
Transcriptomic Response of White Lupin Roots to Short-Term Sucrose Treatment


 , ,
, ,
Abstract

1. Introduction
2. Results
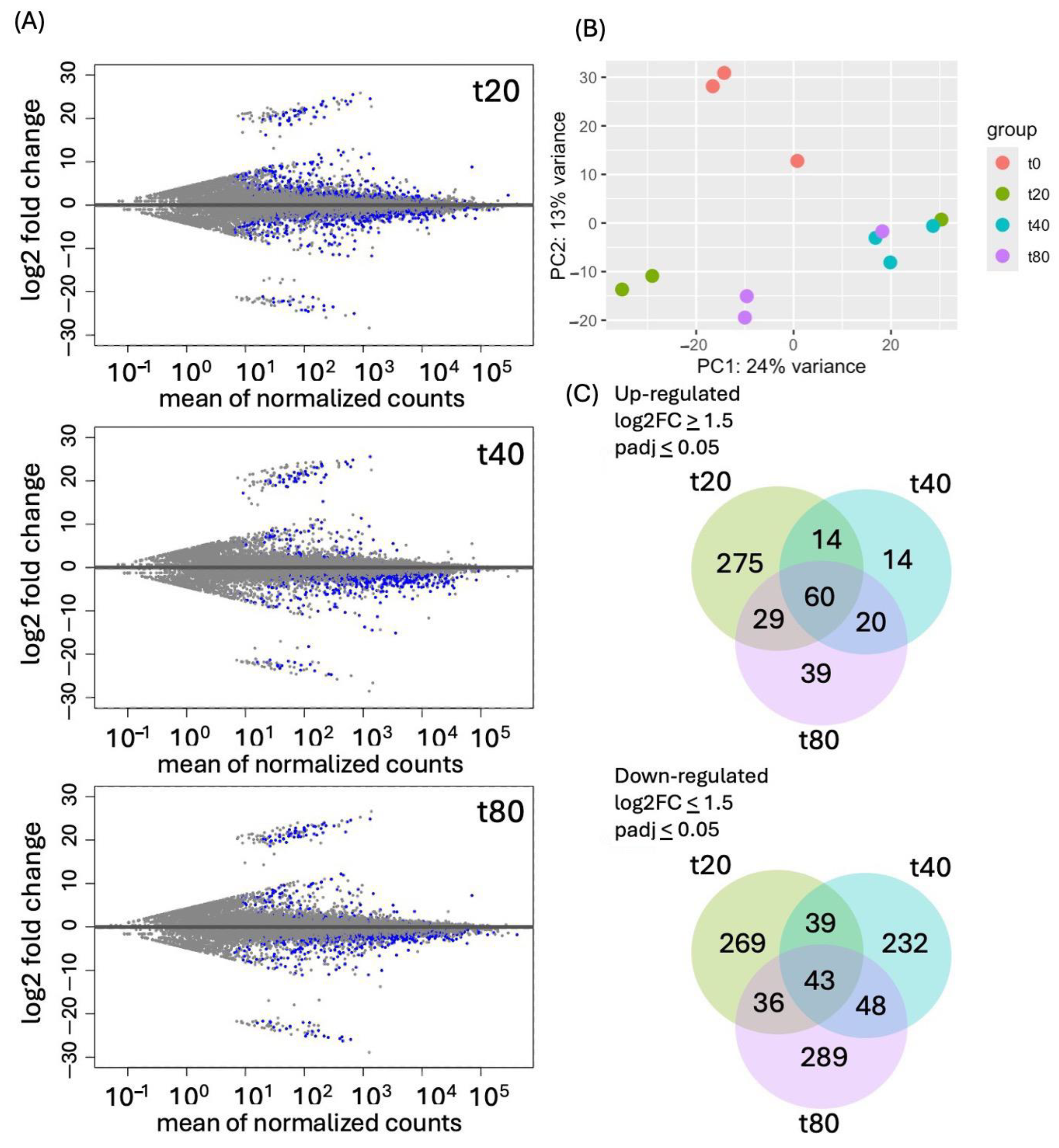
2.1. RNA-Seq Resulted in 382 Million Paired-End Reads
2.2. Short-Term (20 min) Sucrose Exposure Changed Expression of More Genes than Longer Exposure (40 and 80 min)
2.3. Several Hormone-Responsive Genes and Transcription Factors Are Expressed at 20 min of Sucrose Treatment
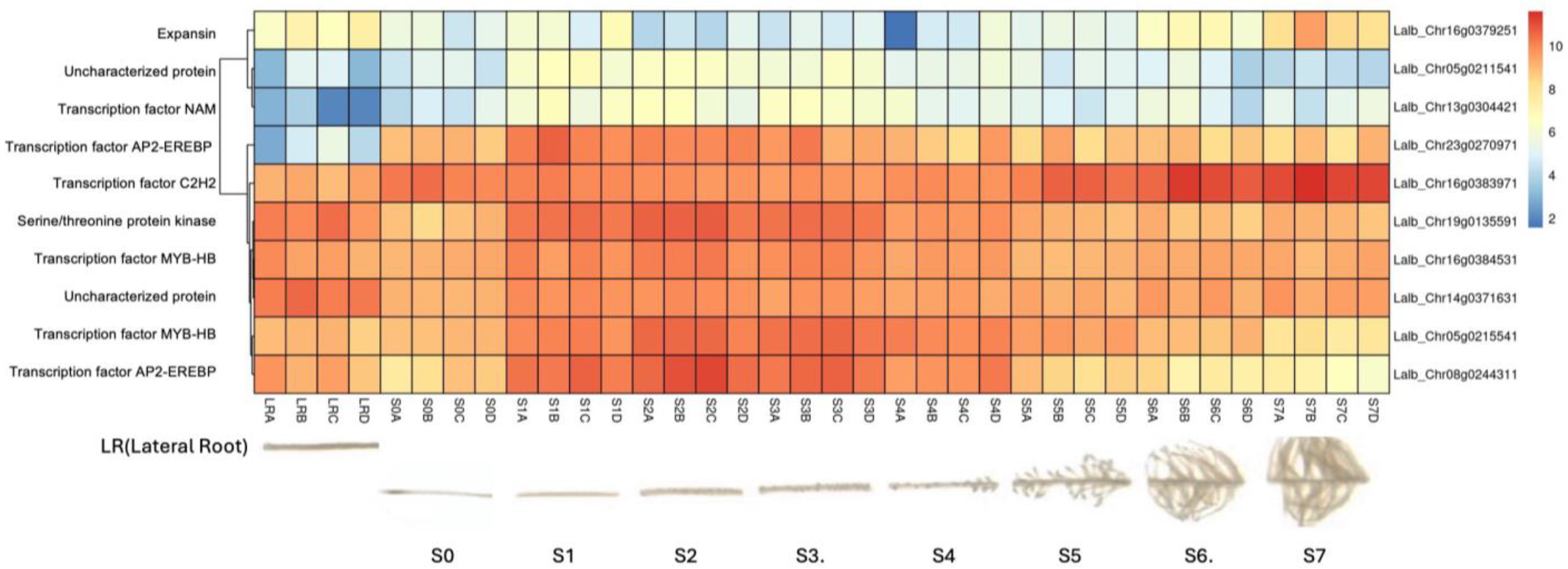
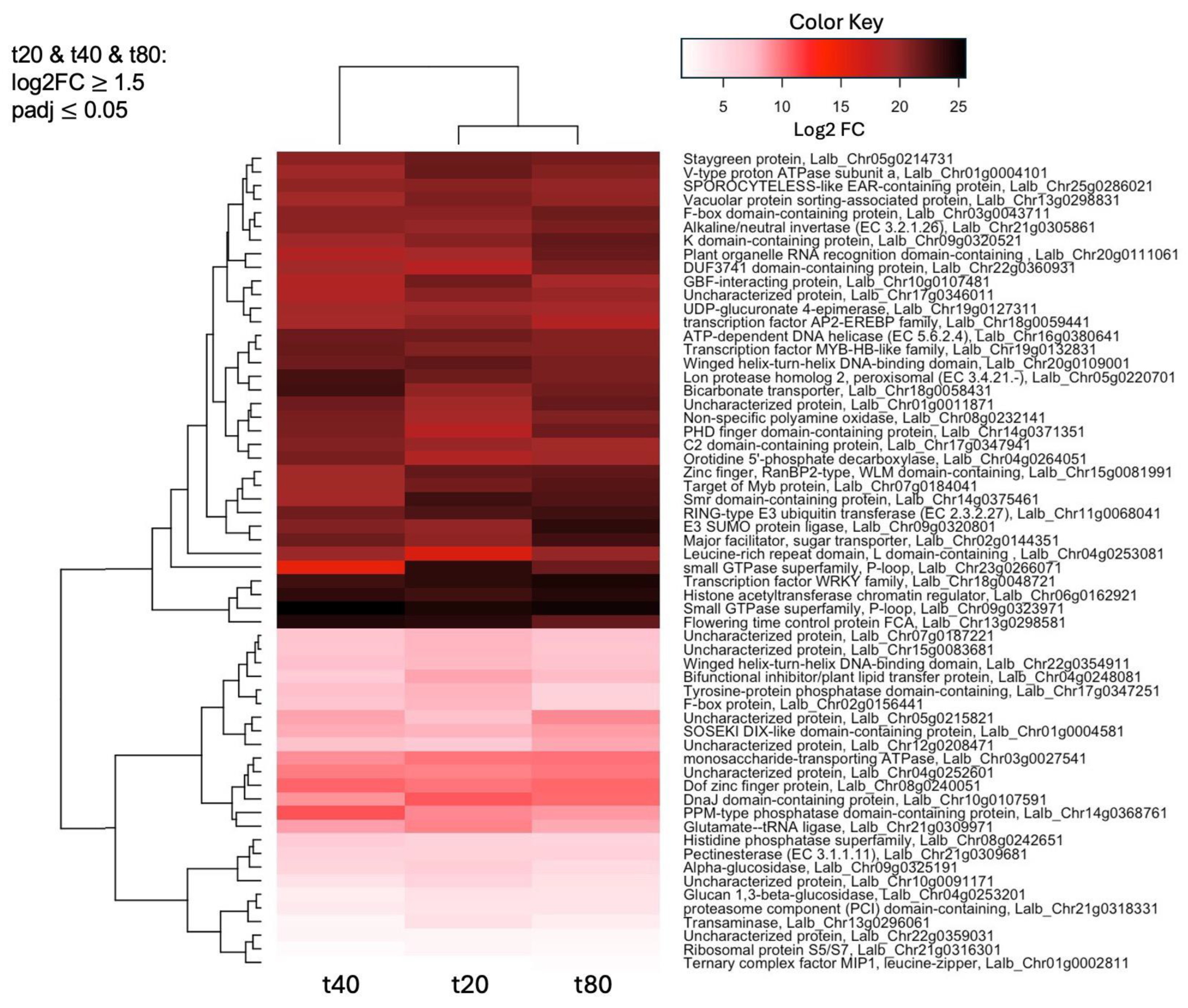
2.4. Small GTPases and a WRKY Transcription Factor Are Among the Most Up-Regulated Genes in Response to Sucrose at All Three Timepoints
2.5. Gene Ontology (GO) Analysis Reveals Early Enrichment of Ethylene-Activated Signaling
2.6. Promoter Analysis Reveals Enrichment of Putative Transcription Factor-Binding Motifs
3. Discussion
3.1. Reference Genome 1 Is a Good Option for Mapping White Lupin Transcripts
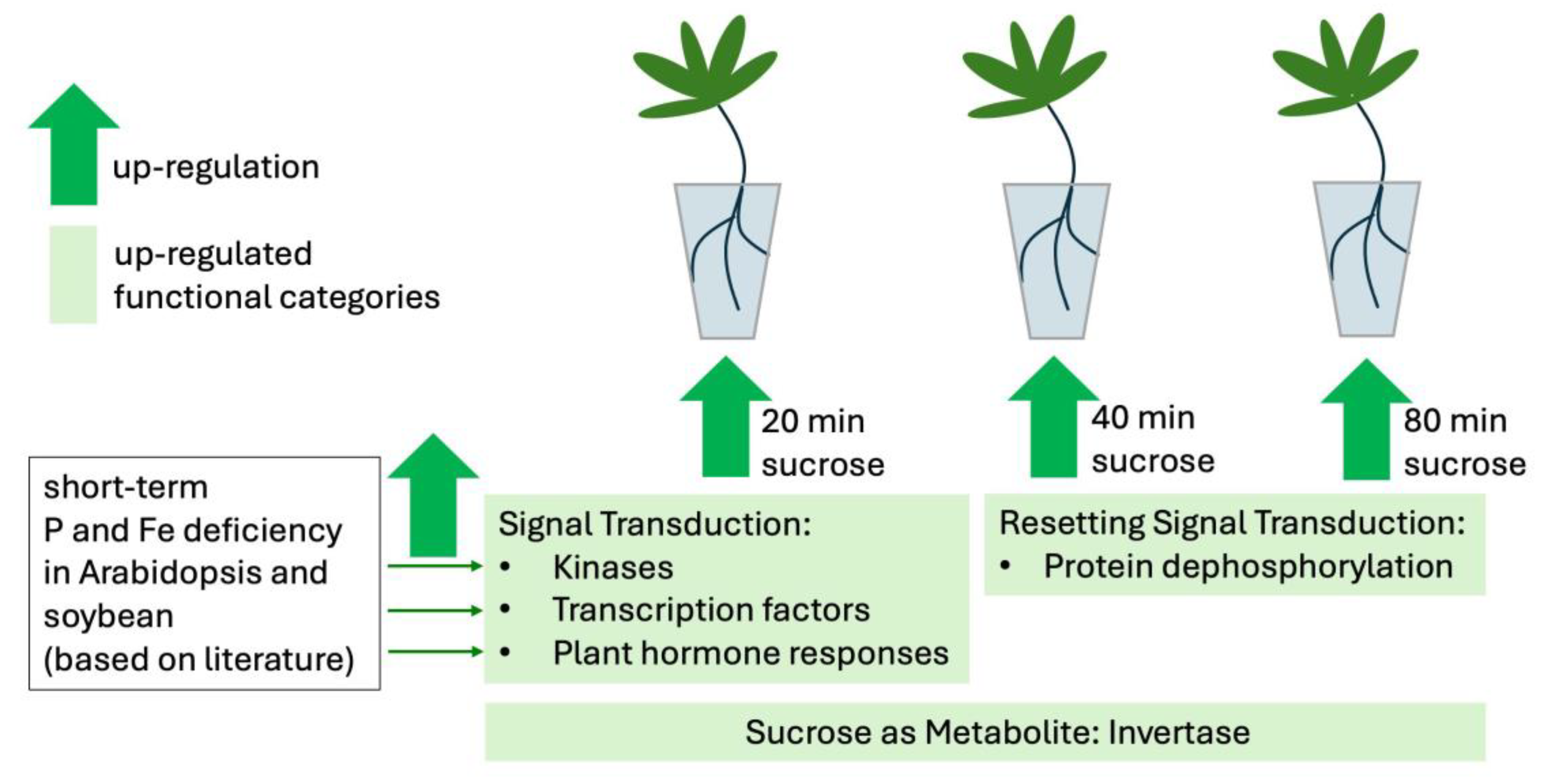
3.2. Hormone Responses Are Enriched at 20 min of Sucrose Treatment
3.3. Kinases Were Up-Regulated at 20 min of Sucrose Treatment, While Protein Dephosphorylation Is Enriched at 40 and 80 min
3.4. Promoter Analysis Reveals Candidate Binding Sites for Sucrose-Responsive Transcription Factors
3.5. Pathway Analysis Summarizes Responses to Sucrose at 20, 40, and 80 min
3.6. Sucrose Can Act as Signaling Molecule, but So Can Its Cleavage Products
3.7. Short-Term Responses to Sucrose and P or Fe Deficiency Display Considerable Overlap
4. Materials and Methods
4.1. Seed Germination, Treatments, and Harvest
4.2. RNA Isolation and Quality Check
4.3. cDNA Library Preparation and RNA-Sequencing
4.4. RNA-Seq Data Analysis
4.5. Differential Expression Analysis
4.6. Promoter Analysis
4.7. MapMan Analysis of Metabolic Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vance, C.P.; Uhde-Stone, C.; Allan, D.L. Phosphorus acquisition and use: Critical adaptations by plants for securing a nonrenewable resource. New Phytol. 2003, 157, 423–447. [Google Scholar] [CrossRef] [PubMed]
- Colombo, C.; Palumbo, G.; He, J.-Z.; Pinton, R.; Cesco, S. Review on iron availability in soil: Interaction of Fe minerals, plants, and microbes. J. Soils Sediments 2014, 14, 538–548. [Google Scholar] [CrossRef]
- Ahmad, N.; Usman, M.; Ahmad, H.R.; Sabir, M.; Farooqi, Z.U.R.; Shehzad, M.T. Environmental implications of phosphate-based fertilizer industrial waste and its management practices. Environ. Monit. Assess. 2023, 195, 1326. [Google Scholar] [CrossRef]
- Yuan, Z.; Jiang, S.; Sheng, H.; Liu, X.; Hua, H.; Liu, X.; Zhang, Y. Human Perturbation of the Global Phosphorus Cycle: Changes and Consequences. Environ. Sci. Technol. 2018, 52, 2438–2450. [Google Scholar] [CrossRef]
- Withers, P.J.A.; Forber, K.G.; Lyon, C.; Rothwell, S.; Doody, D.G.; Jarvie, H.P.; Martin-Ortega, J.; Jacobs, B.; Cordell, D.; Patton, M.; et al. Towards resolving the phosphorus chaos created by food systems. Ambio 2019, 49, 1076–1089. [Google Scholar] [CrossRef]
- Uhde-Stone, C. White lupin: A model system for understanding plant adaptation to low phosphorus availability. In Legume Nitrogen Fixation in Soils with Low Phosphorus Availability; Springer: Berlin/Heidelberg, Germany, 2017; pp. 243–280. [Google Scholar]
