
Transcriptomic Analysis of Broussonetia papyrifera Fruit Under Manganese Stress and Mining of Flavonoid Synthesis Genes
 ,
, 
Abstract
1. Introduction
2. Results
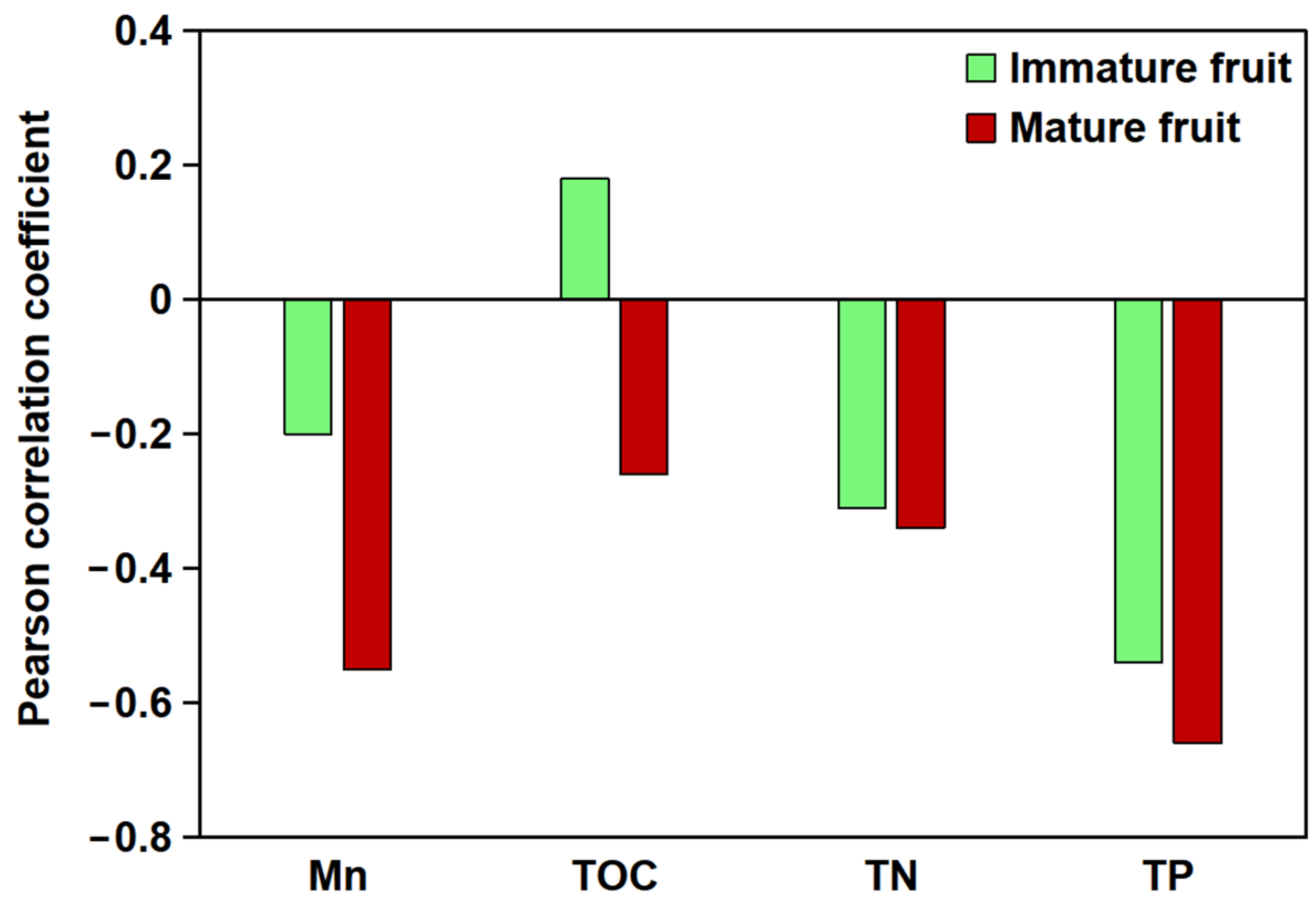
2.1. The Relationship Between the Flavonoid Content of B. papyrifera Fruit and the Soil Environment
2.2. Transcriptomic Data Analysis
2.3. Annotation of Gene Functions
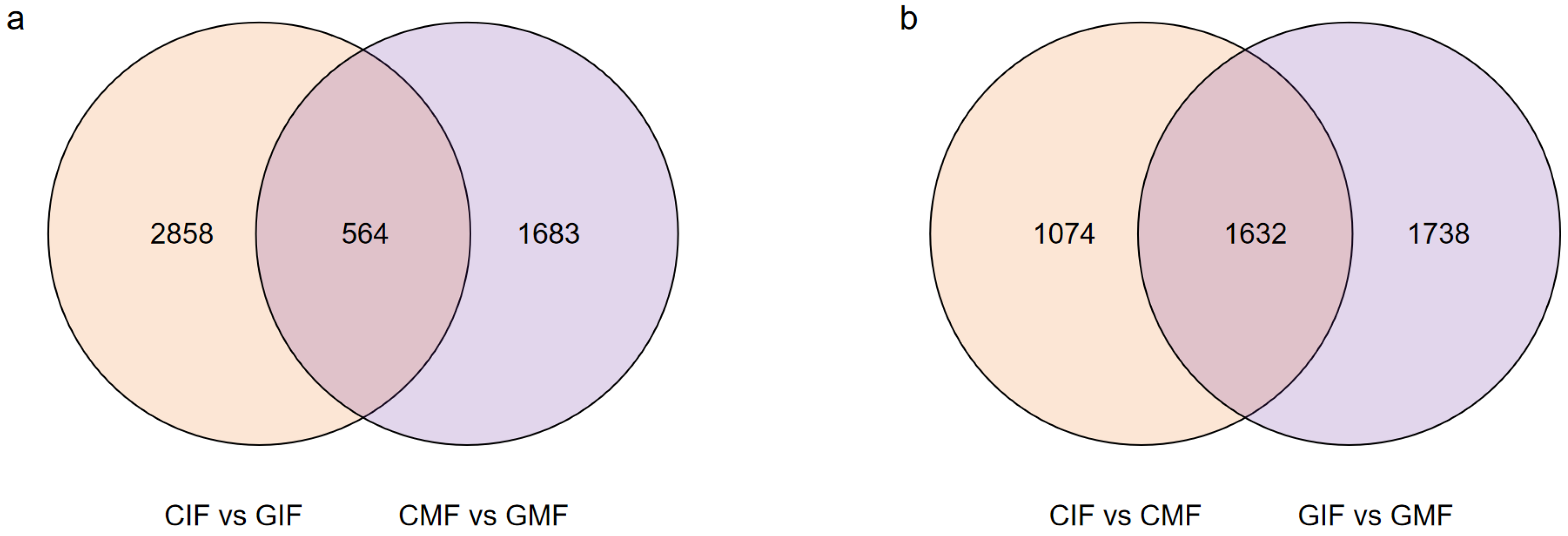
2.4. Screening of DEGs
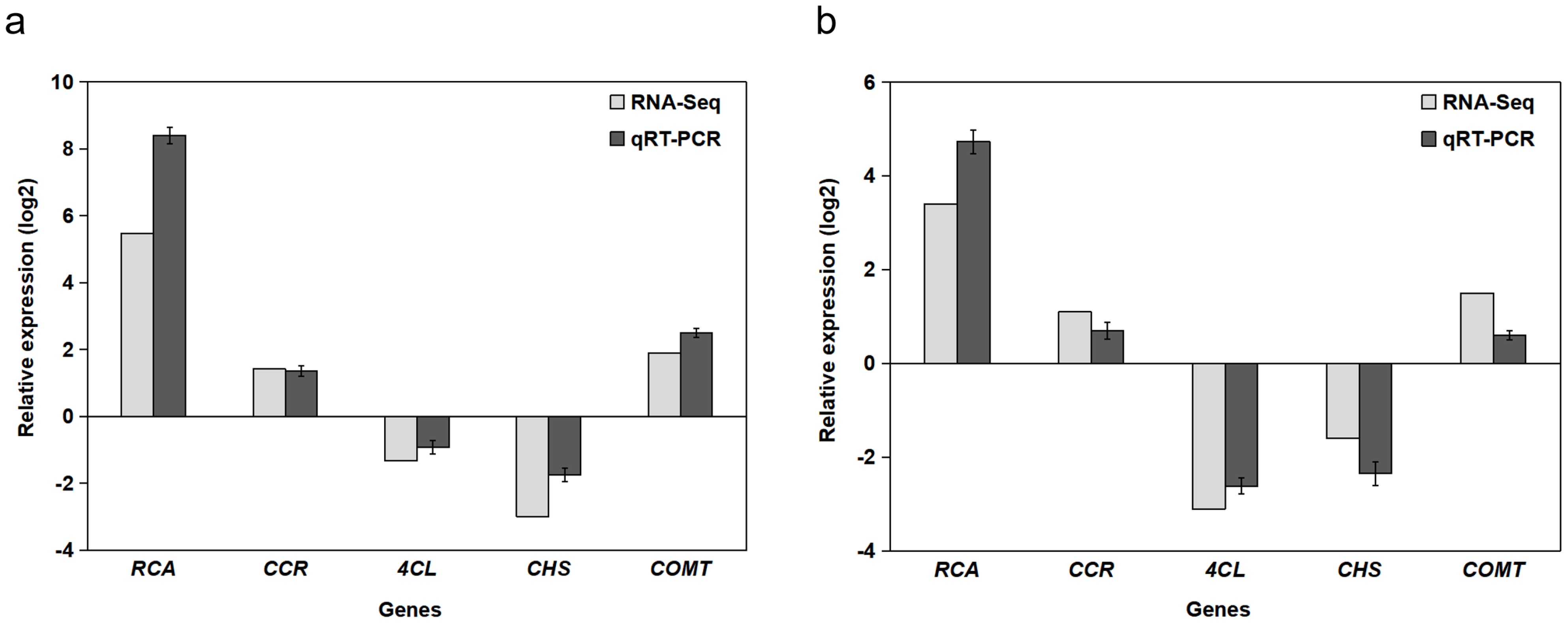
2.5. RT-qPCR Validation of DEGs
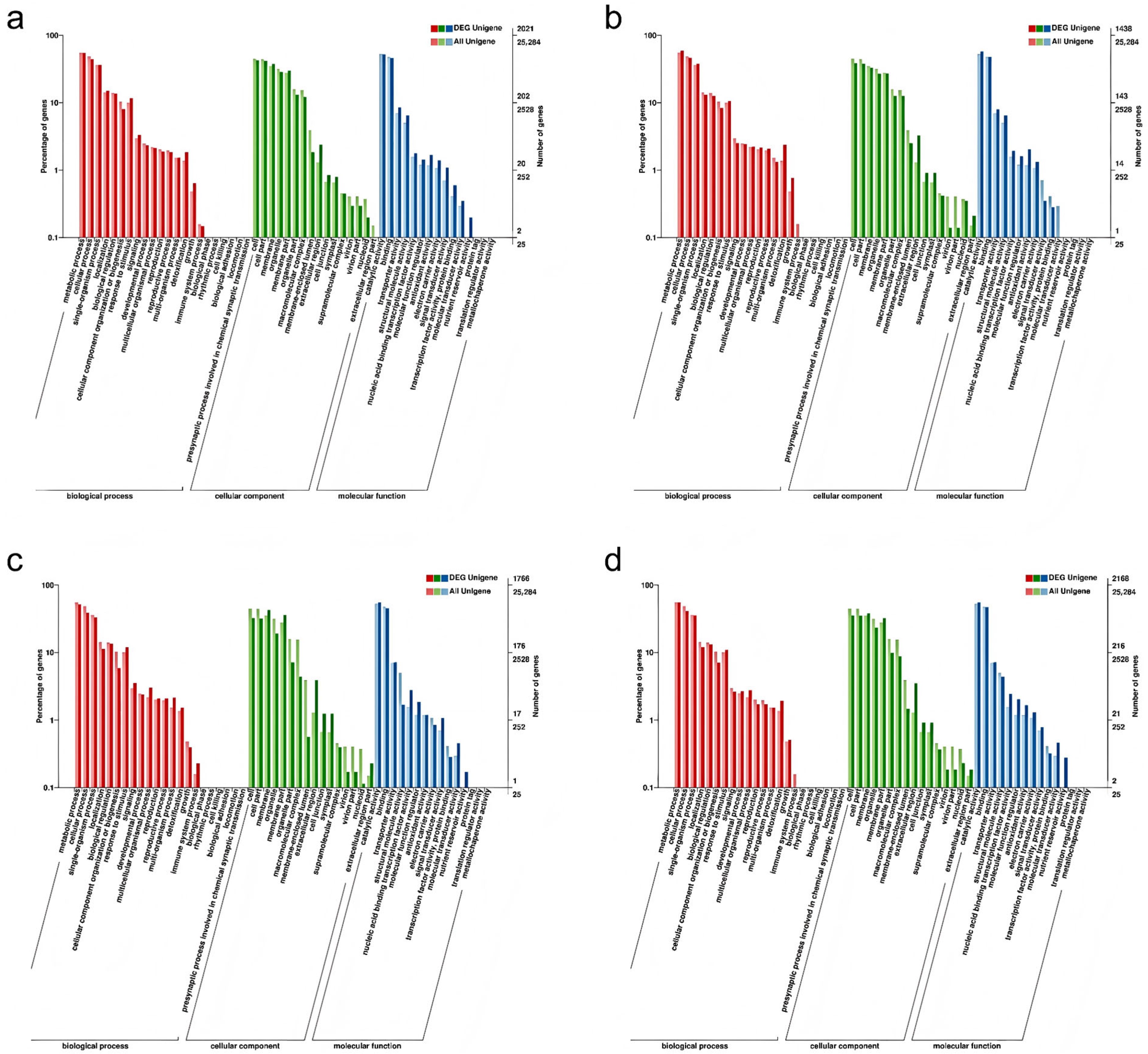
2.6. GO Annotation of DEGs
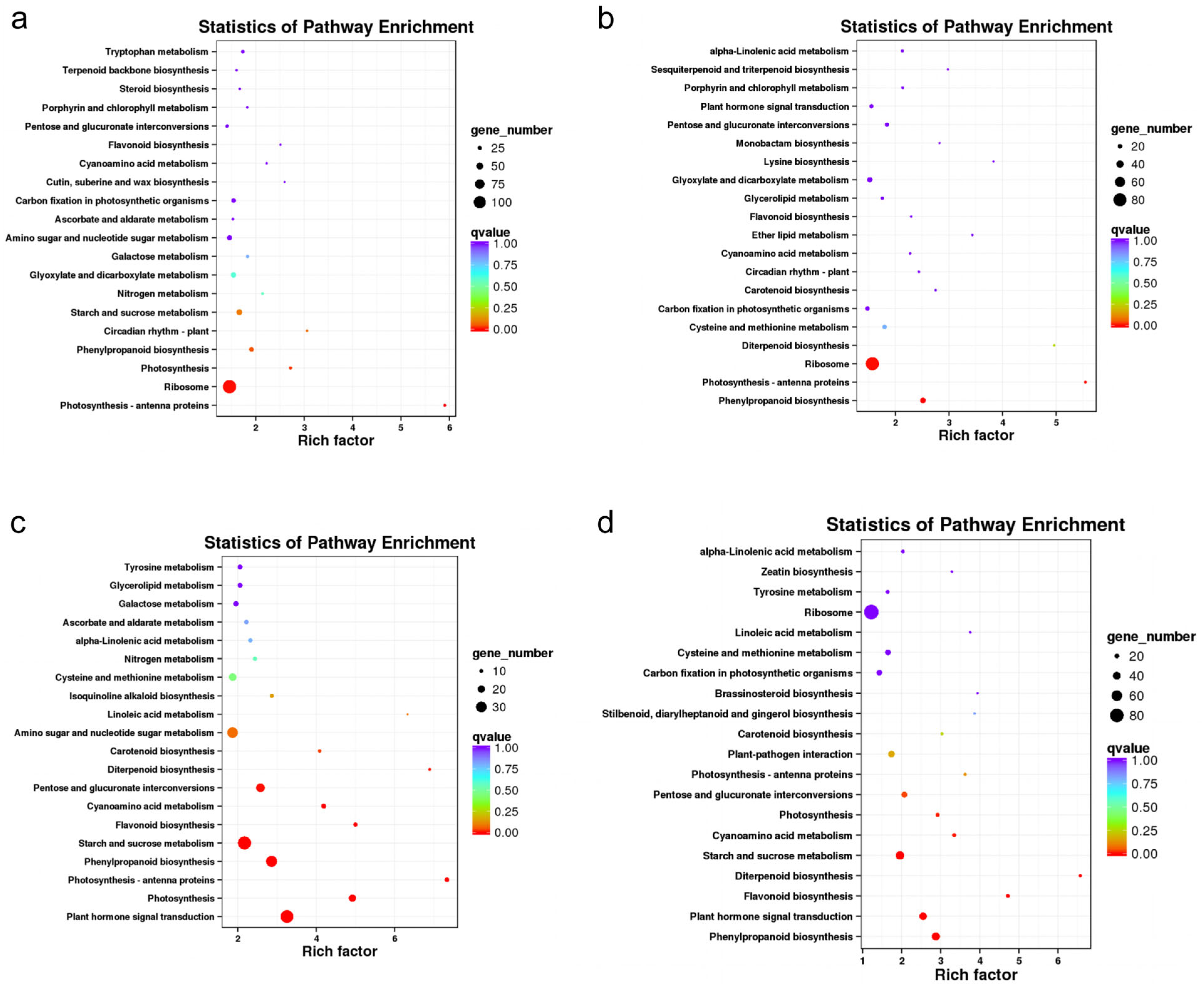
2.7. KEGG Pathway Analysis of DEGs
2.8. Analysis of DEGs Related to Flavonoid Synthesis
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Soil Analysis
4.2. RNA Extraction, Library Construction, and Sequencing
4.3. Data Assembly and Gene Function Annotation
4.4. Quantitative Gene Expression Level and Differential Expression Analysis
4.5. RT-qPCR to Verify DEGs
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xu, Z.; Yang, S.; Li, C.; Xie, M.; He, Y.; Chen, S.; Tang, Y.; Li, D.; Wang, T.; Yang, G. Characterization of metallothionein genes from Broussonetia papyrifera: Metal binding and heavy metal tolerance mechanisms. BMC Genom. 2024, 25, 563. [Google Scholar] [CrossRef]
- Zhou, F.; Xu, L.; Shi, C.; Wu, F.; Yang, S. Identification of the Optimal Quantitative RT-PCR Reference Gene for Paper Mulberry (Broussonetia papyrifera). Curr. Issues Mol. Biol. 2024, 46, 10779–10794. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, B.; Zou, J.; Luo, Z.; Yang, H.; Zhou, P.; Chen, X.; Zhou, W. Induction of tetraploids in Paper Mulberry (Broussonetia papyrifera (L.) L’Hér. ex Vent.) by colchicine. BMC Plant Biol. 2023, 23, 574. [Google Scholar] [CrossRef]
- Tang, T.; Bai, J.; Ao, Z.; Wei, Z.; Hu, Y.; Liu, S. Effects of dietary paper mulberry (Broussonetia papyrifera) on growth performance and muscle quality of grass carp (Ctenopharyngodon idella). Animals 2021, 11, 1655. [Google Scholar] [CrossRef] [PubMed]
- Nong, H.; Liu, J.; Chen, J.; Zhao, Y.; Wu, L.; Tang, Y.; Liu, W.; Yang, G.; Xu, Z. Woody plants have the advantages in the phytoremediation process of manganese ore with the help of microorganisms. Sci. Total Environ. 2023, 863, 160995. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-H.; Xiao, X.-Y.; Guo, Z.-H.; Peng, C.; Wang, X.-Y. Potential of Intercropping Pennisetum purpureum Schum with Melia azedarach L. and Broussonetia papyrifera for Phytoremediation of Heavy-metal Contaminated Soil around Mining Areas. Huan Jing Ke Xue Huanjing Kexue 2023, 44, 426–435. [Google Scholar] [PubMed]
