]
- You, J.; Chan, Z.L. ROS Regulation During Abiotic Stress Responses in Crop Plants. Front. Plant Sci. 2015, 6, 36–50. [Google Scholar] [CrossRef]
- Davey, M.W.; Stals, E.; Panis, B.; Keulemans, J.; Swennen, R.L. High-throughput determination of malondialdehyde in plant tissues. Anal. Biochem. 2005, 347, 201–207. [Google Scholar] [CrossRef]
- Liu, Z.B.; Zhang, W.J.; Gong, X.D.; Zhang, Q.; Zhou, L.R. A Cu/Zn superoxide dismutase from Jatropha curcas enhances salt tolerance of Arabidopsis thaliana. Genet. Mol. Res. 2015, 14, 2086–2098. [Google Scholar] [CrossRef]
- Yang, H.B. Effects of osmotic and salt stress on some physiological indexes of wheat seedlings. Res. Crops 2013, 14, 404–408. [Google Scholar]
- Masuta, C.; Inaba, J.I.; Shimura, H. The 2b proteins of Cucumber mosaic virus generally have the potential to differentially induce necrosis on Arabidopsis. Plant Signal. Behav. 2012, 7, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Long, R.C.; Gao, Y.L.; Sun, H.; Zhang, T.J.; Li, X.; Li, M.N.; Sun, Y.; Kang, J.M.; Wang, Z.; Ding, W.; et al. Quantitative proteomic analysis using iTRAQ to identify salt-responsive proteins during the germination stage of two Medicago species. Sci. Rep. 2018, 8, 15701. [Google Scholar] [CrossRef]
- Jennifer, Y.C.; Scott, J.D. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar]
- Sara, R.; Leonardo, M.; Lello, Z. Redox proteomics: Basic principles and future perspectives for the detection of protein oxidation in plants. J. Exp. Bot. 2008, 59, 3781–3801. [Google Scholar]
- Yao, M.; Xu, F.R.; Yao, Y.J.; Wang, H.O.; Ju, X.R.; Wang, L.F. Assessment of Novel Oligopeptides from Rapeseed Napin (Brassica napus) in Protecting HepG2 Cells from Insulin Resistance and Oxidative Stress. J. Agric. Food Chem. 2022, 58, 12418–12429. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Shi, Y.J.; Mu, C.S.; Wang, J.F. Differences in Organic Solute and Metabolites of Leymus chinensis in Response to Different Intensities of Salt and Alkali Stress. Plants 2023, 12, 1916. [Google Scholar] [CrossRef]
- Bao, J.B.; Liu, Z.Y.; Ding, Z.J.; Gulbar, Y.; Wang, Q.Y.; Tian, X.M. Metabolomic analysis reveals key metabolites and metabolic pathways in Suaeda salsa under salt and drought stress. Funct. Plant Biol. 2023, 9, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Obata, T.; Fernie, A.R. The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 2012, 69, 3225–3243. [Google Scholar] [CrossRef]
- Hernández, J.A.; Jiménez, A.; Mullineaux, P.; Sevilia, F. Tolerance of pea (Pisum sativum L.) to long-term salt stress is associated with induction of antioxidant defences. Plant Cell Environ. 2000, 23, 853–862. [Google Scholar] [CrossRef]
- Meng, X.Q.; Zhou, J.; Sui, N. Mechanisms of salt tolerance in halophytes: Current understanding and recent advances. Open Life Sci. 2018, 13, 149–154. [Google Scholar] [CrossRef]
- Yang, D.S.; Zhang, J.; Li, M.X.; Shi, L.X. Metabolomics Analysis Reveals the Salt-Tolerant Mechanism in Glycine soja. J. Plant Growth Regul. 2017, 36, 460–471. [Google Scholar] [CrossRef]
