
Sixteen New Complete Plastid Genomes in the Tribe Loteae (Leguminosae): Structure and Phylogenetic Analysis

 , and
, and
Abstract
1. Introduction
2. Results
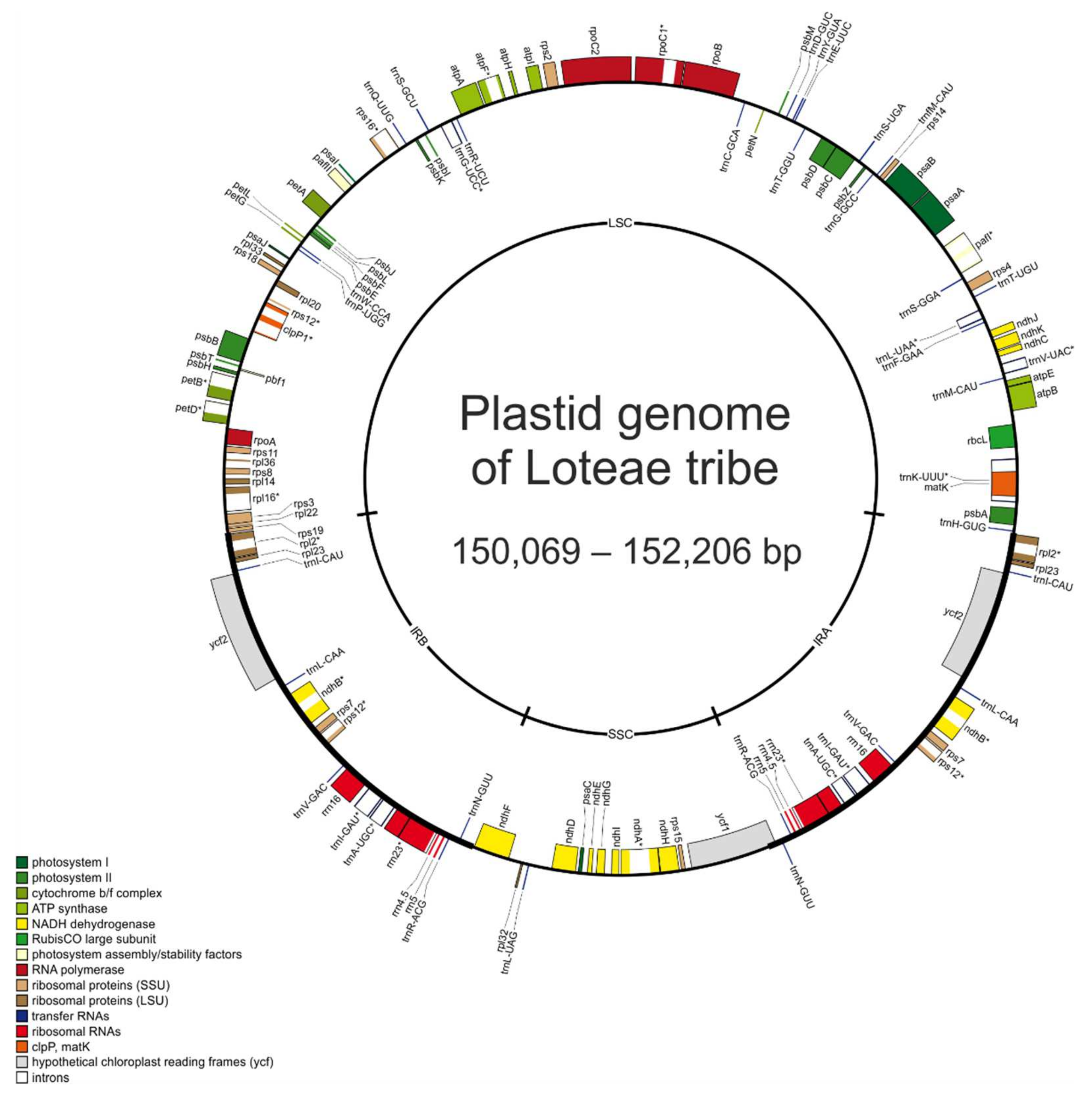
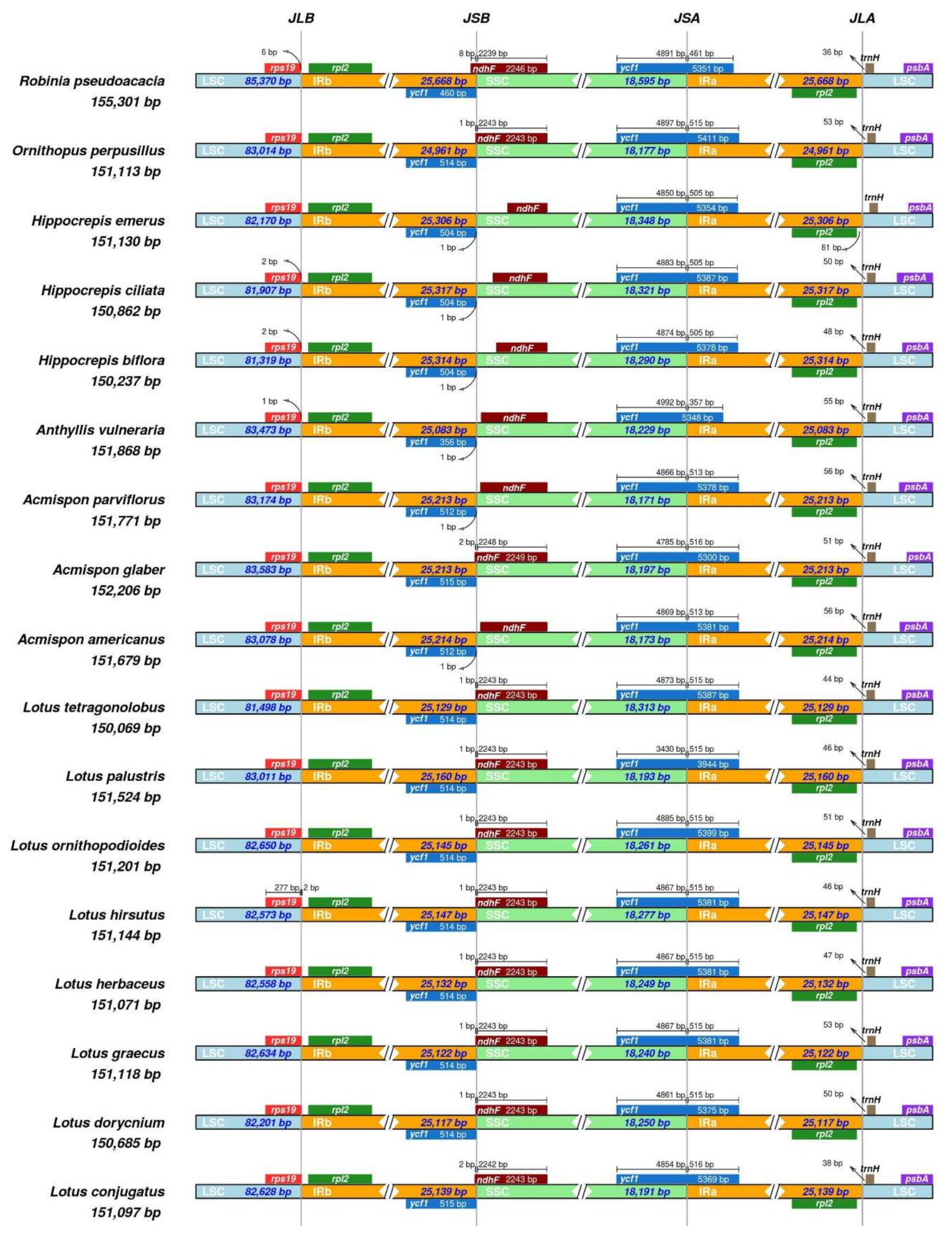
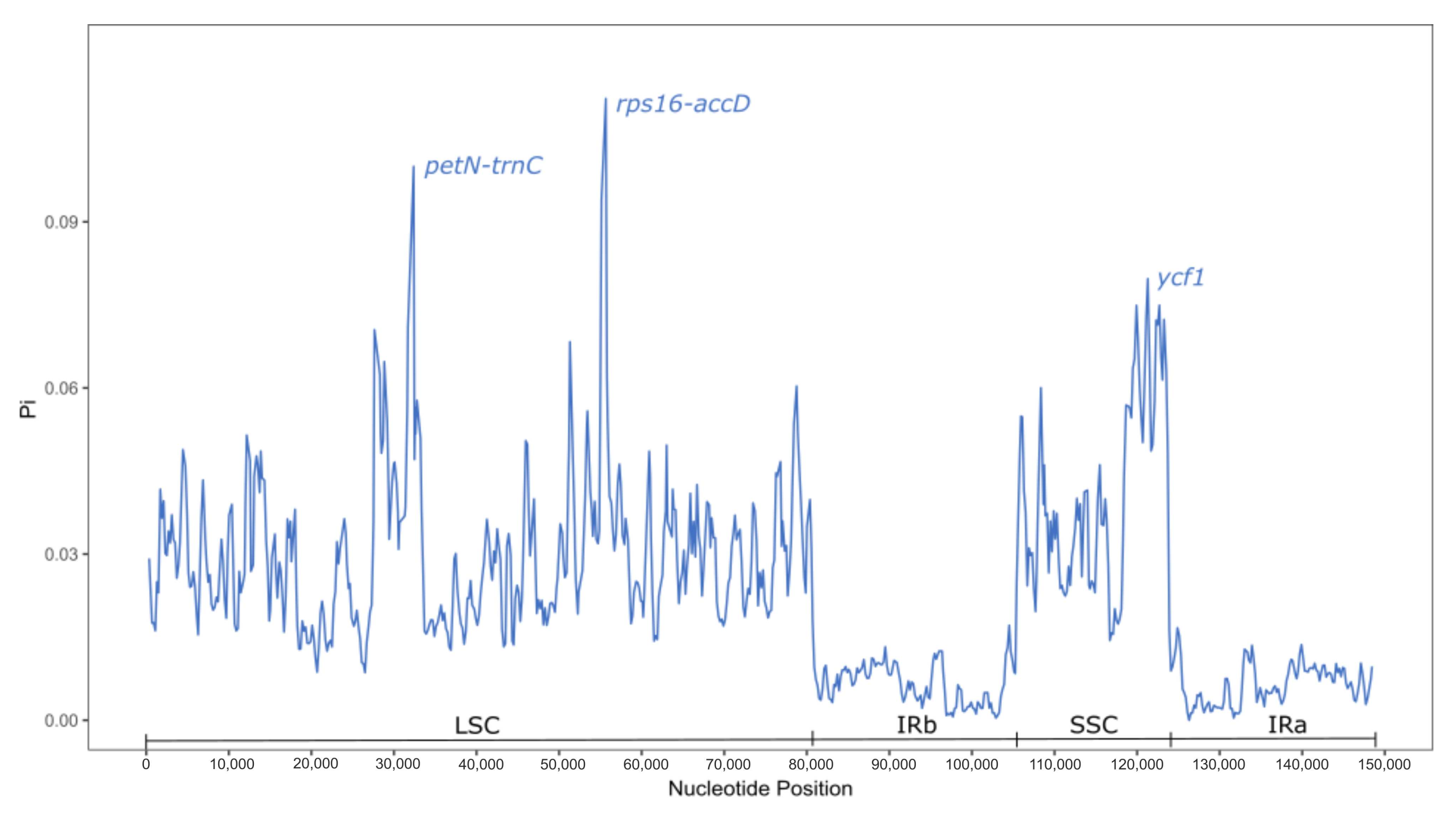
2.1. Comparative Analysis of the Loteae Plastomes
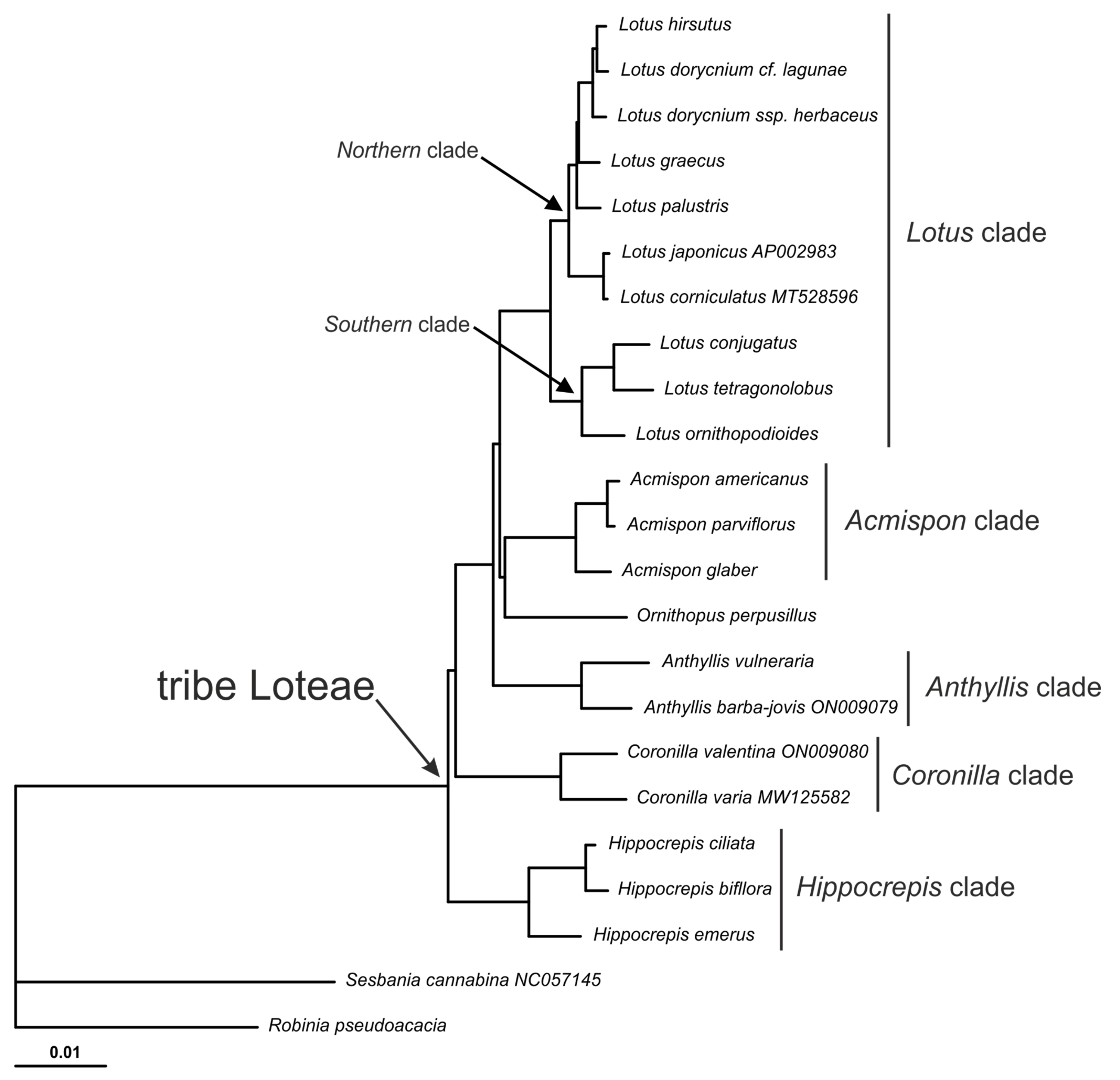
2.2. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Taxon Sampling and Sequencing
4.2. Chloroplast Genome Assembly and Annotation
4.3. Comparative Analysis of the Loteae Plastomes
4.4. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allan, G.J.; Porter, J.M. Tribal delimitation and phylogenetic relationships of Loteae and Coronilleae (Faboideae: Fabaceae) with special reference to Lotus: Evidence from nuclear ribosomal ITS sequences. Amer. J. Bot. 2000, 87, 1871–1881. [Google Scholar] [CrossRef]
- Legume Phylogeny Working Group (LPWG); Andrella, G.C.; Atahuachi Burgos, M.; Bagnatori Sartori, Â.L.; Balan, A.; Bandyopadhyay, S.; Barbosa Pinto, R.; Barrett, R.; Boatwright, J.S.; Borges, L.M.; et al. The World Checklist of Vascular Plants (WCVP): Fabaceae, 2023v.4; Govaerts, R., Ed.; Royal Botanic Gardens Kew: Kew, UK, 2023. [Google Scholar] [CrossRef]
- Sokoloff, D.D. On taxonomy and phylogeny of the tribe Loteae DC. (Leguminosae). Byull. Moskovsk. Obshch. Isp. Prir. Otd. Biol. 2003, 108, 35–48. (In Russian) [Google Scholar]
- Sokoloff, D.D.; Degtjareva, G.V.; Endress, P.K.; Remizowa, M.V.; Samigullin, T.H.; Valiejo-Roman, C.M. Inflorescence and early flower development in Loteae (Leguminosae) in a phylogenetic and taxonomic context. Int. J. Plant Sci. 2007, 168, 801–833. [Google Scholar] [CrossRef]
- Allan, G.J.; Zimmer, E.A.; Wagner, W.L.; Sokoloff, D.D. Molecular phylogenetic analyses of tribe Loteae (Leguminosae): Implications for classification and biogeography. In Advances in Legume Systematics; Klitgaard, B.B., Bruneau, A., Eds.; Royal Botanic Gardens: Kew, UK, 2003; Volume 10, pp. 371–393. [Google Scholar]