- Zanin, L.; Venuti, S.; Marroni, F.; Franco, A.; Morgante, M.; Pinton, R.; Tomasi, N. Physiological and RNA sequencing data of white lupin plants grown under Fe and P deficiency. Data Brief. 2019, 25, 104069. [Google Scholar] [CrossRef]
- Neumann, G.; Massonneau, A.; Langlade, N.; Dinkelaker, B.; Hengeler, C.; Römheld, V.; Martinoia, E. Physiological aspects of cluster root function and development in phosphorus-deficient white lupin (Lupinus albus L.). Ann. Bot. 2000, 85, 909–919. [Google Scholar] [CrossRef]
- Massonneau, A.; Langlade, N.; Léon, S.; Smutny, J.; Vogt, E.; Neumann, G.; Martinoia, E. Metabolic changes associated with cluster root development in white lupin (Lupinus albus L.): Relationship between organic acid excretion, sucrose metabolism and energy status. Planta 2001, 213, 534–542. [Google Scholar] [CrossRef]
- Sas, L.; Rengel, Z.; Tang, C. Excess cation uptake, and extrusion of protons and organic acid anions by Lupinus albus under phosphorus deficiency. Plant Sci. 2001, 160, 1191–1198. [Google Scholar] [CrossRef]
- Wang, B.; Tang, X.; Cheng, L.; Zhang, A.; Zhang, W.; Zhang, F.; Liu, J.; Cao, Y.; Allan, D.; Vance, C. Nitric oxide is involved in phosphorus deficiency-induced cluster-root development and citrate exudation in white lupin. New Phytol. 2010, 187, 1112–1123. [Google Scholar] [CrossRef]
- Johnson, J.; Allan, D.; Vance, C. Phosphorus Stress-Induced Proteoid Roots Show Altered Metabolism in Lupinus albus. Plant Physiol. 1994, 104, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Olt, P.; Neuhauser, B.; Moradtalab, N.; Bautista, W.; Uhde-Stone, C.; Neumann, G.; Ludewig, U. Loss of LaMATE impairs isoflavonoid release from cluster roots of phosphorus-deficient white lupin. Physiol. Plant 2021, 173, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Uhde-Stone, C.; Zinn, K.E.; Ramirez-Yanez, M.; Li, A.; Vance, C.P.; Allan, D.L. Nylon filter arrays reveal differential gene expression in proteoid roots of white lupin in response to phosphorus deficiency. Plant Physiol. 2003, 131, 1064–1079. [Google Scholar] [CrossRef]
- O’Rourke, J.A.; Yang, S.S.; Miller, S.S.; Bucciarelli, B.; Liu, J.; Rydeen, A.; Bozsoki, Z.; Uhde-Stone, C.; Tu, Z.J.; Allan, D.; et al. An RNA-Seq transcriptome analysis of orthophosphate-deficient white lupin reveals novel insights into phosphorus acclimation in plants. Plant Physiol. 2013, 161, 705–724. [Google Scholar] [CrossRef]
- Secco, D.; Shou, H.; Whelan, J.; Berkowitz, O. RNA-seq analysis identifies an intricate regulatory network controlling cluster root development in white lupin. BMC Genom. 2014, 15, 230. [Google Scholar] [CrossRef]
- Wang, Z.; Straub, D.; Yang, H.; Kania, A.; Shen, J.; Ludewig, U.; Neumann, G. The regulatory network of cluster-root function and development in phosphate-deficient white lupin (Lupinus albus) identified by transcriptome sequencing. Physiol. Plant. 2014, 151, 323–338. [Google Scholar] [CrossRef]
- Shu, L.; Shen, J.; Rengel, Z.; Tang, C.; Zhang, F. Cluster root formation by Lupinus albus is modified by stratified application of phosphorus in a split-root system. J. Plant Nutr. 2007, 30, 271–288. [Google Scholar] [CrossRef]
- Tian, L.; Liu, H.; Ren, L.; Ku, L.; Wu, L.; Li, M.; Wang, S.; Zhou, J.; Song, X.; Zhang, J.; et al. MicroRNA 399 as a potential integrator of photo-response, phosphate homeostasis, and sucrose signaling under long day condition. BMC Plant Biol. 2018, 18, 290. [Google Scholar] [CrossRef]
