
Adaptive Defense Mechanism During Flowering Period of Rhododendron decorum Revealed by Comparative Transcriptomic Analysis


Abstract
1. Introduction
2. Results
2.1. Sequencing and Transcription Data Statistics
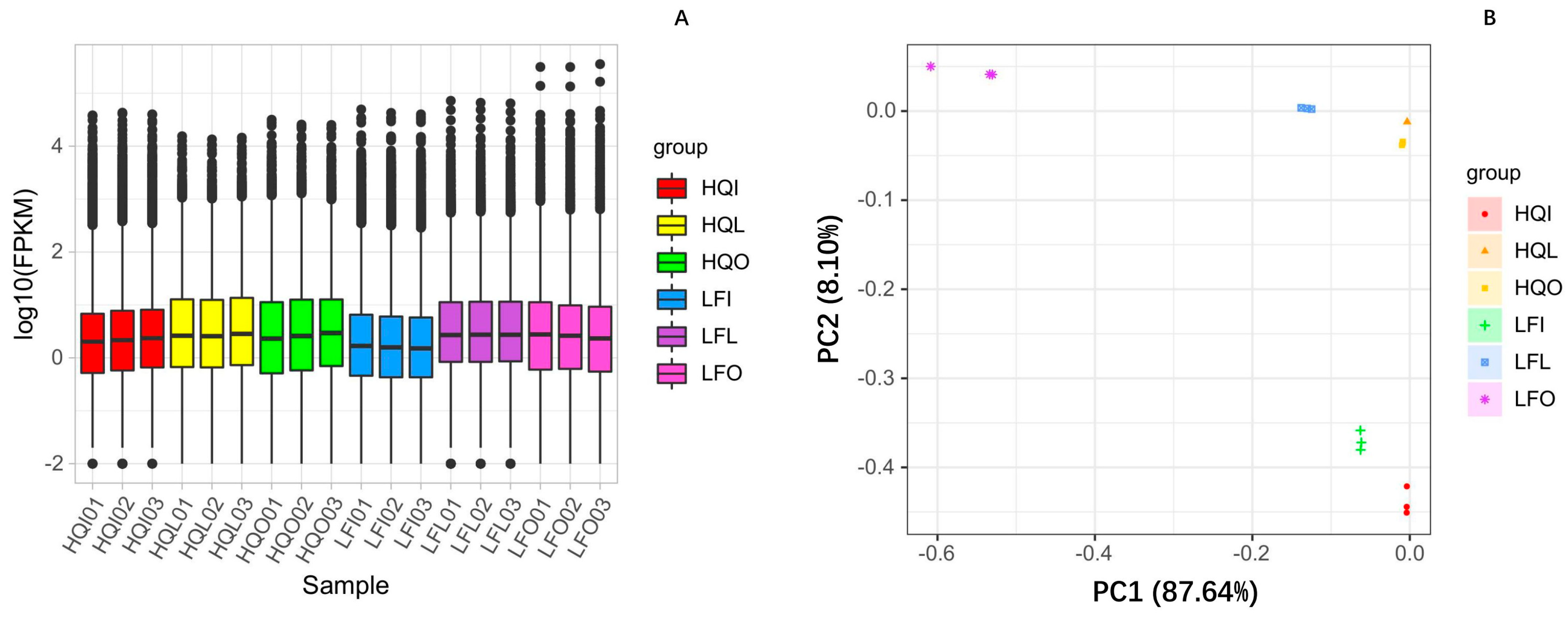
2.2. Gene Functional Annotation and Expression Analysis
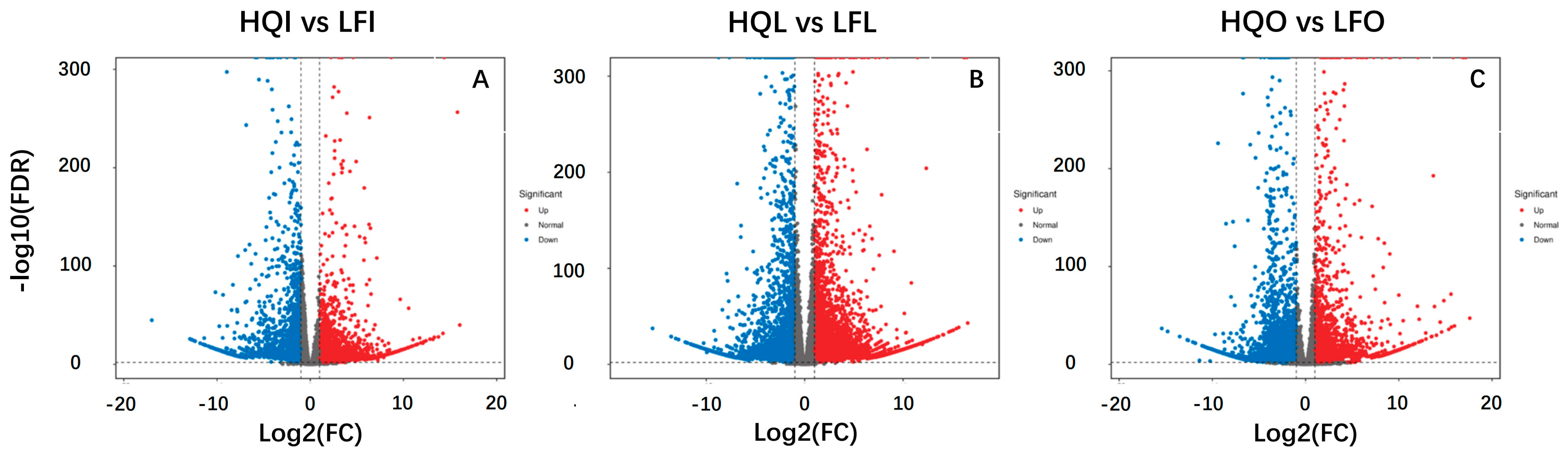
2.3. The Differences in Transcriptome Gene Expression
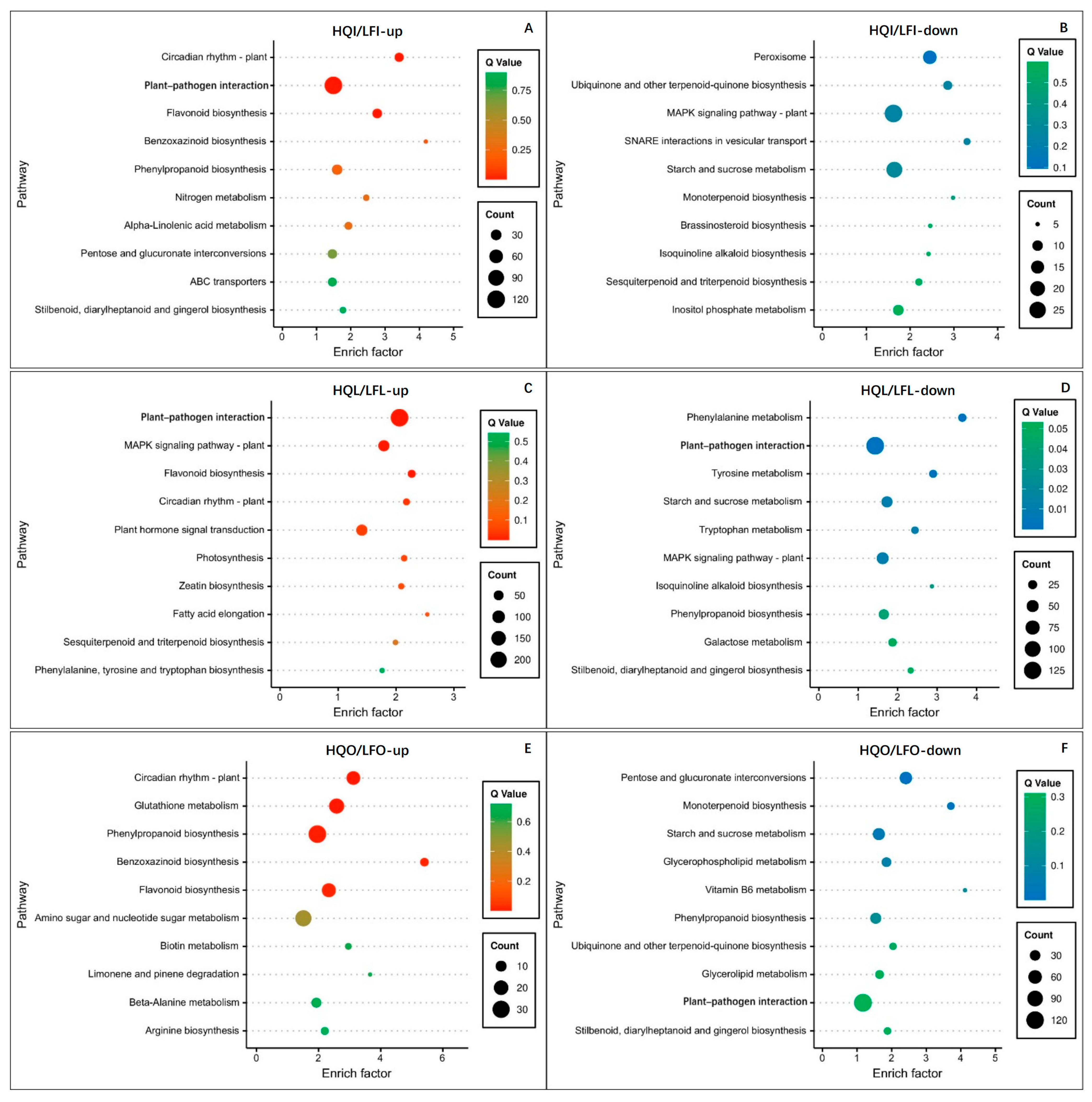
2.4. Enrichment Analysis of Differentially Expressed Transcriptome Genes
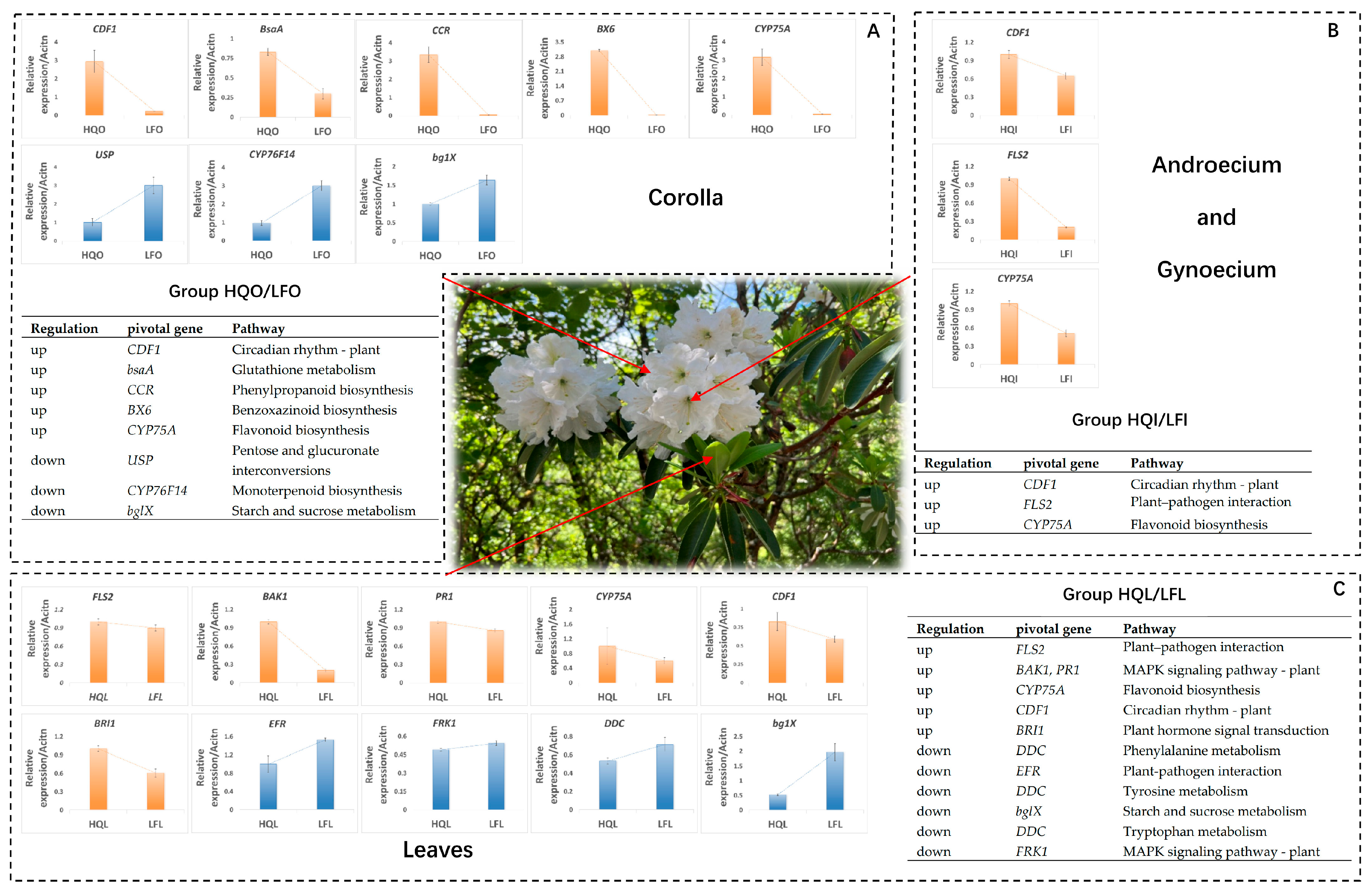
2.5. Identification and Validation of Pivotal Genes in Differentially Enriched Pathways
3. Discussion
3.1. The Transcriptional Characteristics of Leaves Exhibit Regional Variations
3.2. Transcriptional Variations Among Floral Organs from Distinct Regions Elucidate the Underlying Factors Contributing to Adaptive Defense
4. Materials and Methods
4.1. Sample Collection and RNA Extraction
4.2. cDNA Library Construction and Transcriptome Sequencing
4.3. Transcriptome Assembly and Annotation
4.4. Functional Annotation and Differential Expression Analysis
4.5. Validation of Pivotal Gene Expression by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HQ | Heqing County, Dali Bai Autonomous Prefecture, Yunnan Province |
| LF | Yulong Naxi Autonomous County, Lijiang City, Yunnan Province |
| CDSs | Coding sequences |
| COG | The database of Clusters of Orthologous Groups of proteins |
| GO | Gene Ontology database |
| KEGG | The database of Kyoto Encyclopedia of Genes and Genomes |
| KOG | The database of Clusters of Protein homology |
| Pfam | The database of Homologous protein family |
| Swissprot | A manually annotated, non-redundant protein sequence database |
| TrEMBL | Translation of EMBL Nucleotide Sequence Database |
| eggNOG | A database of orthologous groups of genes |
| nr | Non-redundant protein sequence database |
| FPKM | Fragments Per Kilobase of exon model per Million mapped fragments |
| PCA | Principal component analysis |
| HQI | Androecium/gynoecium group in Heqing County |
| LFI | Androecium/gynoecium group in Yulong Naxi Autonomous County |
| HQL | Leaf group in Heqing County |
| LFL | Leaf group in Yulong Naxi Autonomous County |
| HQO | Corolla group in Heqing County |
| LFO | Corolla group in Yulong Naxi Autonomous County |
| DEGs | Differentially expressed genes |
| MAPK | Mitogen-activated protein kinase |
| SNARE | Soluble N-ethylmaleimide-sensitive factor attachment protein receptor |
| CO | A circadian clock-regulated gene encoding a transcription factor |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
References
- Mo, Z.Q.; Fu, C.N.; Zhu, M.S.; Milne, R.; Yang, J.B.; Cai, J.; Qin, H.T.; Zheng, W.; Hollingsworth, P.M.; Li, D.Z.; et al. Resolution, conflict and rate shifts: Insights from a densely sampled plastome phylogeny for Rhododendron (Ericaceae). Ann. Bot. 2022, 130, 687–701. [Google Scholar] [CrossRef]