- Degtjareva, G.V.; Valiejo-Roman, C.M.; Kramina, T.E.; Mironov, E.M.; Samigullin, T.H.; Sokoloff, D.D. Taxonomic and phylogenetic relationships between Old World and New World members of the tribe Loteae (Leguminosae): New insights from molecular and morphological data, with special emphasis on Ornithopus. Wulfenia 2003, 10, 15–50. [Google Scholar]
- Degtjareva, G.; Valiejo-Roman, C.; Samigullin, T.; Sokoloff, D. On generic rank and phylogenetic relationships of Dorycnopsis Boiss. (Leguminosae, Loteae). An. Jardín Botánico Madr. 2006, 63, 41–50. [Google Scholar] [CrossRef]
- Degtjareva, G.V.; Samigullin, T.H.; Vallejo-Roman, C.M.; Sokoloff, D.D. Phylogenetic placement of Podolotus suggests independent origin of lomentaceous fruits in Coronilla and Hippocrepis (Leguminosae: Loteae). Pakistan J. Bot. 2010, 42, 11–25. [Google Scholar]
- Nanni, L.; Ferradini, N.; Taffetani, F.; Papa, R. Molecular phylogeny of Anthyllis spp. Plant Biol. 2004, 6, 454–464. [Google Scholar] [CrossRef]
- Degtjareva, G.V.; Valiejo-Roman, C.M.; Samigullin, T.H.; Guara-Requena, M.; Sokoloff, D.D. Phylogenetics of Anthyllis (Leguminosae: Papilionoideae: Loteae): Partial incongruence between nuclear and plastid markers, a long branch problem and implications for morphological evolution. Mol. Phylogenet. Evol. 2012, 62, 693–707. [Google Scholar] [CrossRef]
- Cano, A.L.; Gómez, P.S.; Martínez, J.F.J. A New Species of Coronilla (Loteae, Fabaceae) from Southeastern Spain: Evidence from Morphological and Molecular Data. Folia Geobot. 2012, 47, 317–335. [Google Scholar] [CrossRef]
- Degtjareva, G.V.; Kramina, T.E.; Sokoloff, D.D.; Samigullin, T.H.; Valiejo-Roman, C.M.; Antonov, A.S. Phylogeny of the genus Lotus (Leguminosae, Loteae): Evidence from nrITS sequences and morphology. Can. J. Bot. 2006, 84, 813–830. [Google Scholar] [CrossRef]
- Degtjareva, G.V.; Kramina, T.E.; Sokoloff, D.D.; Samigullin, T.H.; Sandral, G.; Valiejo-Roman, C.M. New data on nrITS phylogeny of Lotus (Leguminosae, Loteae). Wulfenia 2008, 15, 35–49. [Google Scholar]