- Lin, X.Y.; Ye, Y.Q.; Fan, S.K.; Jin, C.W.; Zheng, S.J. Increased Sucrose Accumulation Regulates Iron-Deficiency Responses by Promoting Auxin Signaling in Arabidopsis Plants. Plant Physiol. 2016, 170, 907–920. [Google Scholar] [CrossRef]
- Hammond, J.; White, P. Sucrose transport in the phloem: Integrating root responses to phosphorus starvation. J. Exp. Bot. 2008, 59, 93–109. [Google Scholar] [CrossRef]
- Poirier, Y.; Jaskolowski, A.; Clua, J. Phosphate acquisition and metabolism in plants. Curr. Biol. CB 2022, 32, R623–R629. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.Y.; Huang, T.K.; Leong, S.J.; Chiou, T.J. Long-distance call from phosphate: Systemic regulation of phosphate starvation responses. J. Exp. Bot. 2014, 65, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Y.; Huang, T.K.; Tseng, C.Y.; Lai, Y.S.; Lin, S.I.; Lin, W.Y.; Chen, J.W.; Chiou, T.J. PHO2-dependent degradation of PHO1 modulates phosphate homeostasis in Arabidopsis. Plant Cell 2012, 24, 2168–2183. [Google Scholar] [CrossRef]
- Lin, S.; Chiang, S.; Lin, W.; Chen, J.; Tseng, C.; Wu, P.; Chiou, T. Regulatory network of microRNA399 and PHO2 by systemic signaling. Plant Physiol. 2008, 147, 732–746. [Google Scholar] [CrossRef]
- Pant, B.D.; Buhtz, A.; Kehr, J.; Scheible, W.R. MicroRNA399 is a long-distance signal for the regulation of plant phosphate homeostasis. Plant J. Cell Mol. Biol. 2008, 53, 731–738. [Google Scholar] [CrossRef]
- Liu, J.; Samac, D.; Bucciarelli, B.; Allan, D.; Vance, C. Signaling of phosphorus deficiency-induced gene expression in white lupin requires sugar and phloem transport. Plant J. Cell Mol. Biol. 2005, 41, 257–268. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, J.; Ludewig, U.; Neumann, G. A re-assessment of sucrose signaling involved in cluster-root formation and function in phosphate-deficient white lupin (Lupinus albus). Physiol. Plant. 2015, 154, 407–419. [Google Scholar] [CrossRef]
- Zhou, K.; Yamagishi, M.; Osaki, M.; Masuda, K. Sugar signalling mediates cluster root formation and phosphorus starvation-induced gene expression in white lupin. J. Exp. Bot. 2008, 59, 2749–2756. [Google Scholar] [CrossRef]
- Xia, T.; Zhu, X.; Zhan, Y.; Liu, B.; Zhou, X.; Zhang, Q.; Xu, W. The white lupin trehalase gene LaTRE1 regulates cluster root formation and function under phosphorus deficiency. Plant Physiol. 2024, 196, 2184–2198. [Google Scholar] [CrossRef]
- Liu, J.; Vance, C.P. Crucial roles of sucrose and microRNA399 in systemic signaling of P deficiency: A tale of two team players? Plant Signal Behav. 2010, 5, 1556–1560. [Google Scholar] [CrossRef]
- Lei, M.; Liu, D. Sucrose regulates plant responses to deficiencies in multiple nutrients. Plant Signal Behav. 2011, 6, 1247–1249. [Google Scholar] [CrossRef] [PubMed]
- Ruffel, S. Nutrient-related Long-Distance Signals: Common players and possible crosstalk. Plant Cell Physiol. 2018, 59, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Keurentjes, J.; Bentsink, L.; Koornneef, M.; Smeekens, S. Sucrose-specific induction of anthocyanin biosynthesis in Arabidopsis requires the MYB75/PAP1 gene. Plant Physiol. 2005, 139, 1840–1852. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Cho, L.H.; Tun, W.; Jeon, J.S.; An, G. Sucrose signaling in higher plants. Plant Sci. 2021, 302, 110703. [Google Scholar] [CrossRef]