- Xu, Z.; Wang, T.; Hou, S.; Ma, J.; Li, D.; Chen, S.; Gao, X.; Zhao, Y.; He, Y.; Yang, G. A R2R3-MYB, BpMYB1, from paper mulberry interacts with DELLA protein BpGAI1 in soil cadmium phytoremediation. J. Hazard. Mater. 2024, 463, 132871. [Google Scholar] [CrossRef]
- Pang, S.; Huang, B.; Zhang, Q. Advances in research on chemical constituents of Broussonetia plants and their pharmacological activities. Pharm. Care Res. 2006, 6, 98. [Google Scholar]
- Guo, P.; Huang, Z.; Li, X.; Zhao, W.; Wang, Y. Transcriptome Sequencing of Broussonetia papyrifera Leaves Reveals Key Genes Involved in Flavonoids Biosynthesis. Plants 2023, 12, 563. [Google Scholar] [CrossRef]
- Peng, X.; Liu, H.; Chen, P.; Tang, F.; Hu, Y.; Wang, F.; Pi, Z.; Zhao, M.; Chen, N.; Chen, H.; et al. A Chromosome-Scale Genome Assembly of Paper Mulberry (Broussonetia papyrifera) Provides New Insights into Its Forage and Papermaking Usage. Mol. Plant 2019, 12, 661–677. [Google Scholar] [CrossRef]
- Hou, D.; O’Connor, D.; Igalavithana, A.D.; Alessi, D.S.; Luo, J.; Tsang, D.C.W.; Sparks, D.L.; Yamauchi, Y.; Rinklebe, J.; Ok, Y.S. Metal contamination and bioremediation of agricultural soils for food safety and sustainability. Nat. Rev. Earth Environ. 2020, 1, 366–381. [Google Scholar] [CrossRef]
- Zhenggang, X.; Yiwang, T.; Jiaying, W.; Chongxuan, H.; Tianyu, W.; Jiakang, Z.; Guiyan, Y. Broussonetia papyrifera fruits as a potential source of functional materials to develop the phytoremediation strategy. Environ. Chall. 2022, 7, 100478. [Google Scholar] [CrossRef]
- Kumar, S.; Korra, T.; Thakur, R.; Arutselvan, R.; Kashyap, A.S.; Nehela, Y.; Chaplygin, V.; Minkina, T.; Keswani, C. Role of plant secondary metabolites in defence and transcriptional regulation in response to biotic stress. Plant Stress 2023, 8, 100154. [Google Scholar]
- Al-Khayri, J.M.; Rashmi, R.; Toppo, V.; Chole, P.B.; Banadka, A.; Sudheer, W.N.; Nagella, P.; Shehata, W.F.; Al-Mssallem, M.Q.; Alessa, F.M. Plant secondary metabolites: The weapons for biotic stress management. Metabolites 2023, 13, 716. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, W.; Zhu, Y.-M.; Chen, Y.; Qiu, C.-W.; Zhu, S.; Wu, F. Genotypic differences in leaf secondary metabolism, plant hormones and yield under alone and combined stress of drought and salinity in cotton genotypes. Physiol. Plant. 2019, 165, 343–355. [Google Scholar] [CrossRef]