- Shi, Y.X.; Zhou, M.; Zhang, Y.; Fu, Y.; Li, J.W.; Yang, X.F. Poisonous delicacy: Market-oriented surveys of the consumption of Rhododendron flowers in Yunnan, China. J. Ethnopharmacol. 2021, 265, 113320. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.J.; Möller, M.; Luo, Y.H.; Zou, J.Y.; Zheng, W.; Wang, Y.H.; Liu, J.; Zhu, A.D.; Hu, J.Y.; Li, D.Z.; et al. Differential expressions of anthocyanin synthesis genes underlie flower color divergence in a sympatric Rhododendron sanguineum complex. BMC Plant Biol. 2021, 21, 204. [Google Scholar] [CrossRef] [PubMed]
- Vengryte, M.; Raudone, L. Phytochemical profiling and biological activities of Rhododendron subsect. Ledum: Discovering the medicinal potential of labrador tea species in the northern hemisphere. Plants 2024, 13, 901. [Google Scholar] [CrossRef]
- Jha, A.K.; Khalid, M.A.; Labh, S.N. In vitro antioxidant and antibacterial activities of medicinal flower laligurans Rhododendron arboreum collected from Kathmandu valley, Nepal. Int. J. Food Sci. 2024, 2024, 6073042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Xu, Y.; Sun, D.J.; Li, Y.X.; Li, H.; Chen, L.X. Chromene meroterpenoids from Rhododendron dauricum L. and their anti-inflammatory effects. Phytochemistry 2024, 225, 114200. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Li, Y.; Huang, X.; Li, S.; Sangji, K.Z.; Gu, R. Traditional Tibetan medicine: Therapeutic potential in lung diseases. Front. Pharmacol. 2024, 15, 1365911. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guo, W.J.; Zhang, B.; Xu, H.X.; Yang, Q.Y.; Zhao, J.Y.; Feng, Y.; Yang, J.; Zhang, J.Q. Evaluation of Rhododendri Mollis Flos and its representative component as a potential analgesic. J. Nat. Med. 2024, 78, 753–767. [Google Scholar] [CrossRef]
- Guo, X.H.; Wu, W.H.; Ran, Q.; Wang, L.J.; Li, Y.Y.; Chen, J.; Chen, L.; Yang, M.; Geng, Z.; Liu, Y.P. Exploring the pharmacological mechanisms of the flower of Rhododendron molle in rheumatoid arthritis rats based on metabolomics integrated network pharmacology. J. Ethnopharmacol. 2024, 334, 118524. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Ding, R.P.; Zhang, Y.Y.; Liao, Y.Y.; Zhao, J.Y.; Jia, Y.; Tan, M.P.; Zeng, X.X. The phenotypic variation mechanisms of Atractylodes lancea post-cultivation revealed by conjoint analysis of rhizomic transcriptome and metabolome. Plant Physiol. Biochem. 2023, 203, 108025. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.R.; Zhao, W.Q.; Shi, P.Y.; Zhou, M.H.; Liu, Z.Y.; Wang, Y.C. Soil differentiation and soil comprehensive evaluation of in wild and cultivated Fritillaria pallidiflora Schrenk. Sci. Total Environ. 2023, 872, 162049. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.S.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef]
- Liu, W.W.; Wang, L.; Yu, C.H.; Fan, Z.Y.; Yang, K.Y.; Mo, X.C. Drug or Toxic? A brief understanding of the edible corolla of Rhododendron decorum Franch. by Bai nationality with comparative metabolomics analysis. Metabolites 2024, 14, 484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, M.F.; Wang, X.L.; Dai, J.; Zhang, X.; Zhang, Z.D.; Zhang, X.M.; Tang, M.; Tang, J.; Gong, J.Y.; et al. Transcriptome analysis of Rhododendron liliiflorum H. Lev. flower colour differences. Horticulturae 2023, 9, 82. [Google Scholar] [CrossRef]