- Lalonde, S.; Boles, E.; Hellmann, H.; Barker, L.; Patrick, J.W.; Frommer, W.B.; Ward, J.M. The dual function of sugar carriers: Transport and sugar sensing. Plant Cell 1999, 11, 707–726. [Google Scholar] [CrossRef]
- Nidumolu, L.C.M.; Lorilla, K.M.; Chakravarty, I.; Uhde-Stone, C. Soybean Root Transcriptomics: Insights into Sucrose Signaling at the Crossroads of Nutrient Deficiency and Biotic Stress Responses. Plants 2023, 12, 2117. [Google Scholar] [CrossRef]
- Tognetti, J.A.; Pontis, H.G.; Martinez-Noel, G.M. Sucrose signaling in plants: A world yet to be explored. Plant Signal Behav. 2013, 8, e23316. [Google Scholar] [CrossRef]
- Shammi, T.; Lee, Y.; Trivedi, J.; Sierras, D.; Mansoor, A.; Maxwell, J.M.; Williamson, M.; McMillan, M.; Chakravarty, I.; Uhde-Stone, C. Transcriptomics Provide Insights into Early Responses to Sucrose Signaling in Lupinus albus, a Model Plant for Adaptations to Phosphorus and Iron Deficiency. Int. J. Mol. Sci. 2024, 25, 7692. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, Q.; Yuan, W.; Xu, F.; Muhammad Aslam, M.; Miao, R.; Li, Y.; Wang, Q.; Li, X.; Zhang, X.; et al. The genome evolution and low-phosphorus adaptation in white lupin. Nat. Commun. 2020, 11, 1069. [Google Scholar] [CrossRef]
- Hufnagel, B.; Marques, A.; Soriano, A.; Marques, L.; Divol, F.; Doumas, P.; Sallet, E.; Mancinotti, D.; Carrere, S.; Marande, W.; et al. High-quality genome sequence of white lupin provides insight into soil exploration and seed quality. Nat. Commun. 2020, 11, 492. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Frazee, A.C.; Pertea, G.; Jaffe, A.E.; Langmead, B.; Salzberg, S.L.; Leek, J.T. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 2015, 33, 243–246. [Google Scholar] [CrossRef]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef]
- Liu, H.; Yang, H.; Wu, C.; Feng, J.; Liu, X.; Qin, H.; Wang, D. Overexpressing HRS1 confers hypersensitivity to low phosphate-elicited inhibition of primary root growth in Arabidopsis thaliana. J. Integr. Plant Biol. 2009, 51, 382–392. [Google Scholar] [CrossRef]
- Dadras, A.; Furst-Jansen, J.M.R.; Darienko, T.; Krone, D.; Scholz, P.; Sun, S.; Herrfurth, C.; Rieseberg, T.P.; Irisarri, I.; Steinkamp, R.; et al. Environmental gradients reveal stress hubs pre-dating plant terrestrialization. Nat. Plants 2023, 9, 1419–1438. [Google Scholar] [CrossRef]
- Pontis, H.G. On the scent of the riddle of sucrose. Trends Biochem. Sci. 1978, 3, 137–139. [Google Scholar] [CrossRef]
- Granot, D.; Kelly, G.; Stein, O.; David-Schwartz, R. Substantial roles of hexokinase and fructokinase in the effects of sugars on plant physiology and development. J. Exp. Bot. 2014, 65, 809–819. [Google Scholar] [CrossRef]
- Kall, L.; Krogh, A.; Sonnhammer, E.L. Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wind, J.J.; Shi, X.; Zhang, H.; Hanson, J.; Smeekens, S.C.; Teng, S. Fructose sensitivity is suppressed in Arabidopsis by the transcription factor ANAC089 lacking the membrane-bound domain. Proc. Natl. Acad. Sci. USA 2011, 108, 3436–3441. [Google Scholar] [CrossRef] [PubMed]
- Venuti, S.; Zanin, L.; Marroni, F.; Franco, A.; Morgante, M.; Pinton, R.; Tomasi, N. Physiological and transcriptomic data highlight common features between iron and phosphorus acquisition mechanisms in white lupin roots. Plant Sci. 2019, 285, 110–121. [Google Scholar] [CrossRef]