- Ngo, V.-D.; Jang, B.-E.; Park, S.-U.; Kim, S.-J.; Kim, Y.-J.; Chung, S.-O. Estimation of functional components of Chinese cabbage leaves grown in a plant factory using diffuse reflectance spectroscopy. J. Sci. Food Agric. 2019, 99, 711–718. [Google Scholar] [CrossRef]
- Yanzi, L.; Peng, L.; Yan, Z.; Kangkang, J.; Meng, D.; Zhiyuan, H.; Yunlin, Z.; Guiyan, Y.; Zhenggang, X. Utilization of Gynostemma pentaphyllum and Houttuynia cordata medicinal plants to make Jiaosu: A healthy food. CyTA-J. Food 2022, 20, 143–148. [Google Scholar] [CrossRef]
- Teklić, T.; Parađiković, N.; Špoljarević, M.; Zeljković, S.; Lončarić, Z.; Lisjak, M. Linking abiotic stress, plant metabolites, biostimulants and functional food. Ann. Appl. Biol. 2021, 178, 169–191. [Google Scholar] [CrossRef]
- Karmakar, R.; Mukhopadhyay, C. Green synthesis of bioactive flavonoids as cardioprotective and anticancer drug agents. In Green Approaches in Medicinal Chemistry for Sustainable Drug Design; Elsevier: Amsterdam, The Netherlands, 2024; pp. 305–343. [Google Scholar]
- Rao, M.J.; Duan, M.; Eman, M.; Yuan, H.; Sharma, A.; Zheng, B. Comparative Analysis of Citrus Species’ Flavonoid Metabolism, Gene Expression Profiling, and Their Antioxidant Capacity under Drought Stress. Antioxidants 2024, 13, 1149. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, X.; Cui, D.; Zhu, Y.G.; Zhang, Y.; Zhang, Z. Mechanism of flavonols on detoxification, migration and transformation of indium in rhizosphere system. Sci. Total Environ. 2024, 929, 172693. [Google Scholar] [CrossRef]
- Oluwole, O.; Fernando, W.B.; Lumanlan, J.; Ademuyiwa, O.; Jayasena, V. Role of phenolic acid, tannins, stilbenes, lignans and flavonoids in human health—A review. Int. J. Food Sci. Technol. 2022, 57, 6326–6335. [Google Scholar] [CrossRef]
- Gupta, R.; Min, C.W.; Kramer, K.; Agrawal, G.K.; Rakwal, R.; Park, K.-H.; Wang, Y.; Finkemeier, I.; Kim, S.T. A Multi-Omics Analysis of Glycine max Leaves Reveals Alteration in Flavonoid and Isoflavonoid Metabolism Upon Ethylene and Abscisic Acid Treatment. Proteomics 2018, 18, 1700366. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The Flavonoid Biosynthesis Network in Plants. Int. J. Mol. Sci. 2021, 22, 12824. [Google Scholar] [CrossRef]
- Wang, F.; Su, Y.; Chen, N.; Shen, S. Genome-wide analysis of the UGT gene family and identification of flavonoids in Broussonetia papyrifera. Molecules 2021, 26, 3449. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.-L.; Liu, T.-L.; Xue, J.-J.; Hong, W.; Zhang, Y.; Zhang, D.-X.; Cui, C.-C.; Liu, M.-C.; Niu, S.-L. Flavanoids derivatives from the root bark of Broussonetia papyrifera as a tyrosinase inhibitor. Ind. Crops Prod. 2019, 138, 111445. [Google Scholar] [CrossRef]