- Huang, T.F.; Georg, J. Abscisic acid-regulated protein degradation causes osmotic stress-induced accumulation of branched-chain amino acids in Arabidopsis thaliana. Planta 2017, 4, 737–747. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, X.; Zhao, C.F.; Fang, Z.M.; Jiao, P.P. Transcriptomic and metabolomic analysis reveals the role of CoA in the salt tolerance of Zygophyllum spp. BMC Plant Biol. 2020, 20, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Jens, F.; John, F.A. Protein tyrosine phosphorylation in the transition to light state 2 of chloroplast thylakoids. Photosynth. Res. 2001, 68, 71–79. [Google Scholar]
- Fougère, F.; Le Rudulier, D.; Streeter, J.G. Effects of Salt Stress on Amino Acid, Organic Acid, and Carbohydrate Composition of Roots, Bacteroids, and Cytosol of Alfalfa (Medicago sativa L.). Plant Physiol. 1991, 96, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- López-Gómez, M.; Palma, F.; Lluch, C. Strategies of salt tolerance in the rhizobia-legume symbiosis. In Beneficial Plant-Microbial Interactions: Ecology and Applications; CRC Press: Boca Raton, FL, USA, 2013; pp. 99–121. [Google Scholar]
- Prell, J.; White, J.P.; Bourdes, A.; Bunnewell, S.; Bongaerts, R.J.; Poole, P.S. Legumes regulate Rhizobium bacteroid development and persistence by the supply of branched-chain amino acids. Proc. Natl. Acad. Sci. USA 2009, 106, 12477–12482. [Google Scholar] [CrossRef] [PubMed]
- Fàbregas, N.; Fernie, A.R. The metabolic response to drought. J. Exp. Bot. 2019, 70, 1077–1085. [Google Scholar] [CrossRef]
- Podlešáková, K.; Ugena, L.; Spíchal, L.; Doležal, K.; De Diego, N. Phytohormones and polyamines regulate plant stress responses by altering gaba pathway. New Biotechnol. 2019, 48, 53–65. [Google Scholar] [CrossRef]
- Bertrand, A.; Gatzke, C.; Bipfubusa, M.; Lévesque, V.; Chalifour, F.P.; Claessens, A.; Rocher, S.; Tremblay, G.F.; Beauchamp, C.J. Physiological and Biochemical Responses to Salt Stress of Alfalfa Populations Selected for Salinity Tolerance and Grown in Symbiosis with Salt-Tolerant Rhizobium. Agronomy 2020, 10, 569. [Google Scholar] [CrossRef]
- Jiang, L.X.; Matthys, C.; Marquez-Garcia, B.; De Cuyper, C.; Smet, L.; De Keyser, A.; Boyer, F.D.; Beeckman, T.; Depuydt, S.; Goormachtig, S. Strigolactones spatially influence lateral root development through the cytokinin signaling network. J. Exp. Bot. 2016, 67, 379–389. [Google Scholar] [CrossRef]
- Ragaey, M.M.; Sadak, M.S.; Dawood, M.F.A.; Mousa, N.H.S.; Hanafy, R.S.; Latef, A.A.H.A. Role of Signaling Molecules Sodium Nitroprusside and Arginine in Alleviating Salt-Induced Oxidative Stress in Wheat. Plants 2022, 11, 1786. [Google Scholar] [CrossRef] [PubMed]
- Scarponi, L.; Martinett, L.; Nemat Alla, M.M. Growth response and changes in starch formation as a result of imazethapyr treatment of soybean (Glycine max L.). J. Agric. Food Chem. 1996, 44, 1572–1577. [Google Scholar] [CrossRef]
- Jitendra, M.; Tahmish, F.; Naveen, K.A. Role of Secondary Metabolites from Plant Growth-Promoting Rhizobacteria in Combating Salinity Stress. In Plant Microbiome: Stress Response; Springer: Singapore, 2018; Volume 5, pp. 127–163. [Google Scholar]
- Pavel, F.; Liubov, S.; Anastasiia, R.; Artem, P.; Elina, T.; Pavel, M.; Galina, C. Phenylalanine and Tyrosine as Exogenous Precursors of Wheat (Triticum aestivum L.) Secondary Metabolism through PAL-Associated Pathways. Plants 2020, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2015, 102, 15545–15550. [Google Scholar] [CrossRef]