- Kramina, T.E.; Degtjareva, G.V.; Samigullin, T.H.; Valiejo-Roman, C.M.; Kirkbride, J.H., Jr.; Volis, S.; Deng, T.; Sokoloff, D.D. Phylogeny of Lotus (Leguminosae: Loteae): Partial incongruence between nrITS, nrETS and plastid markers and biogeographic implications. Taxon 2016, 65, 997–1018. [Google Scholar] [CrossRef]
- Jaén-Molina, R.; Marrero-Rodríguez, Á.; Caujapé-Castells, J.; Ojeda, D.I. Molecular phylogenetics of Lotus (Leguminosae) with emphasis in the tempo and patterns of colonization in the Macaronesian region. Mol. Phylogenet. Evol. 2021, 154, 106970. [Google Scholar] [CrossRef]
- Xiao, T.-W.; Xu, Y.; Jin, L.; Liu, T.-J.; Yan, H.-F.; Ge, X.-J. Conflicting phylogenetic signals in plastomes of the tribe Laureae (Lauraceae). PeerJ 2020, 8, e10155. [Google Scholar] [CrossRef]
- Choi, I.-S.; Cardoso, D.; de Queiroz, L.P.; de Lima, H.C.; Lee, C.; Ruhlman, T.A.; Jansen, R.K.; Wojciechowski, M.F. Highly Resolved Papilionoid Legume Phylogeny Based on Plastid Phylogenomics. Front. Plant Sci. 2022, 13, 823190. [Google Scholar] [CrossRef]
- Lee, C.; Choi, I.-S.; Cardoso, D.; de Lima, H.C.; de Queiroz, L.P.; Wojciechowski, M.F.; Jansen, R.K.; Ruhlman, T.A. The chicken or the egg? Plastome evolution and an independent loss of the inverted repeat in papilionoid legumes. Plant J. 2021, 107, 861–875. [Google Scholar] [CrossRef]
- Cardoso, D.; de Queiroz, L.P.; Pennigton, R.T.; de Lima, H.C.; Fonty, E.; Wojciechowski, M.F.; Lavin, M. Revisiting the phylogeny of papilionoid legumes: New insight from comprehensively sampled early-branching lineages. Am. J. Bot. 2012, 99, 1991–2013. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, D.; Pennington, R.T.; de Queiroz, L.P.; Boatwright, J.S.; VanWyk, B.-E.; Wojciechowski, M.F.; Lavin, M. Reconstructing the deep-branching relationships of the papilionoid legumes. S. Arf. J. Bot. 2013, 89, 58–75. [Google Scholar] [CrossRef]
