
Unveiling Stage-Specific Flavonoid Dynamics Underlying Drought Tolerance in Sweet Potato (Ipomoea batatas L.) via Integrative Transcriptomic and Metabolomic Analyses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
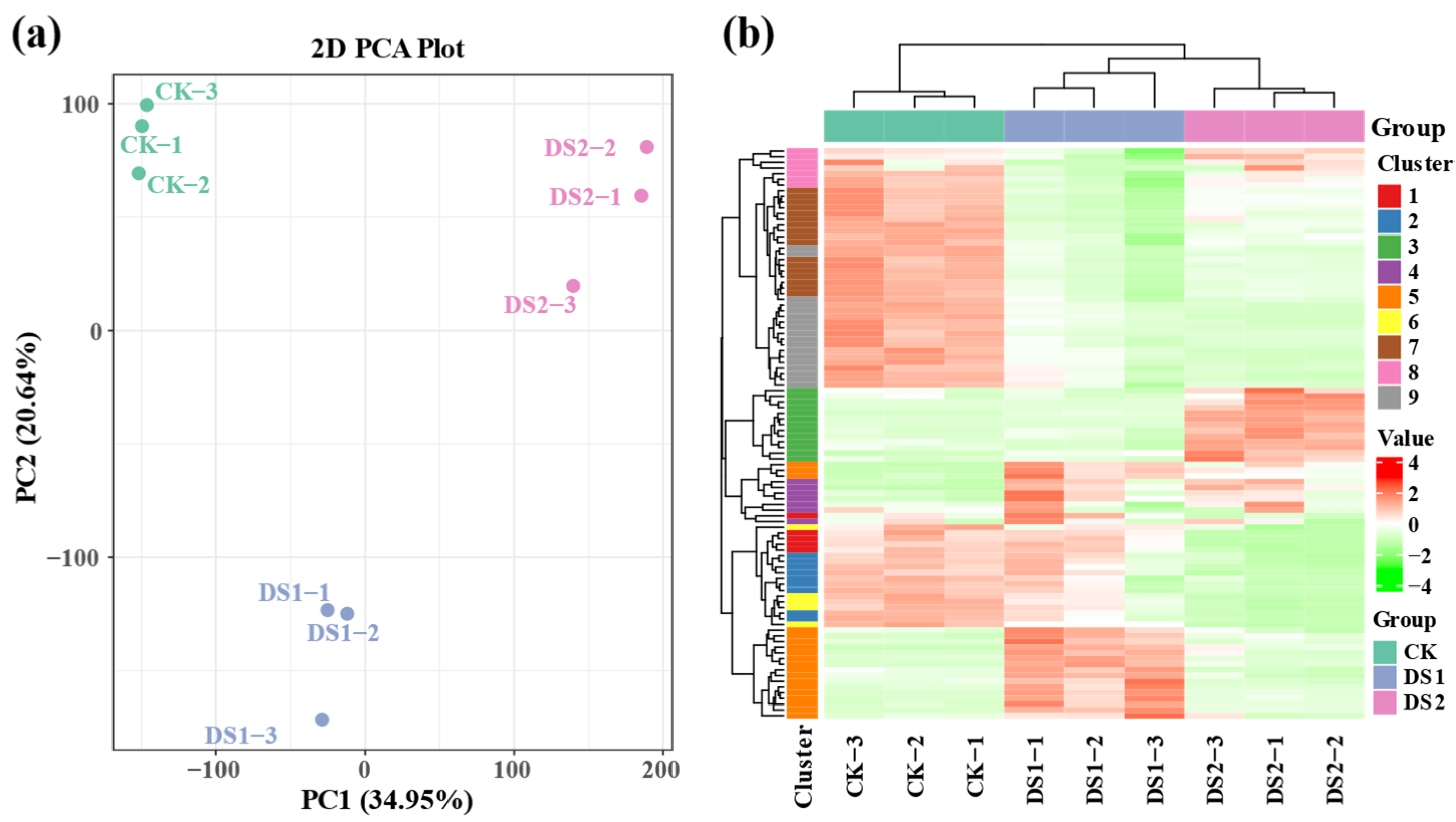
2.1. Transcriptomic Analysis of Sweet Potato Leaves Under Drought Stress
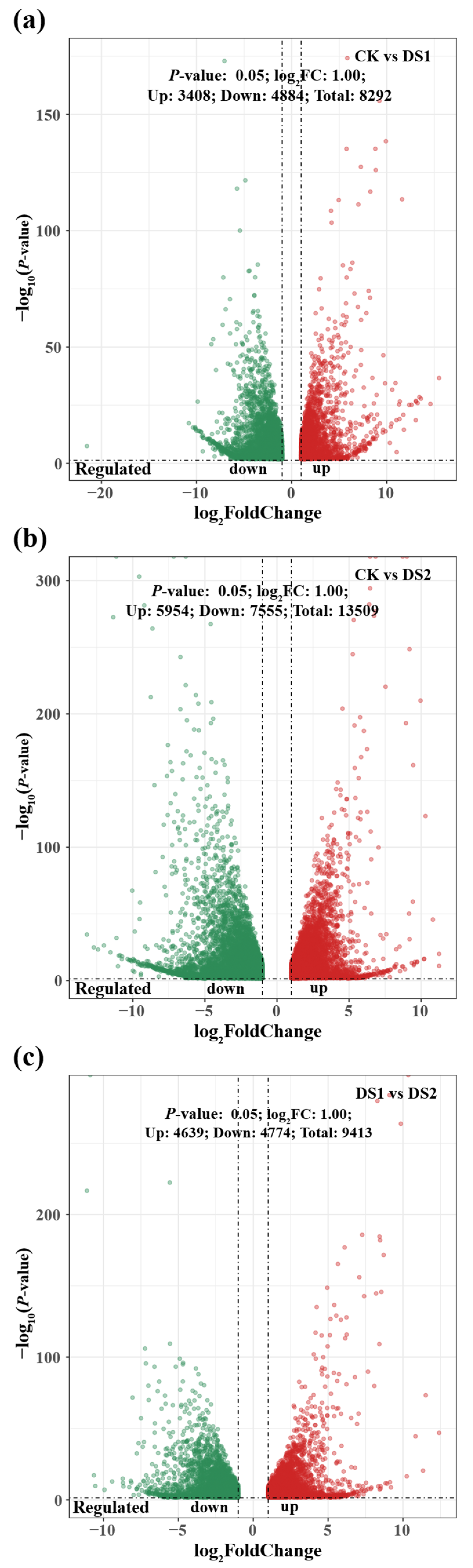
2.2. Differentially Expressed Genes Analysis in Response to Drought Stress
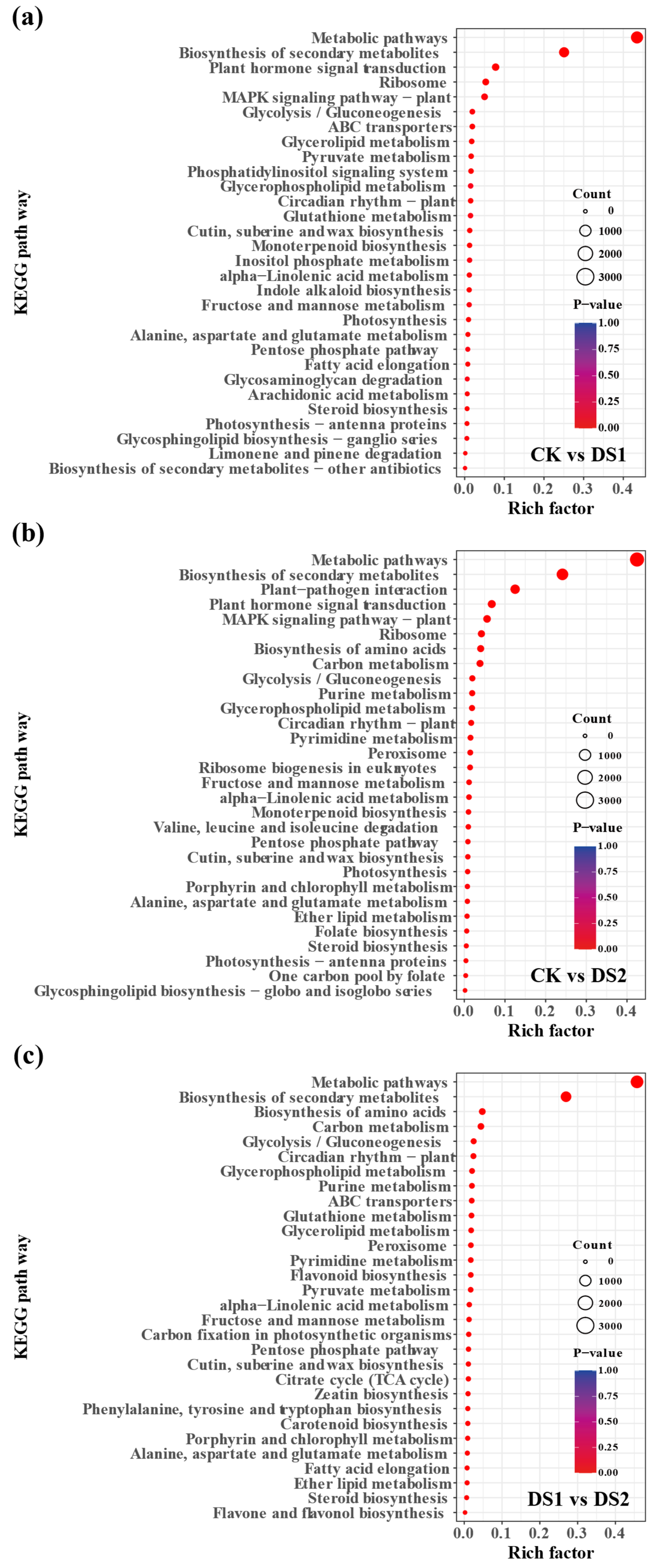
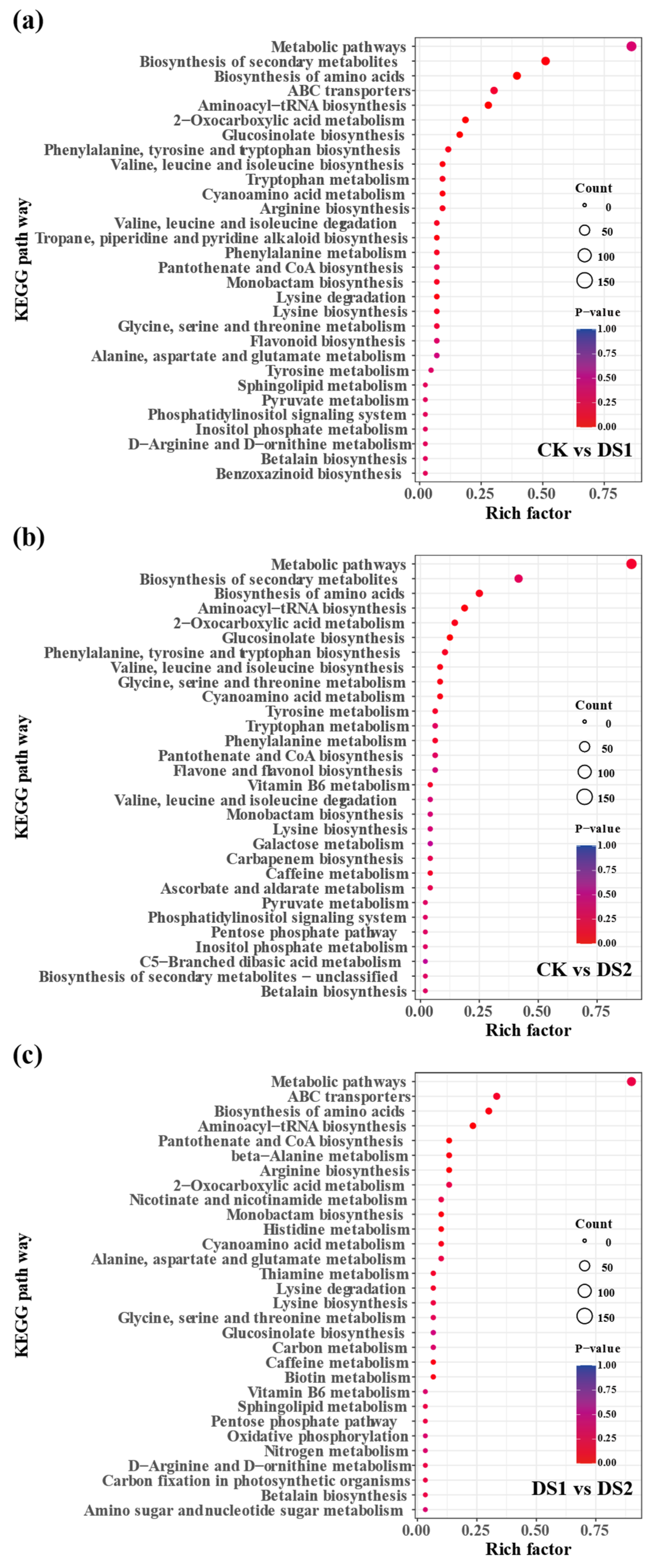
2.3. KEGG Pathway Enrichment Analysis of Differentially Expressed Genes
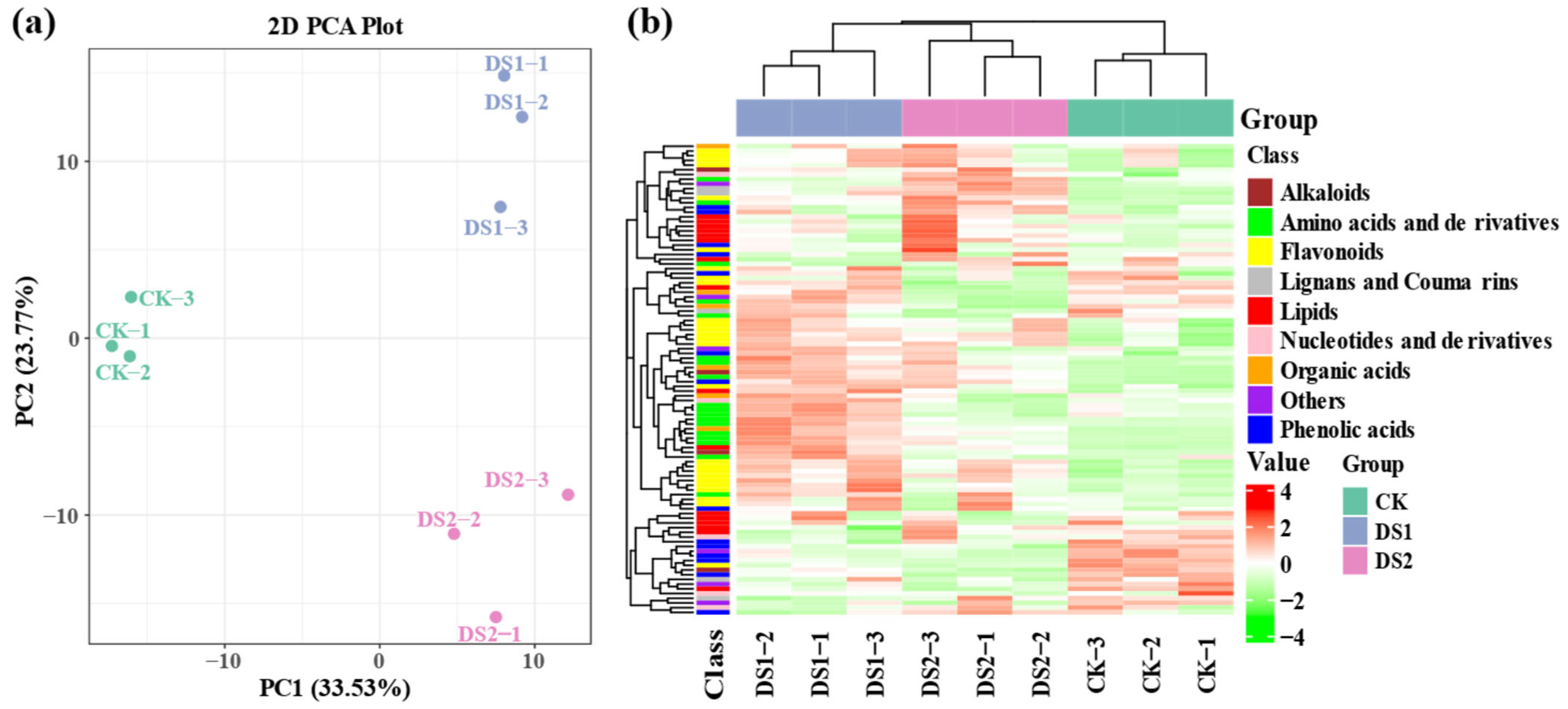
2.4. Metabolomic Analysis of Sweet Potato Leaves Under Drought Stress
2.5. Classification and Distribution of Differentially Accumulated Metabolites
2.6. Identification of Differentially Accumulated Metabolites Under Drought Stress
2.7. KEGG Enrichment Analysis of Differentially Accumulated Metabolites
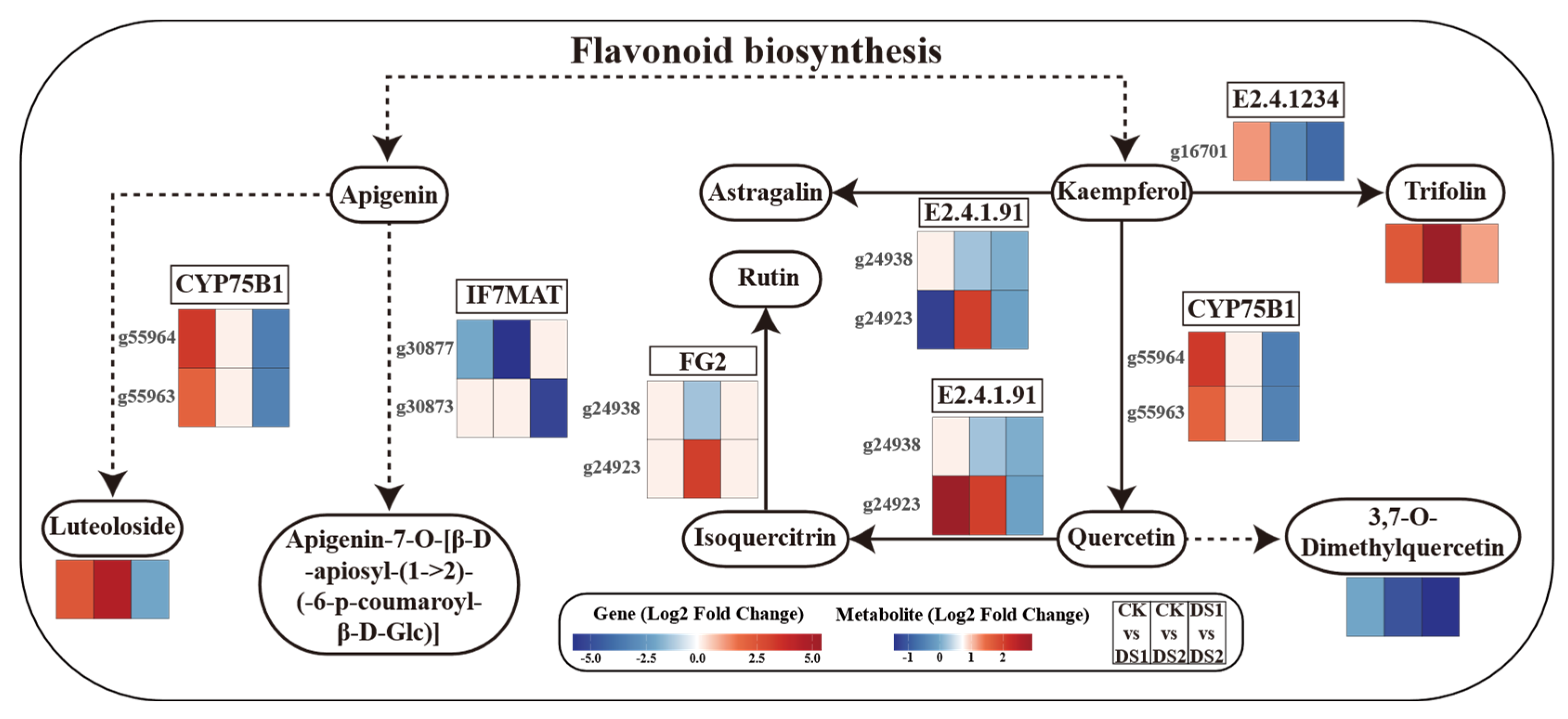
2.8. Integrated Transcriptomic and Metabolomic Analysis of Flavonoid and Flavonol Biosynthesis Pathways
3. Discussion
3.1. Stage-Specific Transcriptomic Responses to Drought Stress