- Jiao, P.; Chaoyang, L.; Wenhan, Z.; Jingyi, D.; Yunlin, Z.; Zhenggang, X. Integrative Metabolome and Transcriptome Analysis of Flavonoid Biosynthesis Genes in Broussonetia papyrifera Leaves from the Perspective of Sex Differentiation. Front. Plant Sci. 2022, 13, 900030. [Google Scholar] [CrossRef]
- Chang, C.-S.; Liu, H.-L.; Moncada, X.; Seelenfreund, A.; Seelenfreund, D.; Chung, K.-F. A holistic picture of Austronesian migrations revealed by phylogeography of Pacific paper mulberry. Proc. Natl. Acad. Sci. USA 2015, 112, 13537. [Google Scholar] [CrossRef]
- Xu, Z.; Dong, M.; Peng, X.; Ku, W.; Zhao, Y.; Yang, G. New insight into the molecular basis of cadmium stress responses of wild paper mulberry plant by transcriptome analysis. Ecotoxicol. Environ. Saf. 2019, 171, 301–312. [Google Scholar] [CrossRef]
- Vidović, N.; Pasković, I.; Lukić, I.; Žurga, P.; Majetić Germek, V.; Grozić, K.; Cukrov, M.; Marcelić, Š.; Ban, D.; Talhaoui, N. Biophenolic profile modulations in olive tissues as affected by manganese nutrition. Plants 2021, 10, 1724. [Google Scholar] [CrossRef]
- Chen, H.; Yang, J.; Deng, X.; Lei, Y.; Xie, S.; Guo, S.; Ren, R.; Li, J.; Zhang, Z.; Xu, T. Foliar-sprayed manganese sulfate improves flavonoid content in grape berry skin of Cabernet Sauvignon (Vitis vinifera L.) growing on alkaline soil and wine chromatic characteristics. Food Chem. 2020, 314, 126182. [Google Scholar] [CrossRef]
- Li, J.; Ackah, M.; Amoako, F.K.; Cui, Z.; Sun, L.; Li, H.; Tsigbey, V.E.; Zhao, M.; Zhao, W. Metabolomics and physio-chemical analyses of mulberry plants leaves response to manganese deficiency and toxicity reveal key metabolites and their pathways in manganese tolerance. Front. Plant Sci. 2024, 15, 1349456. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Pan, G.; Li, X.; Kuang, X.; Wang, W.; Liu, W. Effects of exogenous manganese on its plant growth, subcellular distribution, chemical forms, physiological and biochemical traits in Cleome viscosa L. Ecotoxicol. Environ. Saf. 2020, 198, 110696. [Google Scholar] [CrossRef] [PubMed]
- Santos, E.F.; Santini, J.M.K.; Paixão, A.P.; Júnior, E.F.; Lavres, J.; Campos, M.; Dos Reis, A.R. Physiological highlights of manganese toxicity symptoms in soybean plants: Mn toxicity responses. Plant Physiol. Biochem. 2017, 113, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xiao, Y.; Bai, C.; Liu, H.; Su, X.; Jin, P.; Xu, H.; Cao, L.; Yao, L. The physiological and biochemical responses to dark pericarp disease induced by excess manganese in litchi. Plant Physiol. Biochem. 2024, 206, 108269. [Google Scholar] [CrossRef]