- Cai, Y.F.; Wang, J.H.; Zhang, L.; Song, J.; Peng, L.C.; Zhang, S.B. Physiological and transcriptomic analysis highlight key metabolic pathways in relation to drought tolerance in Rhododendron delavayi. Physiol. Mol. Biol. Plants 2019, 25, 991–1008. [Google Scholar] [CrossRef]
- Cheng, H.F.; Wan, Z.Y.; Xu, Y.X.; Shen, J.S.; Li, X.Q.; Jin, S.H. Transcriptome and photosynthetic analyses provide new insight into the molecular mechanisms underlying heat stress tolerance in Rhododendron x pulchrum Sweet. Tree Physiol. 2024, 44, tpad133. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.P.; Mao, X.; Chen, Y.; Li, L.; Hu, Y.R.; Zhang, H.H.; Zhang, R.; Wang, Y.K. Transcriptome analysis reveals salt stress-related genes in Rhododendron simii and RsWRKY40 is referred to salt tolerance. Environ. Exp. Bot. 2024, 220, 105678. [Google Scholar] [CrossRef]
- He, J.; Shang, X.F.; Dai, L.X.; Yang, X.R.; Li, B.; Wei, Y.M.; Zhang, J.Y.; Pan, H. Chemical constituents, antibacterial, acaricidal and anti-inflammatory activities of the essential oils from four Rhododendron species. Front. Vet. Sci. 2022, 9, 882060. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; Su, H.G.; Peng, X.R.; Bi, H.C.; Qiu, M.H. An updated review of the genus Rhododendron since 2010: Traditional uses, phytochemistry, and pharmacology. Phytochemistry 2024, 217, 113899. [Google Scholar] [CrossRef] [PubMed]
- Deepak, H.V.; Swamy, M.M.M.; Murai, Y.; Suga, Y.; Anetai, M.; Yo, T.; Kuragano, M.; Uwai, K.; Tokuraku, K.; Monde, K. Daurichromenic Acid from the Chinese traditional medicinal plant Rhododendron dauricum inhibits sphingomyelin synthase and Aβ aggregation. Molecules 2020, 25, 4077. [Google Scholar] [CrossRef]
- Shen, J.S.; Rong, X.L.; Li, X.Q.; Ma, Y.L.; Cheng, H.F.; Sheng, J.R.; Huang, L.; Jin, S.H. Transcriptome and flavonoid compounds metabolome analyses reveal the mechanisms of heat stress in Rhododendron with exogenously applied calcium. Agronomy 2024, 14, 1282. [Google Scholar] [CrossRef]
- Chinchilla, D.; Bauer, Z.; Regenass, M.; Boller, T.; Felix, G. The Arabidopsis receptor kinase FLS2 binds flg22 and determines the specificity of flagellin perception. Plant Cell 2006, 18, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, C.; Kunze, G.; Chinchilla, D.; Caniard, A.; Jones, J.D.G.; Boller, T.; Felix, G. Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts Agrobacterium-mediated transformation. Cell 2006, 125, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wen, J.Q.; Lease, K.A.; Doke, J.T.; Tax, F.E.; Walker, J.C. BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 2002, 110, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Gao, Y.; Zhan, Y.Y.; Kui, H.; Liu, H.Y.; Yan, L.; Kemmerling, B.; Zhou, J.M.; He, K.; Li, J. Loss of the common immune coreceptor BAK1 leads to NLR-dependent cell death. Proc. Natl. Acad. Sci. USA 2020, 117, 27044–27053. [Google Scholar] [CrossRef]
- Han, Z.; Xiong, D.G.; Schneiter, R.; Tian, C.M. The function of plant PR1 and other members of the CAP protein superfamily in plant-pathogen interactions. Mol. Plant Pathol. 2023, 24, 651–668. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, M.X.; Shen, D.Y.; Liu, T.L.; Chen, Y.Y.; Zhou, J.M.; Dou, D.L. A Phytophthora sojae effector PsCRN63 forms homo-/heterodimers to suppress plant immunity via an inverted association manner. Sci. Rep. 2016, 6, 26951. [Google Scholar] [CrossRef]