- Lavin, M.; Herendeen, P.S.; Wojciechowski, M.F. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the Tertiary. Syst. Biol. 2005, 54, 530–549. [Google Scholar] [CrossRef]
- Farruggia, F.T.; Lavin, M.; Wojciechowski, M.F. Phylogenetic systematics and biogeography of the pantropical genus Sesbania (Leguminosae). Syst. Bot. 2018, 43, 414–429. [Google Scholar] [CrossRef]
- Schwarz, E.N.; Ruhlman, T.A.; Sabir, J.S.; Hajrah, N.H.; Alharbi, N.S.; Al-Malki, A.L.; Bailey, C.D.; Jansen, R.K. Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. J. Syst. Evol. 2015, 53, 458–468. [Google Scholar] [CrossRef]
- Kato, T.; Kaneko, T.; Sato, S.; Nakamura, Y.; Tabata, S. Complete structure of the chloroplast genome of a legume, Lotus japonicus. DNA Res. 2000, 7, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ma, L.; Yi, D.; Pang, Y. The complete chloroplast genome sequence of Lotus corniculatus L. Mitochondr. DNA Part B 2021, 6, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xi, Q.; Wei, X.; Fu, J.; Wang, Q.; He, H.; Ling, C.; Chang, T.; Zhao, Y. The complete chloroplast genome of Securigera varia (L.) Lassen. Mitochondr. DNA Part B 2021, 6, 900–901. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-X.; Wang, D.-J.; Yi, T.-S. Does IR-loss promote plastome structural variation and sequence evolution? Front. Plant Sci. 2022, 13, 888049. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.; Tewari, K.K. Inverted repeats in chloroplast DNA from higher plants. Proc. Natl. Acad. Sci. USA 1979, 76, 41–45. [Google Scholar] [CrossRef]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef]
- Ruhlman, T.A.; Zhang, J.; Blazier, J.C.; Sabir, J.S.M.; Jansen, R.K. Recombination-dependent replication and gene conversion homogenize repeat sequences and diversify plastid genome structure. Am. J. Bot. 2017, 104, 559–572. [Google Scholar] [CrossRef]
- Plunkett, G.M.; Downie, S.R. Expansion and contraction of the chloroplast inverted repeat in Apiaceae subfamily Apioideae. Syst. Bot. 2000, 25, 648–667. [Google Scholar] [CrossRef]