- Bai, Y.; Zhou, Y.; Gong, J. Physiological mechanisms of the tolerance response to manganese stress exhibited by Pinus massoniana, a candidate plant for the phytoremediation of Mn-contaminated soil. Environ. Sci. Pollut. Res. 2021, 28, 45422–45433. [Google Scholar] [CrossRef]
- Chen, Q.; Song, D.; Sun, X.; Tian, Y.; Yan, Z.; Min, T.; Wang, H.; Wang, L. Functional Characterization of F3H Gene and Optimization of Dihydrokaempferol Biosynthesis in Saccharomyces cerevisiae. Molecules 2024, 29, 2196. [Google Scholar] [CrossRef]
- Dai, M.; Kang, X.; Wang, Y.; Huang, S.; Guo, Y.; Wang, R.; Chao, N.; Liu, L. Functional characterization of Flavanone 3-Hydroxylase (F3H) and its role in anthocyanin and flavonoid biosynthesis in mulberry. Molecules 2022, 27, 3341. [Google Scholar] [CrossRef]
- Malaník, M.; Treml, J.; Leláková, V.; Nykodýmová, D.; Oravec, M.; Marek, J.; Šmejkal, K. Anti-inflammatory and antioxidant properties of chemical constituents of Broussonetia papyrifera. Bioorg. Chem. 2020, 104, 104298. [Google Scholar] [CrossRef]
- Liang, J.; Wang, Z.; Ren, Y.; Jiang, Z.; Chen, H.; Hu, W.; Tang, M. The alleviation mechanisms of cadmium toxicity in Broussonetia papyrifera by arbuscular mycorrhizal symbiosis varied with different levels of cadmium stress. J. Hazard. Mater. 2023, 459, 132076. [Google Scholar] [CrossRef]
- Beyer Jr, W.F.; Fridovich, I. In vivo competition between iron and manganese for occupancy of the active site region of the manganese-superoxide dismutase of Escherichia coli. J. Biol. Chem. 1991, 266, 303–308. [Google Scholar] [CrossRef]
- Katayama-Ikegami, A.; Sakamoto, T.; Shibuya, K.; Katayama, T.; Gao-Takai, M. Effects of abscisic acid treatment on berry coloration and expression of flavonoid biosynthesis genes in grape. Am. J. Plant Sci. 2016, 7, 1325. [Google Scholar] [CrossRef]
- Costa, G.B.; Simioni, C.; Ramlov, F.; Maraschin, M.; Chow, F.; Bouzon, Z.L.; Schmidt, É.C. Effects of manganese on the physiology and ultrastructure of Sargassum cymosum. Environ. Exp. Bot. 2017, 133, 24–34. [Google Scholar] [CrossRef]
- He, L.; Su, R.; Chen, Y.; Zeng, P.; Du, L.; Cai, B.; Zhang, A.; Zhu, H. Integration of manganese accumulation, subcellular distribution, chemical forms, and physiological responses to understand manganese tolerance in Macleaya cordata. Environ. Sci. Pollut. Res. 2022, 29, 39017–39026. [Google Scholar] [CrossRef]