- de Wit, P.J.G.M. How plants recognize pathogens and defend themselves. Cell. Mol. Life Sci. 2007, 64, 2726–2732. [Google Scholar] [CrossRef]
- Cheng, C.; Gao, X.Q.; Feng, B.M.; Sheen, J.; Shan, L.B.; He, P. Plant immune response to pathogens differs with changing temperatures. Nat. Commun. 2013, 4, 2726–2732. [Google Scholar] [CrossRef]
- Chen, D.V.; Slowinski, S.P.; Kido, A.K.; Bruns, E.L. High temperatures reduce growth, infection, and transmission of a naturally occurring fungal plant pathogen. Ecology 2024, 105, e4373. [Google Scholar] [CrossRef]
- Jia, Y.; Li, B.; Zhang, Y.J.; Zhang, X.Q.; Xu, Y.H.; Li, C.D. Evolutionary dynamic analyses on monocot flavonoid 3′-hydroxylase gene family reveal evidence of plant-environment interaction. BMC Plant Biol. 2019, 19, 347. [Google Scholar] [CrossRef]
- Pan, Y.Y.; Yan, Z.B.; Xue, S.Y.; Xiao, C.F.; Li, G.J.; Lou, W.Y.; Huang, M.T. Optimizing the biosynthesis of dihydroquercetin from naringenin in saccharomyces cerevisiae. J. Agric. Food Chem. 2024, 72, 4880–4887. [Google Scholar] [CrossRef]
- Sunil, C.; Xu, B.J. An insight into the health-promoting effects of taxifolin (dihydroquercetin). Phytochemistry 2019, 166, 112066. [Google Scholar] [CrossRef] [PubMed]
- Stafford, H.A.; Lester, H.H. Flavan-3-ol biosynthesis—The conversion of (+)-dihydroquercetin and flavan-3,4-cis-diol (leucocyanidin) to (+)-catechin by reductases extracted from cell-suspension cultures of douglas-fir. Plant Physiol. 1984, 76, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.W.; Zhang, N.; Jiang, C.Y.; Wu, S.X.; Feng, X.Y.; Feng, X.Y. Exploring the genes involved in biosynthesis of dihydroquercetin and dihydromyricetin in Ampelopsis grossedentata. Sci. Rep. 2021, 11, 15596. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, J.; Shi, B.; Qian, J.; Guo, H. Antihyperlipidemic activity of myricetin. Acta Aliment. Hung. 2024, 53, 292–304. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, Y.W.; Wang, X.; Shi, T.Z.; Lv, P.; Li, Q.X.; Hua, R.M. Myricetin inhibits photodegradation of profenofos in water: Pathways and mechanisms. Agronomy 2024, 14, 399. [Google Scholar] [CrossRef]
- Aksoy, S.; Kuloglu, N.; Karabulut, D.; Yakan, B. Investigation of the effect of myricetin on cisplatin-induced liver hepatotoxicity. Rev. Assoc. Med. Bras. 2024, 70, e20240136. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Q.; Zhang, G.W.; Hu, M.M.; Pan, J.H.; Li, A.; Zhang, Y. Molecular characteristics of gallocatechin gallate affecting protein glycation. Food Hydrocoll. 2020, 105, 782. [Google Scholar] [CrossRef]
- Park, D.; Park, J.Y.; Kang, K.S.; Hwang, G.S. Neuroprotective effect of gallocatechin gallate on glutamate-induced oxidative stress in hippocampal HT22 cells. Molecules 2021, 26, 1387. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Youdim, M.B.H. Catechin polyphenols: Neurodegeneration and neuroprotection in neurodegenerative diseases. Free Radic. Biol. Med. 2004, 37, 304–317. [Google Scholar] [CrossRef]
- Ganeshpurkar, A.; Saluja, A. The pharmacological potential of catechin. Indian J. Biochem. Biophys. 2020, 57, 505–511. [Google Scholar] [CrossRef]