- Samigullin, T.; Logacheva, M.; Terentieva, E.; Degtjareva, G.; Pimenov, M.; Valiejo-Roman, C. Plastid phylogenomic analysis of Tordylieae tribe (Apiaceae, Apioideae). Plants 2022, 11, 709. [Google Scholar] [CrossRef]
- Haberle, R.C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA genes. J. Mol. Evol. 2008, 66, 350–361. [Google Scholar] [CrossRef]
- Lee, H.L.; Jansen, R.K.; Chumley, T.W.; Kim, K.J. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 2007, 24, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yu, X.; Yang, Y.; Wei, P.; Zhang, W.; Li, X.; Liu, C.; Zhao, S.; Li, X.; Liu, X. Comparative Analysis of Chloroplast Genomes within Saxifraga (Saxifragaceae) Takes Insights into Their Genomic Evolution and Adaption to the High-Elevation Environment. Genes 2022, 13, 1673. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Zhu, J.W.; Ma, X.D.; Li, H.-M.; Wu, B.-C.; Zhou, W.; Yang, J.-X.; Song, C.-F. Phylogenomics and adaptive evolution of hydrophytic umbellifers (tribe Oenantheae, Apioideae) revealed from chloroplast genomes. BMC Plant Biol. 2024, 24, 1140. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 1, e102. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Bouras, G.; Judd, L.M.; Edwards, R.A.; Vreugde, S.; Stinear, T.P.; Wick, R.R. How low can you go? Short-read polishing of Oxford Nanopore bacterial genome assemblies. Microb. Genom. 2024, 10, 001254. [Google Scholar] [CrossRef]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet—Next generation sequence assembly visualization. Bioinformatics 2010, 26, 401–402. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Carver, T.; Berriman, M.; Tivey, A.; Patel, C.; Böhme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.-A. Artemis and ACT: Viewing, Annotating and Comparing Sequences Stored in a Relational Database. Bioinformatics 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) Version 1.3.1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Total cpDNA Size (bp) | Length of LSC Region (bp) | Length of IR Region (bp) | Length of SSC Region (bp) | Total GC Content (%) |