- Shen, C.; Du, H.L.; Chen, Z.; Lu, H.W.; Zhu, F.G.; Chen, H.; Meng, X.Z.; Liu, Q.W.; Liu, P.; Zheng, L.H.; et al. The Chromosome-Level Genome Sequence of the Autotetraploid Alfalfa and Resequencing of Core Germplasms Provide Genomic Resources for Alfalfa Research. Mol. Plant 2020, 13, 1250–1261. [Google Scholar] [CrossRef]
- Raja, S.N.; Wen, J.Q.; Kirankumar, S.M. Medicago truncatula resources to study legume biology and symbiotic nitrogen fixation. Fundam. Res. 2023, 3, 219–224. [Google Scholar]
- Jones, P.; Binns, D.; Chang, H.Y. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S. STRING V9.1: Protein-Protein Interaction Networks, with Increased Coverage and Integration. Nucleic Acids Res. 2012, 41, D1. [Google Scholar] [CrossRef]
- Peskin, A.V.; Winterbourn, C.C. A microtiter plate assay for superoxide dismutase using a water-soluble tetrazolium salt (WST-1). Clin. Chim. Acta 2000, 293, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.F.; Wang, X.Y.; Li, Y.Y.; Peng, F.L.; Wang, L.S. Regulation of POD activity by pelargonidin during vegetative growth in radish (Raphanus sativus L.). Sci. Hortic. 2014, 174, 105–111. [Google Scholar] [CrossRef]
- Sima, Y.H.; Yao, J.H.; Hou, Y.S.; Wang, L.; Zhao, L.C. Variations of hydrogen peroxide and catalase expression in Bombyx eggs during diapause initiation and termination. Arch. Insect Biochem. Physiol. 2011, 77, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Sofia, E.C.; Anatoly, K.Y. Cyclodextrin enhanced fluorimetric determination of malonaldehyde by the thiobarbituric acid method. Talanta 1997, 44, 951–957. [Google Scholar]
- Ma, H.Z.; Liu, C.; Li, Z.X.; Ran, Q.J.; Xie, G.N.; Wang, B.M.; Fang, S.; Chu, J.F.; Zhang, J.R. ZmbZIP4 Contributes to Stress Resistance in Maize by Regulating ABA Synthesis and Root Development. Plant Physiol. 2018, 178, 753–770. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M. sativa (Medicago sativa L.) | M. truncatula (Medicago truncatula L.) | |||||||
|---|---|---|---|---|---|---|---|---|
| Treatments | Control | NaCl | Leu | Leu + NaCl | Control | NaCl | Leu | Leu + NaCl |
| Root | 2.10 c ± 0.03 | 1.16 f ± 0.01 | 2.51 a ± 0.01 | 1.71 e ± 0.02 | 1.94 d ± 0.02 | 0.84 g ± 0.02 | 2.39 b ± 0.00 | 1.22 f ± 0.03 |
| Seedling | 3.7 3c ± 0.05 | 2.42 e ± 0.02 | 4.10 b ± 0.01 | 2.79 d ± 0.01 | 3.87 d ± 0.01 | 2.31 d ± 0.01 | 4.41 a ± 0.04 | 2.79 d ± 0.01 |
| SOD | 57.36 c ± 1.51 | 72.65 a ± 0.64 | 45.19 d ± 0.41 | 65.53 b ± 0.76 | 52.81 b ± 0.76 | 63.66 b ± 0.76 | 40.45 e ± 0.73 | 57.53 e ± 1.12 |
| CAT | 7.29 e ± 0.16 | 28.73 a ± 0.29 | 4.90 f ± 0.08 | 10.66 d ± 0.18 | 15.83 d ± 0.18 | 31.65 d ± 0.18 | 12.92 c ± 0.06 | 20.46 b ± 0.15 |
| POD | 0.63 bc ± 0.01 | 0.82 a ± 0.02 | 0.41 e ± 0.02 | 0.63 bc ± 0.01 | 0.66 bc ± 0.01 | 0.79 bc ± 0.01 | 0.53 d ± 0.01 | 0.62 c ± 0.01 |
| TBARS | 0.41 c ± 0.03 | 0.52 b ± 0.04 | 0.22 d ± 0.01 | 0.52 b ± 0.03 | 0.42 b ± 0.03 | 0.72 b ± 0.03 | 0.23 d ± 0.01 | 0.71 a ± 0.01 |
| M. sativa (Medicago sativa L.) | M. truncatula (Medicago truncatula L.) | |||||||
|---|---|---|---|---|---|---|---|---|