- Lin, W.D.; Liao, Y.Y.; Yang, T.J.; Pan, C.Y.; Buckhout, T.J.; Schmidt, W. Coexpression-based clustering of Arabidopsis root genes predicts functional modules in early phosphate deficiency signaling. Plant Physiol. 2011, 155, 1383–1402. [Google Scholar] [CrossRef]
- Moran Lauter, A.N.; Peiffer, G.A.; Yin, T.; Whitham, S.A.; Cook, D.; Shoemaker, R.C.; Graham, M.A. Identification of candidate genes involved in early iron deficiency chlorosis signaling in soybean (Glycine max) roots and leaves. BMC Genom. 2014, 15, 702. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, X.; Lu, C.; Zeng, X.; Li, Y.; Fu, D.; Wu, G. Non-specific lipid transfer proteins in plants: Presenting new advances and an integrated functional analysis. J. Exp. Bot. 2015, 66, 5663–5681. [Google Scholar] [CrossRef]
- Lauter, A.N.M.; Rutter, L.; Cook, D.; O’Rourke, J.A.; Graham, M.A. Examining Short-Term Responses to a Long-Term Problem: RNA-Seq Analyses of Iron Deficiency Chlorosis Tolerant Soybean. Int. J. Mol. Sci. 2020, 21, 3591. [Google Scholar] [CrossRef]
- Hoagland, D.R.; Arnon, D.I. The water-culture method for growing plants without soil. Circular. Calif. Agric. Exp. Stn. 1950, 347, 39. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bailey, T.L. STREME: Accurate and versatile sequence motif discovery. Bioinformatics 2021, 37, 2834–2840. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cDNA Library, Biological Replicate (rep) | Number of Paired Sequences (in Millions) | Overall Mapping Rate to Reference Genome 1 [41] | Overall Mapping Rate to Reference Genome 2 [40] |
|---|---|---|---|
| Control (t0), rep 1 | 24.1 | 86.63% | 86.67% |
| Control (t0), rep 2 | 27.8 | 91.64% | 91.65% |
| Control (t0), rep 3 | 28.3 | 90.54% | 90.58% |
| 20 min sucrose (t20), rep 1 | 38.1 | 79.33% | 79.42% |
| 20 min sucrose (t20), rep 2 | 18.8 | 94.04% | 94.26% |
| 20 min sucrose (t20), rep 3 | 37.4 | 91.37% | 91.38% |
| 40 min sucrose (t40), rep 1 | 25.8 | 90.88% | 90.91% |
| 40 min sucrose (t40), rep 2 | 29.4 | 91.10% | 91.14% |
| 40 min sucrose (t40), rep 3 | 34.4 | 87.78% | 87.81% |
| 80 min sucrose (t40), rep 1 | 40.7 | 83.84% | 83.87% |
| 80 min sucrose (t40), rep 2 | 46.2 | 91.20% | 91.22% |
| 80 min sucrose (t40), rep 3 | 30.5 | 92.63% | 92.67% |
| Total | 381.5 | 89.25% | 89.3% |
| Features | Mapping Statistics for Reference Genome 1 [41] | Mapping Statistics for Reference Genome 2 [40] |
|---|---|---|
| Total transcripts in reference genome | 41,385 (30,479 multi-exon) | 47,603 (38,154 multi-exon) |
| Mapped genes in our RNA-seq analysis | 33,390 (80.7%) | 31,842 (66.9%) |
| Novel exons | 6457/25,1260 (2.6%) | 12,152/30,6263 (4.0%) |
| Novel introns | 6724/17,9038 (3.8%) | 7400/21,8573 (3.4%) |
| Novel loci | 1260/40,617 (3.1%) | 1979/48,624 (4.1%) |
| Annotation, Gene ID | Putative Function | t20 Log2FC * (padj) | t40 Log2FC ** (padj) | t80 Log2FC ** (padj) |
|---|---|---|---|---|
| Catechol O-methyltransferase, Lalb_Chr19g0135061 | Catalyzing methylation of various phenolic compounds, including lignin monomers | 4.4 (3.2 × 10−08) | −0.9 (0.9) | −1.7 (0.2) |
| Shikimate O-hydroxy-cinnamoyl transferase, Lalb_Chr15g0084261 | Involved in generating building blocks of lignin | 3.3 (4.1 × 10−02) | −0.2 (1.0) | 0.0 (1.0) |