3.2. Metabolic Reprogramming Under Drought Stress
3.3. Coordinated Regulation of Flavonoid Biosynthesis: An Integrated Omics Perspective
4. Materials and Methods
4.1. Plant Material and Stress Treatment
4.2. Widely Targeted Metabolomics Analysis
4.2.1. Sample Preparation and Metabolite Extraction
4.2.2. UPLC-MS/MS Analysis
4.2.3. Mass Spectrometry Parameters
4.2.4. Metabolite Identification and Quantification
4.2.5. Statistical Analysis of Metabolomic Data
4.2.6. Metabolic Pathway Analysis
4.3. Transcriptome Sequencing and Data Analysis
4.3.1. RNA Extraction and Quality Assessment
4.3.2. Library Construction and RNA Sequencing
4.3.3. Data Processing and Quality Control
4.3.4. Gene Expression Quantification and Differential Expression Analysis
4.3.5. Functional Annotation and Enrichment Analysis
4.4. Integrating Transcriptome and Metabolome Analysis
4.5. Metabolome and Transcriptome Data Visualization
4.5.1. Principal Component Analysis
4.5.2. Heatmap Visualization
4.5.3. Metabolite Class Distribution
4.5.4. Volcano Plots
4.5.5. KEGG Pathway Enrichment Analysis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Low, J.W.; Thiele, G. Understanding innovation: The development and scaling of orange-fleshed sweetpotato in major African food systems. Agric. Syst. 2020, 179, 102770. [Google Scholar] [CrossRef]
- Motsa, M.M.; Modi, A.T.; Mabhaudhi, T. Sweet potato (Ipomoea batatas L.) as a drought tolerant and food security crop. S. Afr. J. Sci. 2015, 111, 1–8. [Google Scholar] [CrossRef]
- Neela, S.; Fanta, S.W. Review on nutritional composition of orange-fleshed sweet potato and its role in management of vitamin A deficiency. Food Sci. Nutr. 2019, 7, 1920–1945. [Google Scholar] [CrossRef]
- Zhou, M.; Hu, S.; Wang, S.; Yin, T.; Liu, Q.; Li, H. Sweet potato yield and quality characteristics affected by different late-season irrigation levels. J. Sci. Food Agric. 2024, 104, 5207–5218. [Google Scholar] [CrossRef]
- Gajanayake, B.; Reddy, K.R.; Shankle, M.W.; Arancibia, R.A. Early-season soil moisture deficit reduces sweetpotato storage root initiation and development. HortScience 2013, 48, 1457–1462. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Liu, Q. Photosynthesis product allocation and yield in sweet potato with spraying exogenous hormones under drought stress. J. Plant Physiol. 2020, 253, 153265. [Google Scholar] [CrossRef] [PubMed]