- Thiesen, L.A.; Brunetto, G.; Trentin, E.; da Silva, A.A.K.; Tabaldi, L.A.; Schwalbert, R.; Birck, T.P.; Machado, L.C.; Nicoloso, F.T. Subcellular distribution and physiological responses of native and exotic grasses from the Pampa biome subjected to excess manganese. Chemosphere 2023, 310, 136801. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Guo, Z.; Zhang, Y.; Xiao, X.; Peng, C. Potential of Pyrolysis for the Recovery of Heavy Metals and Bioenergy from Contaminated Broussonetia papyrifera Biomass. BioResources 2018, 13, 2932–2944. [Google Scholar] [CrossRef]
- Xu, Z.; Ding, Y.; Huang, H.; Wu, L.; Zhao, Y.; Yang, G. Biosorption Characteristics of Mn (II) by Bacillus cereus Strain HM-5 Isolated from Soil Contaminated by Manganese Ore. Pol. J. Environ. Stud. 2019, 28, 463–472. [Google Scholar] [CrossRef]
- Yue, Y.; Wang, H.; Wang, T.; Zheng, D.; Thomas, H.R.; Yang, X.; Wang, L. Integrated physiological, hormonal, and transcriptomic analyses reveal a novel E3 ubiquitin ligase-mediated cold acclimation mechanism for the acquisition of cold tolerance in sweet osmanthus. Ind. Crops Prod. 2024, 220, 119171. [Google Scholar] [CrossRef]
- Almalki, S.; Kiss-Toth, E.; Ridger, V. BS07 Micro RNA sequencing analysis of neutrophil derived microvesicles produced in response to proatherogenic stimuli. Heart 2024, 110, 248–249. [Google Scholar]
- Xu, L.; Berninger, A.; Lakin, S.M.; O’Donnell, V.; Pierce, J.L.; Pauszek, S.J.; Barrette, R.W.; Faburay, B. Direct RNA sequencing of foot-and-mouth disease virus genome using a flongle on MinION. Bio-Protocol 2024, 14, e5017. [Google Scholar] [CrossRef]
- de Sena Brandine, G.; Smith, A.D. Falco: High-speed FastQC emulation for quality control of sequencing data. F1000Research 2021, 8, 1874. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Nasiri, J.; Soorni, A.; van Dijk, A.; Naghavi, M. Terpenoid Biosynthetic Pathway in Ferula persica Using Transcriptome Analysis and Metabolome Data. J. Agric. Sci. Technol. (JAST) 2024, 26, 177–192. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Langdon, W.B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015, 8, 1. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Liu, L.; Wen, J.; Liu, J.; Li, D.; Zhang, T.; Peng, C.; He, Y. Rhizosphere metagenomics provides insights into the environmental effect on the secondary metabolism of Ligusticum chuanxiong. Ind. Crops Prod. 2024, 217, 118779. [Google Scholar] [CrossRef]