- Yi, B.W.; Chew, B.X.Z.; Chen, H.X.; Lee, R.C.H.; Fong, Y.D.; Chin, W.X.; Mok, C.K.; Chu, J.J.H. Antiviral activity of catechin against dengue virus infection. Viruses 2023, 15, 1377. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Schultz, T.F.; Harmon, F.G.; Ho, L.A.; Kay, S.A. FKF1F-BOX protein mediates cyclic degradation of a repressor of CONSTANS in Arabidopsis. Science 2005, 309, 293–297. [Google Scholar] [CrossRef]
- Valverde, F.; Mouradov, A.; Soppe, W.; Ravenscroft, D.; Coupland, G. Photoreceptor regulation of CONSTANS protein in photoperiodic flowering. Science 2004, 303, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Roden, L.C.; Ingle, R.A. Lights, rhythms, infection: The role of light and the circadian clock in determining the outcome of plant-pathogen interactions. Plant Cell 2009, 21, 2546–2552. [Google Scholar] [CrossRef]
- Hua, J. Modulation of plant immunity by light, circadian rhythm, and temperature. Curr. Opin. Plant Biol. 2013, 16, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Q.; Han, Q.; Zhang, D.W. Recent advances in the crosstalk between brassinosteroids and environmental stimuli. Plant Cell Physiol. 2024, 65, 1552–1567. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, C. Pattern-recognition receptors in plant innate immunity. Curr. Opin. Immunol. 2008, 20, 10–16. [Google Scholar] [CrossRef] [PubMed]
- He, Z.Q.; Zhou, M.D.; Feng, X.J.; Di, Q.H.; Meng, D.; Yu, X.C.; Yan, Y.; Sun, M.T.; Li, Y.S. The role of brassinosteroids in plant cold stress response. Life 2024, 14, 1015. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, T.; Nakagawa, A.; Sakurama, H.; Yamamoto, K.; Sakurai, N.; Takagi, Y.; Minami, H.; Katayama, T.; Kumagai, H. Eukaryotic-type aromatic amino acid decarboxylase from the root colonizer Pseudomonas putida is highly specific for 3,4-dihydroxyphenyl-L-alanine, an allelochemical in the rhizosphere. Microbiology 2012, 158, 2965–2974. [Google Scholar] [CrossRef]
- Du, T.T.; Qu, X.D.; Wang, Y.B.; Li, M.X.; Qie, X.H.; Jin, J.; Gao, Y.X.; Wang, Z.Y.; Lin, K.J.; Yang, C.; et al. Rhizosphere Mortierella strain of alfalfa exerted weed growth inhibition by inducing expression of plant hormone-related genes. Front. Microbiol. 2024, 15, 1385992. [Google Scholar] [CrossRef]
- Duan, B.; Zhang, Y.H.; Feng, Z.; Liu, Z.G.; Tao, N.G. Octanal enhances disease resistance in postharvest citrus fruit by the biosynthesis and metabolism of aromatic amino acids. Pestic. Biochem. Phys. 2024, 200, 105835. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhao, Y.S.; Wei, S.C.; Yu, X.J. Isolation of allelochemicals from Rhododendron capitatum and their allelopathy on three perennial herbaceous plants. Plants 2024, 13, 2585. [Google Scholar] [CrossRef]
- Shafi, A.; Pal, A.K.; Sharma, V.; Kalia, S.; Kumar, S.; Ahuja, P.S.; Singh, A.K. Transgenic potato plants overexpressing SOD and APX exhibit enhanced lignification and starch biosynthesis with improved salt stress tolerance. Plant Mol. Biol. Rep. 2017, 35, 504–518. [Google Scholar] [CrossRef]