|---|---|---|---|---|---|
| Acmispon americanus | 151,679 | 83,078 | 25,214 | 18,173 | 35.80 |
| Acmispon parviflorus | 151,771 | 83,174 | 25,213 | 18,171 | 35.80 |
| Acmispon glaber ssp. glaber | 152,206 | 83,583 | 25,213 | 18,197 | 35.80 |
| Ornithopus perpusillus | 151,113 | 83,014 | 24,961 | 18,177 | 36.02 |
| Lotus graecus | 151,118 | 82,634 | 25,122 | 18,240 | 36.04 |
| Lotus palustris | 151,524 | 83,011 | 25,160 | 18,193 | 35.95 |
| Lotus dorycnium ssp. lagunae | 150,685 | 82,201 | 25,117 | 18,250 | 36.02 |
| Lotus dorycnium ssp. herbaceus | 151,071 | 82,558 | 25,132 | 18,249 | 36.01 |
| Lotus hirsutus | 151,144 | 82,573 | 25,147 | 18,277 | 36.00 |
| Lotus tetragonolobus | 150,069 | 81,498 | 25,129 | 18,313 | 36.21 |
| Lotus conjugatus ssp. requienii | 151,097 | 82,628 | 25,139 | 18,191 | 36.11 |
| Lotus ornithopodioides | 151,201 | 82,650 | 25,145 | 18,261 | 36.04 |
| Anthyllis vulneraria | 151,868 | 83,473 | 25,083 | 18,229 | 35.94 |
| Hippocrepis biflora | 150,237 | 81,319 | 25,314 | 18,290 | 36.23 |
| Hippocrepis ciliata | 150,862 | 81,908 | 25,317 | 18,320 | 36.18 |
| Hippocrepis emerus | 151,130 | 82,170 | 25,306 | 18,348 | 36.04 |
| Robinia pseudoacacia | 155,301 | 85,370 | 25,668 | 18,595 | 35.90 |
| Dispersed Repeats Number | Direct/Inverted Repeat Length, bp | Relative Length, % of Plastome Length | ||
|---|---|---|---|---|
| Direct | Inverted | |||
| Acmispon americanus | 4 | 1 | 629/39 | 0.88 |
| Acmispon glaber ssp. glaber | 6 | 2 | 706/77 | 1.02 |
| Acmispon parviflorus | 5 | 4 | 675/140 | 1.08 |
| Anthyllis vulneraria | 5 | 3 | 622/111 | 0.96 |
| Hippocrepis biflora | 6 | 2 | 744/79 | 1.08 |
| Hippocrepis ciliata | 5 | 3 | 698/109 | 1.06 |
| Hippocrepis emerus | 5 | 3 | 721/111 | 1.10 |
| Ornithopus perpusillus | 7 | 2 | 737/77 | 1.06 |
| Lotus conjugatus ssp. requienii | 5 | 2 | 652/77 | 0.96 |
| Lotus dorycnium ssp. lagunae | 5 | 2 | 654/77 | 0.96 |
| Lotus graecus | 5 | 2 | 654/77 | 0.96 |