| Treatments | Control | NaCl | Ile | Ile + NaCl | Control | NaCl | Ile | Ile + NaCl |
| Root | 2.10 d ± 0.03 | 1.16 f ± 0.01 | 2.41 a ± 0.01 | 1.58 e ± 0.02 | 1.94 d ± 0.02 | 0.84 d ± 0.02 | 2.29 c ± 0.00 | 1.16 f ± 0.03 |
| Seedling | 3.73 c ± 0.05 | 2.42 f ± 0.02 | 3.99 b ± 0.01 | 2.69 d ± 0.02 | 3.87 d ± 0.01 | 2.31 d ± 0.01 | 4.39 a ± 0.01 | 2.62 e ± 0.03 |
| SOD | 57.36 d ± 1.51 | 72.65 a ± 0.64 | 49.22 e ± 0.75 | 66.21 b ± 0.97 | 52.81 b ± 0.76 | 63.66 b ± 0.76 | 41.17 f ± 0.83 | 58.79 cd ± 0.58 |
| CAT | 7.29 e ± 0.16 | 28.73 a ± 0.29 | 5.31 f ± 0.03 | 21.19 c ± 0.54 | 15.83 d ± 0.18 | 31.65 d ± 0.18 | 12.90 d ± 0.09 | 24.65 b ± 0.45 |
| POD | 0.63 e ± 0.01 | 0.82 a ± 0.02 | 0.46 g ± 0.01 | 0.68 c ± 0.01 | 0.66 bc ± 0.01 | 0.79 bc ± 0.01 | 0.54 f ± 0.02 | 0.64 de ± 0.01 |
| TBARS | 0.41 c ± 0.03 | 0.52 b ± 0.04 | 0.23 d ± 0.01 | 0.54 b ± 0.02 | 0.42 b ± 0.03 | 0.72 b ± 0.03 | 0.24 d ± 0.01 | 0.68 a ± 0.02 |
| M. sativa (Medicago sativa L.) | M. truncatula (Medicago truncatula L.) | |||||||
|---|---|---|---|---|---|---|---|---|
| Treatments | Control | NaCl | Tyr | Tyr + NaCl | Control | NaCl | Tyr | Tyr + NaCl |
| Root | 2.10 c ± 0.03 | 1.16 f ± 0.01 | 2.35 b ± 0.03 | 1.52 d ± 0.01 | 1.94 d ± 0.02 | 0.84 d ± 0.02 | 2.42 a ± 0.01 | 1.29 e ± 0.01 |
| Seedling | 3.73 c ± 0.05 | 2.42 f ± 0.02 | 3.96 b ± 0.06 | 2.61 e ± 0.02 | 3.87 d ± 0.01 | 2.31 d ± 0.01 | 4.39 a ± 0.02 | 2.88 d ± 0.03 |
| SOD | 57.36 d ± 1.51 | 72.65 a ± 0.64 | 53.76 e ± 1.26 | 62.45 c ± 1.23 | 52.81 b ± 0.76 | 63.66 b ± 0.76 | 34.70 f ± 0.67 | 52.45 e ± 1.08 |
| CAT | 7.29 f ± 0.16 | 28.73 a ± 0.29 | 5.77 g ± 0.05 | 23.83 b ± 0.29 | 15.83 d ± 0.18 | 31.65 d ± 0.18 | 11.53 e ± 0.04 | 19.05 d ± 0.50 |
| POD | 0.63 e ± 0.01 | 0.82 a ± 0.02 | 0.47 g ± 0.01 | 0.75 c ± 0.01 | 0.66 bc ± 0.01 | 0.79 bc ± 0.01 | 0.50 f ± 0.01 | 0.65 de ± 0.01 |
| TBARS | 0.41 d ± 0.03 | 0.52 c ± 0.04 | 0.24 e ± 0.01 | 0.53 c ± 0.01 | 0.42 b ± 0.03 | 0.72 b ± 0.03 | 0.22 e ± 0.01 | 0.65 b ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, S.; Pan, L.; Li, J.; Wang, J.; Ahmad, I.; Liu, H.; Bai, Y.; Kang, B.; Yin, J.; Gao, Y.; et al. The Involvement of Amino Acid Metabolism in the Mechanisms of Salt Tolerance Adaptation in Medicago sativa and Medicago truncatula. Plants 2025, 14, 929. https://doi.org/10.3390/plants14060929
Shen S, Pan L, Li J, Wang J, Ahmad I, Liu H, Bai Y, Kang B, Yin J, Gao Y, et al. The Involvement of Amino Acid Metabolism in the Mechanisms of Salt Tolerance Adaptation in Medicago sativa and Medicago truncatula. Plants. 2025; 14(6):929. https://doi.org/10.3390/plants14060929
Chicago/Turabian StyleShen, Sicong, Ling Pan, Junhao Li, Jing Wang, Irshad Ahmad, Huhu Liu, Yuyu Bai, Bowen Kang, Juncheng Yin, Yang Gao, and et al. 2025. "The Involvement of Amino Acid Metabolism in the Mechanisms of Salt Tolerance Adaptation in Medicago sativa and Medicago truncatula" Plants 14, no. 6: 929. https://doi.org/10.3390/plants14060929
APA StyleShen, S., Pan, L., Li, J., Wang, J., Ahmad, I., Liu, H., Bai, Y., Kang, B., Yin, J., Gao, Y., Lu, Y., & Wang, X. (2025). The Involvement of Amino Acid Metabolism in the Mechanisms of Salt Tolerance Adaptation in Medicago sativa and Medicago truncatula. Plants, 14(6), 929. https://doi.org/10.3390/plants14060929