- Singh, T.; Sandhu, P.S.; Chahal, G.K.; Walia, S.S. Foliar thiourea confers moisture stress tolerance in rainfed maize through elevated antioxidative defence system, osmolyte accumulation and starch synthesis grown under different planting methods. J. Plant Growth Regul. 2023, 42, 199–217. [Google Scholar] [CrossRef]
- Yano, R.; Nakamura, M.; Yoneyama, T.; Nishida, I. Starch-related α-glucan/water dikinase is involved in the cold-induced development of freezing tolerance in Arabidopsis. Plant Physiol. 2005, 138, 837–846. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R. Glutathione peroxidases and redox-regulated transcription factors. Biol. Chem. 2006, 387, 1329–1335. [Google Scholar] [CrossRef]
- Szalai, G.; Kellös, T.; Galiba, G.; Kocsy, G. Glutathione as an antioxidant and regulatory molecule in plants under abiotic stress conditions. J. Plant Growth Regul. 2009, 28, 66–80. [Google Scholar] [CrossRef]
- Popescu, R.; Kopp, B. The genus Rhododendron: An ethnopharmacological and toxicological review. J. Ethnopharmacol. 2013, 147, 42–62. [Google Scholar] [CrossRef] [PubMed]
- Lauvergeat, V.; Lacomme, C.; Lacombe, E.; Lasserre, E.; Roby, D.; Grima-Pettenati, J. Two cinnamoyl-CoA reductase (CCR) genes from Arabidopsis thaliana are differentially expressed during development and in response to infection with pathogenic bacteria. Phytochemistry 2001, 57, 1187–1195. [Google Scholar] [CrossRef]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef] [PubMed]
- Chen, O.; Deng, L.L.; Ruan, C.Q.; Yi, L.H.; Zeng, K.F. Pichia galeiformis induces resistance in postharvest Citrus by activating the phenylpropanoid biosynthesis pathway. J. Agric. Food Chem. 2021, 69, 2619–2631. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, W.B.; Liu, J.C.; Liu, H.; Lv, Z.Z.; Zhang, C.L.; Chen, D.L.; Jiao, Z.G. Postharvest UV-C irradiation increased the flavonoids and anthocyanins accumulation, phenylpropanoid pathway gene expression, and antioxidant activity in sweet cherries (Prunus avium L.). Postharvest Biol. Technol. 2021, 175, 111490. [Google Scholar] [CrossRef]
- Golawska, S.; Lukasik, I.; Chojnacki, A.A. Luteolin and quercetin affect aphid feeding behavior. Eur. Zool. J. 2024, 91, 318–331. [Google Scholar] [CrossRef]
- Shi, X.Q.; Wang, Y.Z.; Gong, S.; Liu, X.L.; Tang, M.; Tang, J.; Sun, W.; Yi, Y.; Gong, J.Y.; Zhang, X.M. The preliminary analysis of flavonoids in the petals of Rhododendron delavayi, Rhododendron agastum and Rhododendron irroratum infected with Neopestalotiopsis clavispora. Int. J. Mol. Sci. 2024, 25, 9605. [Google Scholar] [CrossRef]
- He, B.; Zhou, Y.; Peng, Y.; Xu, D.Y.; Tong, J.; Dong, Y.F.; Fang, L.C.; Mao, J. Comparative metabolomic responses of three rhododendron cultivars to the azalea lace bug (Stephanitis pyrioides). Plants 2024, 13, 2569. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.L.; Wang, P.; Pan, Z.X.; Tu, X.J.; Dai, L.; Xin, Y.Q.; Hao, Y.J.; Chen, P.Q. Repellence and insecticidal activity of Rhododendron anthopogonoides EO and head transcriptome analysis. Arthropod-Plant Interact. 2024, 18, 501–517. [Google Scholar] [CrossRef]