| Transcription factor AP2-EREBP family, Lalb_Chr23g0270971 | Regulation of gene expression | 2.8 (3.0 × 10−02) | −1.0 (1.0) | −0.2 (1.0) |
| Transcription factor MYB-HB-like family, Lalb_Chr05g0215541 | Regulation of gene expression | 2.1 (2.0 × 10−06) | 0.0 (1.0) | 0.4 (0.7) |
| Uncharacterized protein, Lalb_Chr05g0211541 | 2.0 (3.4 × 10−04) | 0.2 (1.0) | −0.3 (0.8) | |
| Transcription factor AP2-EREBP family, Lalb_Chr08g0244311 | Regulation of gene expression | 1.9 (2.0 × 10−02) | −0.3 (1.0) | 0.0 (1.0) |
| Tetratricopeptide-like helical domain-containing protein, Lalb_Chr13g0300041 | Mediates protein interactions | 1.9 (4.6 × 10−02) | −0.2 (1.0) | 0.4 (0.8) |
| Uncharacterized protein, Lalb_Chr23g0267501 | 1.8 (4.9 × 10−02) | 0.3 (1.0) | −0.2 (0.9) | |
| Transcription factor NAM family, Lalb_Chr13g0304421 | Regulation of gene expression | 1.7 (3.1 × 10−02) | −0.6 (1.0) | 0.2 (0.9) |
| RING-type E3 ubiquitin transferase, Lalb_Chr10g0104201 | Ubiquitination of proteins | 1.7 (3.4 × 10−02) | 0.3 (1.0) | −1.3 (0.2) |
| F-box domain-containing protein, Lalb_Chr18g0047021 | Ubiquitination of proteins | 1.7 (1.3 × 10−02) | 0.2 (1.0) | 0.4 (0.8) |
| Non-specific serine/threonine protein kinase, Lalb_Chr19g0135591 | Signal transduction | 1.6 (5.8 × 10−04) | 0.5 (0.9) | 0.4 (0.7) |
| Transcription factor C2H2 family, Lalb_Chr16g0383971 | Regulation of gene expression | 1.6 (3.3 × 10−03) | 0.5 (1.0) | 0.1 (1.0) |
| Uncharacterized protein, Lalb_Chr15g0090181 | 1.6 (9.7 × 10−03) | 0.4 (1.0) | 0.0 (1.0) | |
| ATPase, AAA-type, Lalb_Chr07g0180441 | ATPases associated with diverse cellular activities | 1.6 (1.7 × 10−03) | 0.1 (1.0) | 0.1 (0.9) |
| Uncharacterized protein, Lalb_Chr17g0346531 | 1.6 (6.4 × 10−03) | 0.1 (1.0) | 0.0 (1.0) | |
| Transcription factor MYB-HB-like family, Lalb_Chr16g0384531 | Regulation of gene expression | 1.6 (4.6 × 10−04) | 0.1 (1.0) | 0.5 (0.6) |
| Uncharacterized protein, Lalb_Chr14g0371631 | 1.6 (2.3 × 10−03) | 0.1 (1.0) | 0.1 (0.9) | |
| Expansin, Lalb_Chr16g0379251 | Loosening cell walls for cell expansion | 1.5 (3.8 × 10−02) | −0.1 (1.0) | −0.1 (0.9) |
| Transcription factor C3H family, Lalb_Chr22g0350601 | Regulation of gene expression | 1.5 (2.6 × 10−02) | −0.5 (1.0) | −0.4 (0.7) |
| Gene ID/ Annotation, Gene ID | Putative Function | t20 log2FC (padj) | t40 log2FC (padj) | t80 log2FC (padj) |
|---|---|---|---|---|
| Small GTPase superfamily, P-loop containing, Lalb_Chr09g0323971 | RAB family, protein trafficking | 24.5 (3.4 × 10−06) | 25.6 (8.2 × 10−07) | 24.8 (2.8 × 10−06) |
| Flowering time control protein FCA, Lalb_Chr13g0298581 | RNA binding | 24.0 (6.0 × 10−06) | 24.4 (3.5 × 10−06) | 21.9 (6.0 × 10−05) |
| Small GTPase superfamily, P-loop containing, Lalb_Chr23g0266071 | Rac-like GTP-binding protein, signal transduction | 23.9 (6.4 × 10−06) | 15.2 (2.4 × 10−02) | 21.7 (6.8 × 10−05) |
| Transcription factor WRKY family, Lalb_Chr18g0048721 | Similar to WRKY2, regulation of gene expression | 23.9 (4.7 × 10−07) | 23.2 (8.6 × 10−07) | 24.6 (2.0 × 10 −07) |
| Histone acetyltransferase chromatin regulator PHD, Lalb_Chr06g0162921 | PHD (plant homeodomain) zinc fingers, regulation gene expression | 23.3 (1.4 × 10−05) | 23.9 (6.8 × 10−06) | 24.2 (5.8 × 10−06) |