- Amalfitano, A.; Stocchi, N.; Atencio, H.M.; Villarreal, F.; Ten Have, A. Seqrutinator: Scrutiny of large protein superfamily sequence datasets for the identification and elimination of non-functional homologues. Genome Biol. 2024, 25, 230. [Google Scholar] [CrossRef]
- Sami, A.; El-Metwally, S.; Rashad, M. MAC-ErrorReads: Machine learning-assisted classifier for filtering erroneous NGS reads. BMC Bioinform. 2024, 25, 61. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, B.; Lin, L.-L.; Zhao, S. Evaluation and comparison of computational tools for RNA-seq isoform quantification. BMC Genom. 2017, 18, 583. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef]
- Hoyer, A.; Chakraborty, S.; Lilienthal, I.; Konradsen, J.R.; Katayama, S.; Söderhäll, C. The functional role of CST1 and CCL26 in asthma development. Immun. Inflamm. Dis. 2024, 12, e1162. [Google Scholar] [CrossRef]
- Cai, M.M.; He, Z.H.; Lin, Z.R.; Nie, G.B.; Li, X.Y.; Liu, H.S.; Ma, Q. Comparative physiological, transcriptome, and qRT-PCR analysis provide insights into osmotic adjustment in the licorice species Glycyrrhiza inflata under salt stress. Crop Sci. 2023, 63, 1442–1457. [Google Scholar] [CrossRef]
- Chen, M.; Wang, Z.; Hao, Z.; Li, H.; Feng, Q.; Yang, X.; Han, X.; Zhao, X. Screening and Validation of Appropriate Reference Genes for Real-Time Quantitative PCR under PEG, NaCl and ZnSO4 Treatments in Broussonetia papyrifera. Int. J. Mol. Sci. 2023, 24, 15087. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ma, Z.; Qiao, Y.; Wang, Z.; Chen, W.; Zheng, S.; Yu, C.; Song, L.; Lou, H.; Wu, J. Transcriptome sequencing and metabolomics analyses provide insights into the flavonoid biosynthesis in Torreya grandis kernels. Food Chem. 2022, 374, 131558. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Read Number | Base Number | GC Content | %≥Q30 |
|---|---|---|---|---|
| CIF | 21,185,851 | 6,325,128,368 | 47.37% | 92.77% |
| GIF | 22,811,605 | 6,813,841,942 | 47.02% | 92.45% |
| CMF | 24,414,219 | 7,279,736,050 | 48.12% | 93.17% |
| GMF | 22,202,511 | 6,625,058,020 | 47.88% | 92.88% |
| Database | Number of Unigenes | Percentage (%) |
|---|---|---|
| GO | 21,389 | 46.43 |
| KEGG | 10,048 | 21.81 |
| KOG | 16,670 | 36.18 |
| Pfam | 23,501 | 51.01 |
| Swiss-Prot | 18,854 | 40.92 |
| NR | 14,275 | 30.98 |
| COG | 12,044 | 26.14 |
| Egg-NOG | 28,972 | 62.88 |
| Total unigenes | 46,072 | 100 |
| DEG Set | Annotated | COG | GO | KEGG | KOG | Pfam | nr | Swiss-Prot | eggNOG |
|---|---|---|---|---|---|---|---|---|---|