- Jonczyk, R.; Schmidt, H.; Osterrieder, A.; Fiesselmann, A.; Schullehner, K.; Haslbeck, M.; Sicker, D.; Hofmann, D.; Yalpani, N.; Simmons, C.; et al. Elucidation of the final reactions of DIMBOA-glucoside biosynthesis in maize: Characterization of Bx6 and Bx7. Plant Physiol. 2008, 146, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.; Schullehner, K.; Dick, R.; Fiesselmann, A.; Gierl, A. Benzoxazinoid biosynthesis, a model for evolution of secondary metabolic pathways in plants. Phytochemistry 2009, 70, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.Q.; Richter, A.; Jander, G. Beyond defense: Multiple functions of benzoxazinoids in maize metabolism. Plant Cell Physiol. 2018, 59, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Glauser, G.; Marti, G.; Villard, N.; Doyen, G.A.; Wolfender, J.L.; Turlings, T.C.J.; Erb, M. Induction and detoxification of maize 1,4-benzoxazin-3-ones by insect herbivores. Plant J. 2011, 68, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.Y.; Wang, J.; Zhou, F.J.; Li, Z.X.; Qi, Y.Y.; Hu, T.T. The complete chloroplast genome of Rhododendron williamsianum (ericaceae). Mitochondrial DNA B 2024, 9, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Kleczkowski, L.A.; Decker, D.; Wilczynska, M. UDP-Sugar pyrophosphorylase: A new old mechanism for sugar activation. Plant Physiol. 2011, 156, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Kotake, T.; Yamaguchi, D.; Ohzono, H.; Hojo, S.; Kaneko, S.; Ishida, H.K.; Tsumuraya, Y. UDP-sugar pyrophosphorylase with broad substrate specificity toward various monosaccharide 1-phosphates from pea sprouts. J. Biol. Chem. 2004, 279, 45728–45736. [Google Scholar] [CrossRef]
- Elsharif, S.A.; Banerjee, A.; Buettner, A. Structure-odor relationships of linalool, linalyl acetate and their corresponding oxygenated derivatives. Front. Chem. 2015, 3, 57. [Google Scholar] [CrossRef]
- Qian, C.Y.; Quan, W.X.; Li, C.C.; Xiang, Z.M. Analysis of volatile terpenoid compounds in Rhododendron species by multidimensional gas chromatography with quadrupole time-of-flight mass spectrometry. Microchem. J. 2019, 149, 104064. [Google Scholar] [CrossRef]
- Qian, C.Y.; Quan, W.X.; Xiang, Z.M.; Li, C.C. Characterization of volatile compounds in four different rhododendron flowers by GC×GC-QTOFMS. Molecules 2019, 24, 3327. [Google Scholar] [CrossRef]
- Zhou, Y.; He, W.; He, Y.C.; Chen, Q.L.; Gao, Y.; Geng, J.M.; Zhu, Z.R. Formation of 8-hydroxylinalool in tea plant Camellia sinensis var. Assamica ‘Hainan dayezhong’. Food Chem. Mol. Sci. 2023, 6, 100173. [Google Scholar] [CrossRef]
- Kreck, M.; Püschel, S.; Wüst, M.; Mosandl, A. Biogenetic studies in Syringa vulgaris L.: Synthesis and bioconversion of deuterium-labeled precursors into lilac aldehydes and lilac alcohols. J. Agric. Food Chem. 2023, 51, 463–469. [Google Scholar] [CrossRef]
- Peng, B.; Ran, J.G.; Li, Y.Y.; Tang, M.L.; Xiao, H.L.; Shi, S.P.; Ning, Y.Z.; Dark, A.; Li, J.; Guan, X.Q.; et al. Site-directed mutagenesis of VvCYP76F14 (cytochrome P450) unveils its potential for selection in wine grape varieties linked to the development of wine bouquet. J. Agric. Food Chem. 2024, 72, 3683–3694. [Google Scholar] [CrossRef]
- Ongalbek, D.; Sahin, B.; Berdesh, T.; Tas-Küçükaydin, M.; Tokul-Ölmez, O.; Yeskaliyeva, B.; Öztürk, M. Biomarker aroma compounds of monofloral honey from Kazakhstan by gas chromatography-mass spectrometry (GC-MS) and chemometric analysis. Anal. Lett. 2024, 58, 1–18. [Google Scholar] [CrossRef]