| Lotus dorycnium ssp. herbaceus | 6 | 3 | 684/107 | 1.04 |
| Lotus hirsutus | 5 | 3 | 678/107 | 1.04 |
| Lotus ornithopodioides | 5 | 3 | 678/107 | 1.04 |
| Lotus palustris | 5 | 3 | 654/107 | 1.00 |
| Lotus tetragonolobus | 6 | 2 | 688/77 | 1.02 |
| Robinia pseudoacacia | 5 | 0 | 714/0 | 1.00 |
| Taxon Name | Seed or Herbarium Collection Locality | Coordinates | Herbarium Voucher | GenBank Accession Number |
|---|---|---|---|---|
| Acmispon americanus (Nutt.) Rydb. | Cult. in the greenhouse of the MSU, Moscow. Origin: USA, El Cerrito, California, Z. Akulova-Barlow | 37.904 N, 122.303 W | MW | PQ539379 |
| Acmispon glaber (Vogel) Brouillet ssp. glaber | Cult. in the greenhouse of the MSU, Moscow. Origin: USA, Near Briones Reservoir, Contra Costa County, California, Z.Akulova-Barlow | 37.918 N, 122.203 W | MW | PQ539367 |
| Acmispon parviflorus (Benth.) D.D.Sokoloff | Cult. in the greenhouse of the MSU, Moscow. Origin: USA, Near Albion, Mendocino County, California, Z.Akulova-Barlow | 39.205 N, 123.701 W | MW | PQ539368 |
| Hippocrepis biflora Spreng. | Crimea: Sapun-gora hill, Sevastopol, Yu. Kopylov-Guskov, 2023 | 44.559 N, 33.544 E | MW | PQ539370 |
| Hippocrepis ciliata Willd. | Crimea: southern outskirts of Balaklava town, Yu. Kopylov-Guskov, 2023 | 44.492 N, 33.608 E | MW | PQ539371 |
| Lotus graecus L. | Cult. in the greenhouse of the MSU, Moscow. Origin: Crimea: Vinogradnoye, mount Castell, T. Kramina & S. Polevova, 2020 | 44.642 N, 34.384 E | MW | PQ539373 |
| Lotus dorycnium ssp. herbaceus (Vill.) Kramina & D.D. Sokoloff | Cult. in the greenhouse of the MSU, Moscow. Origin: Crimea: Ayu-Dag, T. Kramina & S. Polevova, 2020 | 44.566 N, 34.323 E | MW | PQ539374 |
| L. dorycnium L. ssp. lagunae (Ceresuela & Sanchis) P. P. Ferrer & Rossellу | Cult. in the greenhouse of the MSU, Moscow. Origin: Spain, Alicante, Finestrat | 38.565 N, 0.212 W | MW | PQ539369 |
| Lotus hirsutus L. | Cult. in the greenhouse of the MSU, Moscow. Origin: Spain, Valencia, The Botanical Garden of the University of Valencia. 2019. ES-0-VAL-476-97 | – | MW | PQ539375 |