- Taduri, S.; Bheemanahalli, R.; Wijewardana, C.; Lone, A.A.; Meyers, S.L.; Shankle, M.; Gao, W.; Reddy, K.R. Sweetpotato cultivars responses to interactive effects of warming, drought, and elevated carbon dioxide. Front. Genet. 2023, 13, 1080125. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tang, J.; Cao, Q.; Li, Z.; Ma, D. Differential response of physiology and metabolic response to drought stress in different sweetpotato cultivars. PLoS ONE 2022, 17, e0264847. [Google Scholar] [CrossRef]
- Yin, Y.; Qiao, S.; Kang, Z.; Luo, F.; Bian, Q.; Cao, G.; Zhao, G.; Wu, Z.; Yang, G.; Wang, Y.; et al. Transcriptome and metabolome analyses reflect the molecular mechanism of drought tolerance in sweet potato. Plants 2024, 13, 351. [Google Scholar] [CrossRef]
- Tang, W.; Arisha, M.H.; Zhang, Z.; Yan, H.; Kou, M.; Song, W.; Li, C.; Gao, R.; Ma, M.; Wang, X.; et al. Comparative transcriptomic and proteomic analysis reveals common molecular factors responsive to heat and drought stresses in sweetpotato (Ipomoea batatas). Front. Plant Sci. 2023, 13, 1081948. [Google Scholar] [CrossRef]