- Hosseini, A.; Pourheidar, E.; Rajabian, A.; Asadpour, E.; Hosseinzadeh, H.; Sadeghnia, H.R. Linalool attenuated ischemic injury in PC12 cells through inhibition of caspase-3 and caspase-9 during apoptosis. Food Sci. Nutr. 2022, 11, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Schiestl, F.P.; Glaser, F. Specific ant-pollination in an alpine orchid and the role of floral scent in attracting pollinating ants. Alpine Bot. 2012, 122, 1–9. [Google Scholar] [CrossRef]
- Tao, Z.B.; Ren, Z.X.; Bernhardt, P.; Wang, W.J.; Liang, H.; Li, H.D.; Wang, H. Nocturnal hawkmoth and noctuid moth pollination of Habenaria limprichtii (Orchidaceae) in sub-alpine meadows of the Yulong Snow Mountain (Yunnan, China). Bot. J. Linn. Soc. 2018, 187, 483–498. [Google Scholar] [CrossRef]
- Shi, C.Y.; Xie, Y.J.; Guan, D.L.; Qin, G.L. Transcriptomic analysis reveals adaptive evolution and conservation implications for the endangered Magnolia lotungensis. Genes 2024, 15, 787. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Fulllength transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Boratyn, G.M.; Joukov, V.; Vera Alvarez, R.; Madden, T.L. ElasticBLAST: Accelerating sequence search via cloud computing. BMC Bioinform. 2023, 24, 117. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2015, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2004, 32, D138–D141. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.L.; Jiang, P.; Chen, X.Y.; Fan, P.X.; Wang, X.C.; Li, Y.X. Multiple compartmentalization of sodium conferred salt tolerance in Salicornia europaea. Plant Physiol. Biochem. 2012, 51, 47–52. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotated Database | Annotated Number | Percentage of Annotated Genes (%) |
|---|---|---|
| COG | 8369 | 9.09 |
| GO | 27,221 | 29.58 |
| KEGG | 23,369 | 25.39 |
| KOG | 19,916 | 21.64 |
| Pfam | 25,018 | 27.19 |
| Swissprot | 23,521 | 25.56 |
| TrEMBL | 40,563 | 44.08 |
| eggNOG | 30,146 | 32.76 |
| nr | 40,574 | 44.09 |
| All Annotated | 43,515 | 47.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Yu, C.; Yang, K.; Wang, L.; Gao, L.; Mo, X. Adaptive Defense Mechanism During Flowering Period of Rhododendron decorum Revealed by Comparative Transcriptomic Analysis. Plants 2025, 14, 559. https://doi.org/10.3390/plants14040559
Liu W, Yu C, Yang K, Wang L, Gao L, Mo X. Adaptive Defense Mechanism During Flowering Period of Rhododendron decorum Revealed by Comparative Transcriptomic Analysis. Plants. 2025; 14(4):559. https://doi.org/10.3390/plants14040559
Chicago/Turabian StyleLiu, Weiwei, Chenghua Yu, Kaiye Yang, Ling Wang, Lianming Gao, and Xinchun Mo. 2025. "Adaptive Defense Mechanism During Flowering Period of Rhododendron decorum Revealed by Comparative Transcriptomic Analysis" Plants 14, no. 4: 559. https://doi.org/10.3390/plants14040559
APA StyleLiu, W., Yu, C., Yang, K., Wang, L., Gao, L., & Mo, X. (2025). Adaptive Defense Mechanism During Flowering Period of Rhododendron decorum Revealed by Comparative Transcriptomic Analysis. Plants, 14(4), 559. https://doi.org/10.3390/plants14040559