| Smr domain-containing protein, Lalb_Chr14g0375461 | Polyadenylate-binding protein-interacting; post-transcriptional regulation | 23.2 (1.5 × 10−05) | 19.6 (5.2 × 10−04) | 22.7 (2.9 × 10−05) |
| RING-type E3 ubiquitin transferase, Lalb_Chr11g0068041 | Ubiquitination of proteins | 22.7 (2.6 × 10−05) | 21.4 (8.8 × 10−05) | 23.3 (1.5 × 10−05) |
| Winged helix-turn-helix DNA-binding domain, Lalb_Chr20g0109001 | Regulation of gene expression | 22.1 (5.0 × 10−05) | 21.5 (7.9 × 10−05) | 21.1 (1.2 × 10−04) |
| Zinc finger, RanBP2-type, Lalb_Chr15g0081991 | Interacting with RNA or proteins | 21.9 (5.9 × 10−05) | 19.7 (4.8 × 10−04) | 22.0 (5.2 × 10−05) |
| V-type proton ATPase subunit a, Lalb_Chr01g0004101 | Vacuolar transport of ions and metabolites | 21.8 (2.0 × 10−15) | 19.8 (1.3 × 10−12) | 20.7 (1.1 × 10−13) |
| Staygreen protein, Lalb_Chr05g0214731 | Senescence-induced functions in plastids | 21.7 (7.2 × 10−05) | 20.5 (2.2 × 10−04) | 21.2 (1.1 × 10−04) |
| Lon protease homolog 2, peroxisomal, Lalb_Chr05g0220701/ | Degradation of oxidized proteins | 21.5 (8.5 × 10−05) | 23.0 (1.7 × 10−05) | 21.1 (1.2 × 10−04) |
| ATP-dependent DNA helicase, Lalb_Chr16g0380641 | Unwinding DNA or RNA in replication or transcription | 21.4 (9.1 × 10−05) | 21.7 (7.1 × 10−05) | 20.7 (1.7 × 10−04) |
| Target of Myb protein, Lalb_Chr07g0184041 | Regulated by Myb transcription factor | 21.3 (9.8 × 10−05) | 19.7 (4.8 × 10−04) | 22.4 (3.8 × 10−05) |
| GBF-interacting protein, Lalb_Chr10g0107481 | Enhance binding of G-box binding transcription factors to DNA | 21.3 (9.8 × 10−11) | 18.7 (2.7 × 10−08) | 19.5 (7.5 × 10−09) |
| Vacuolar protein sorting-associated protein, Lalb_Chr13g0298831/ | Sorting and packaging of vacuolar proteins into transport vesicles | 20.9 (3.0 × 10−11) | 19.6 (4.4 × 10−10 | 20.3 (1.3 × 10−10) |
| Transcription factor MYB-HB-like, Lalb_Chr19g0132831 | Regulation of gene expression | 20.9 (1.5 × 10−04) | 21.7 (6.5 × 10−05 | 20.7 (1.7 × 10−04) |
| SPOROCYTELESS-like EAR-containing protein, Lalb_Chr25g0286021 | Transcriptional repressor | 20.6 (1.9 × 10−04) | 20.3 (2.7 × 10−04) | 20.2 (2.8 × 10−04) |
| K domain-containing protein, Lalb_Chr09g0320521 | Nucleic acid-binding protein | 20.6 (3.9 × 10−11) | 19.8 (2.3 × 10−10) | 22.0 (9.9 × 10−13) |
| Uncharacterized protein, Lalb_Chr17g0346011 | Unknown | 20.6 (3.3 × 10−05) | 18.9 (1.8 × 10−04) | 20.0 (6.2 × 10−05) |
| F-box domain-containing protein, Lalb_Chr03g0043711 | Ubiquitination of proteins | 20.6 (2.1 × 10−04) | 20.6 (2.0 × 10−04 | 21.6 (7.5 × 10−05) |
| Transcription factor AP2-EREBP family, Lalb_Chr18g0059441 | Regulation of gene expression | 20.4 (4.1 × 10−05) | 19.5 (9.6 × 10−05) | 18.7 (2.5 × 10−04) |
| Bicarbonate transporter, Lalb_Chr18g0058431 | Active membrane transporter | 20.3 (2.6 × 10−04) | 23.1 (1.6 × 10−05) | 21.4 (9.3 × 10−05) |
| Major facilitator, sugar transporter, Lalb_Chr02g0144351 | Transport of sugar and other substances across membrane | 20.3 (2.7 × 10−04) | 21.6 (7.5 × 10−05) | 23.1 (1.8 × 10−05) |
| E3 SUMO protein ligase, Lalb_Chr09g0320801 | Bridging SUMO and ubiquitin signaling pathways | 20.3 (2.8 × 10−04) | 20.8 (1.6 × 10−04) | 24.0 (7.5 × 10−06) |
| Invertase, Lalb_Chr21g0305861 | Cleaves sucrose into glucose and fructose | 20.1 (3.2 × 10−04) | 20.4 (2.3 × 10−04) | 21.2 (1.2 × 10−04) |