- Tombesi, S.; Nardini, A.; Farinelli, D.; Palliotti, A. Relationships between stomatal behavior, xylem vulnerability to cavitation and leaf water relations in two cultivars of olive trees under drought conditions. Environ. Exp. Bot. 2015, 109, 148–156. [Google Scholar]
- Farooq, M.; Wahid, A.; Kobayashi, N.; Fujita, D.; Basra, S.M.A. Plant drought stress: Effects, mechanisms and management. Agron. Sustain. Dev. 2009, 29, 185–212. [Google Scholar] [CrossRef]
- Flexas, J.; Bota, J.; Loreto, F.; Cornic, G.; Sharkey, T.D. Diffusive and metabolic limitations to photosynthesis under drought and salinity in C3 plants. Plant Biol. 2004, 6, 269–279. [Google Scholar] [CrossRef]
- Lawlor, D.W.; Cornic, G. Photosynthetic carbon assimilation and associated metabolism in relation to water deficits in higher plants. Plant Cell Environ. 2002, 25, 275–294. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, G.F.; Gorni, P.H.; Vitolo, H.F.; Carvalho, M.E.A.; Pacheco, A.C. Sweetpotato tolerance to drought is associated to leaf concentration of total chlorophylls and polyphenols. Theor. Exp. Plant Physiol. 2021, 33, 385–396. [Google Scholar] [CrossRef]