| CIF vs. GIF | 2989 | 1388 | 2021 | 1128 | 1637 | 2427 | 2923 | 2047 | 2775 |
| CMF vs. GMF | 2053 | 941 | 1438 | 750 | 1025 | 1661 | 2042 | 1413 | 1934 |
| CIF vs. CMF | 2451 | 991 | 1766 | 828 | 1126 | 1954 | 2441 | 1853 | 2308 |
| GIF vs. GMF | 3016 | 1314 | 2168 | 1069 | 1446 | 2455 | 2978 | 2232 | 2851 |
| Grouping | Tendency | Genes | Name | Ko ID |
|---|---|---|---|---|
| CIF vs. GIF | Up | Cytochrome P450 | CYP | K05280 |
| Up | Dihydroflavonol/flavanone 4-reductase | DFR | K13082 | |
| Down | Flavone 3-hydroxylase | F3H | K00475 | |
| Down | Flavonoid 3′,5′-O-methyltransferase | FAOMT | K00588 | |
| CMF vs. GMF | Up | Cytochrome P450 | CYP | K00487 |
| Down | Chalcone synthase | CHS | K00660 | |
| Down | Flavonol synthase | FLS | K05278 | |
| CIF vs. CMF | Up | BAHD acyltransferase | BAHD | K13065 |
| Up | Cytochrome P450 | CYP | K00487 | |
| Up | Caffeoyl-CoA-O-methyltransferase | CCoAOMT | K00588 | |
| Up | Chalcone isomerase | CHI | K01859 | |
| Up | Flavone 3-hydroxylase | F3H | K00475 | |
| Up | Flavonol synthase | FLS | K05278 | |
| Up | Anthocyanidin reductase | ANR | K08695 | |
| GIF vs. GMF | Up | Chalcone isomerase | CHI | K01859 |
| Up | Caffeoyl-CoA-O-methyltransferase | CCoAOMT | K00588 | |
| Up | Dihydroflavonol/flavanone 4-reductase | DFR | K13082 | |
| Up | Flavonol synthase | FLS | K05278 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Tang, Y.; Zhang, J.; Li, T.; Wang, Y.; Huang, Y.; Zhao, Y.; Yang, G.; Xu, Z. Transcriptomic Analysis of Broussonetia papyrifera Fruit Under Manganese Stress and Mining of Flavonoid Synthesis Genes. Plants 2025, 14, 883. https://doi.org/10.3390/plants14060883
Hu Z, Tang Y, Zhang J, Li T, Wang Y, Huang Y, Zhao Y, Yang G, Xu Z. Transcriptomic Analysis of Broussonetia papyrifera Fruit Under Manganese Stress and Mining of Flavonoid Synthesis Genes. Plants. 2025; 14(6):883. https://doi.org/10.3390/plants14060883
Chicago/Turabian StyleHu, Zhiyuan, Yiwang Tang, Jihui Zhang, Taotao Li, Yihan Wang, Yani Huang, Yunlin Zhao, Guiyan Yang, and Zhenggang Xu. 2025. "Transcriptomic Analysis of Broussonetia papyrifera Fruit Under Manganese Stress and Mining of Flavonoid Synthesis Genes" Plants 14, no. 6: 883. https://doi.org/10.3390/plants14060883
APA StyleHu, Z., Tang, Y., Zhang, J., Li, T., Wang, Y., Huang, Y., Zhao, Y., Yang, G., & Xu, Z. (2025). Transcriptomic Analysis of Broussonetia papyrifera Fruit Under Manganese Stress and Mining of Flavonoid Synthesis Genes. Plants, 14(6), 883. https://doi.org/10.3390/plants14060883