| C2 domain-containing protein, Lalb_Chr17g0347941 | Various functions, including signal transduction and membrane trafficking | 20.0 (8.5 × 10−05) | 20.8 (3.5 × 10−05) | 19.7 (1.1 × 10−04) |
| UDP-glucuronate 4-epimerase, Lalb_Chr19g0127311 | Conversion of UDP-glucuronate to UDP-D-galacturonate | 19.8 (8.7 × 10−07) | 19.7 (8.2 × 10−07) | 19.6 (1.4 × 10−06) |
| Uncharacterized protein, Lalb_Chr01g0011871 | Unknown | 19.7 (8.3 × 10−17) | 21.5 (1.7 × 10−20) | 21.8 (2.6 × 10−21) |
| Plant organelle RNA recognition domain-containing protein, Lalb_Chr20g0111061 | RNA-binding | 19.5 (5.4 × 10−4) | 5.4 × 10−04 (1.3 × 10−03) | 21.8 (6.3 × 10−5) |
| Most Similar Motif in Arabidopsis (on Top) Compared to Enriched Motif (Bottom Logo) | Protein in Arabidopsis That May Bind to Motif (Similarity p-Value) | Number of Promoters Containing Motif | |
|---|---|---|---|
| 43 genes up-regulated at all timepoints * | 43 genes down-regulated at all timepoints | ||
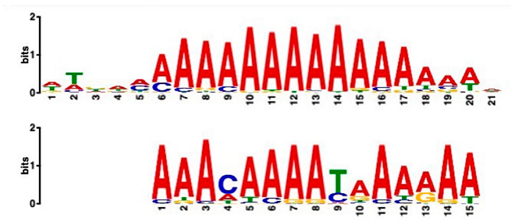 | DNA-binding protein REM (B3 DNA binding domain) (2.28 × 10−6) | 27/43 (enriched, p-value 2.7 × 10−8) | 3/43 |
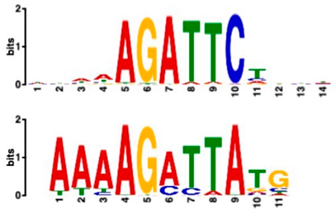 | Transcription factor HHO6, probably involved in phosphate signaling in roots (6.7 × 10−4) | 36/43 (enriched, p-value 6.6 × 10−6) | 15/43 |
| 20 genes up-regulated at all timepoints | 20 genes ONLY up-regulated at t20 | ||
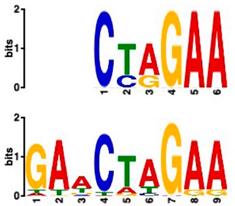 | Heat shock factor (transcription factor) (4.77 × 10−4) | 19/20 (enriched, p-value 2 × 10−7) | 3/20 |
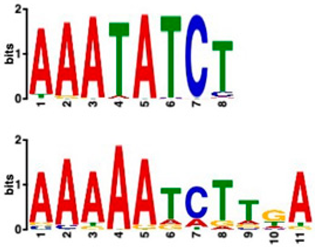 | Transcription factor CCA1 (MYB-related transcription factor) (6.67 × 10−4) | 18/20 (enriched, p-value 2.6 × 10−7) | 2/20 |
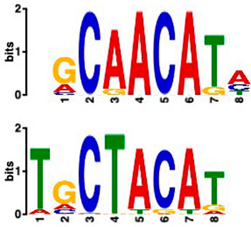 | AP2/ERF and B3 domain-containing transcription factor (3.44 × 10−3) | 1/20 | 15/20 (enriched, p-value 5 × 10−6) |
 | Zinc-finger homeodomain transcription factor (4.65 × 10−7) | 0/20 | 13/20 (enriched, p-value 6.4 × 10−6) |
| 10 genes induced in cluster root development (Figure 3) | 10 genes not induced in cluster root development | ||
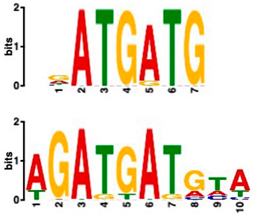 | MADS-box transcription factor (7.84 × 10−6) | 10/10 (enriched, p-value 5.4 × 10−6) | 0/10 |
| Duration of Sucrose Exposure | Count of Up-Regulated * | Ratio of Kinase/Phosphatase | |
|---|---|---|---|
| Kinases | Phosphatases | ||
| 20 min | 17 | 4 | 4.25 |
| 40 min | 4 | 3 | 1.33 |
| 80 min | 3 | 3 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, P.; Sethi, S.; New, J.; Lorilla, K.M.; Maleski, K.; Ancheta, A.; Uhde-Stone, C. Transcriptomic Response of White Lupin Roots to Short-Term Sucrose Treatment. Plants 2025, 14, 381. https://doi.org/10.3390/plants14030381
Roy P, Sethi S, New J, Lorilla KM, Maleski K, Ancheta A, Uhde-Stone C. Transcriptomic Response of White Lupin Roots to Short-Term Sucrose Treatment. Plants. 2025; 14(3):381. https://doi.org/10.3390/plants14030381
Chicago/Turabian StyleRoy, Proyasha, Shrey Sethi, James New, Kristina Mae Lorilla, Karen Maleski, Allan Ancheta, and Claudia Uhde-Stone. 2025. "Transcriptomic Response of White Lupin Roots to Short-Term Sucrose Treatment" Plants 14, no. 3: 381. https://doi.org/10.3390/plants14030381
APA StyleRoy, P., Sethi, S., New, J., Lorilla, K. M., Maleski, K., Ancheta, A., & Uhde-Stone, C. (2025). Transcriptomic Response of White Lupin Roots to Short-Term Sucrose Treatment. Plants, 14(3), 381. https://doi.org/10.3390/plants14030381