| Lotus palustris Willd. | Cult. in the greenhouse of the MSU, Moscow. Origin: Mediterranean Region. Cultivated in Argentina, A.M.Arambarri | – | MW | PQ539377 |
| L. tetragonolobus L. | Cult. in the greenhouse of the MSU, Moscow. Origin: Mainz Bot. Garden, 388, 143-2020 | – | MW | PQ539378 |
| L. conjugatus L. ssp. requienii (Mauri ex Sanguin.) Greuter | Cult. in the greenhouse of the MSU, Moscow. Origin: Mainz Bot. Garden, 386, 143-2020 | – | MW | PQ539372 |
| L. ornithopodioides L. | Cult. in the greenhouse of the MSU, Moscow. Origin: Mainz Bot. Garden, 387, 143-2020 | – | MW | PQ539376 |
| Robinia pseudoacacia L. | Cult. in the MSU, Moscow, Russia. Origin: Eastern N. America. Collected in 2006 | – | MW | PQ636879 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samigullin, T.H.; Kopylov-Guskov, Y.O.; Nikitina, O.V.; Krinitsina, A.A.; Polevova, S.V.; Kramina, T.E. Sixteen New Complete Plastid Genomes in the Tribe Loteae (Leguminosae): Structure and Phylogenetic Analysis. Plants 2025, 14, 618. https://doi.org/10.3390/plants14040618
Samigullin TH, Kopylov-Guskov YO, Nikitina OV, Krinitsina AA, Polevova SV, Kramina TE. Sixteen New Complete Plastid Genomes in the Tribe Loteae (Leguminosae): Structure and Phylogenetic Analysis. Plants. 2025; 14(4):618. https://doi.org/10.3390/plants14040618
Chicago/Turabian StyleSamigullin, Tahir H., Yury O. Kopylov-Guskov, Olga V. Nikitina, Anastasiya A. Krinitsina, Svetlana V. Polevova, and Tatiana E. Kramina. 2025. "Sixteen New Complete Plastid Genomes in the Tribe Loteae (Leguminosae): Structure and Phylogenetic Analysis" Plants 14, no. 4: 618. https://doi.org/10.3390/plants14040618
APA StyleSamigullin, T. H., Kopylov-Guskov, Y. O., Nikitina, O. V., Krinitsina, A. A., Polevova, S. V., & Kramina, T. E. (2025). Sixteen New Complete Plastid Genomes in the Tribe Loteae (Leguminosae): Structure and Phylogenetic Analysis. Plants, 14(4), 618. https://doi.org/10.3390/plants14040618