- Yooyongwech, S.; Samphumphuang, T.; Tisarum, R.; Theerawitaya, C.; Cha-Um, S. Water-deficit tolerance in sweet potato [Ipomoea batatas (L.) Lam.] by foliar application of paclobutrazol: Role of soluble sugar and free proline. Front. Plant Sci. 2017, 8, 1400. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, X.; Dai, M. Improving crop drought resistance with plant growth regulators and rhizobacteria: Mechanisms, applications, and perspectives. Plant Commun. 2022, 3, 100228. [Google Scholar] [CrossRef]
- Zhang, H.; Duan, W.; Xie, B.; Wang, B.; Hou, F.; Li, A.; Dong, S.; Qin, Z.; Wang, Q.; Zhang, L. Root yield, antioxidant capacities, and hormone contents in different drought-tolerant sweet potato cultivars treated with ABA under early drought stress. Acta Physiol. Plant 2020, 42, 132. [Google Scholar] [CrossRef]
- Zhao, D.; Zhao, L.; Liu, Y.; Zhang, A.; Xiao, S.; Dai, X.; Yuan, R.; Zhou, Z.; Cao, Q. Metabolomic and transcriptomic analyses of the flavonoid biosynthetic pathway for the accumulation of anthocyanins and other flavonoids in sweetpotato root skin and leaf vein base. J. Agric. Food Chem. 2022, 70, 2574–2588. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liao, J.; Huang, W.; Qin, N. Salicylic acid protects sweet potato seedlings from drought stress by mediating abscisic acid-related gene expression and enhancing the antioxidant defense system. Int. J. Mol. Sci. 2022, 23, 14819. [Google Scholar] [CrossRef]
- Sheng, M.; Xia, H.; Ding, H.; Pan, D.; He, J.; Li, Z.; Liu, J. Long-term soil drought limits starch accumulation by altering sucrose transport and starch synthesis in sweet potato tuberous root. Int. J. Mol. Sci. 2023, 24, 3053. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Xu, L.; Luo, Z.; Li, Z.; Zhou, G.; Gao, H.; Fang, F.; Tang, J.; Zhao, Y.; Zhou, Z.; et al. Transcriptomic analysis reveals mechanisms for the different drought tolerance of sweet potatoes. Front. Plant Sci. 2023, 14, 1136709. [Google Scholar] [CrossRef]
- Sapakhova, Z.; Raissova, N.; Daurov, D.; Zhapar, K.; Daurova, A.; Zhigailov, A.; Zhambakin, K.; Shamekova, M. Sweet potato as a key crop for food security under the conditions of global climate change: A review. Plants 2023, 12, 2516. [Google Scholar] [CrossRef]
- Nakabayashi, R.; Yonekura-Sakakibara, K.; Urano, K.; Suzuki, M.; Yamada, Y.; Nishizawa, T.; Matsuda, F.; Kojima, M.; Sakakibara, H.; Shinozaki, K.; et al. Enhancement of oxidative and drought tolerance in Arabidopsis by overaccumulation of antioxidant flavonoids. Plant J. 2014, 77, 367–379. [Google Scholar] [CrossRef]
- Ma, D.; Sun, D.; Wang, C.; Li, Y.; Guo, T. Expression of flavonoid biosynthesis genes and accumulation of flavonoid in wheat leaves in response to drought stress. Plant Physiol. Biochem. 2014, 80, 60–66. [Google Scholar] [CrossRef]
- Xiao, J.; Xu, X.; Li, M.; Wu, X.; Guo, H. Regulatory network characterization of anthocyanin metabolites in purple sweetpotato via joint transcriptomics and metabolomics. Front. Plant Sci. 2023, 14, 1030236. [Google Scholar] [CrossRef]
- Wang, J.Q.; Li, H.; Liu, Q.; Xiang, D. Effects of drought stress on root development and physiological characteristics of sweet potato at seedling stage. J. Appl. Ecol. 2019, 30, 3155–3163. [Google Scholar]
- Placide, R.; Shimelis, H.; Laing, M.; Gahakwa, D. Physiological mechanisms and conventional breeding of sweet potato (Ipomoea batatas (L.) Lam.) to drought-tolerance. Afr. J. Agric. Res. 2013, 8, 1837–1846. [Google Scholar] [CrossRef]
- Patil, J.R.; Mhatre, K.J.; Yadav, K.; Yadav, L.S.; Srivastava, S.; Nikalje, G.C. Flavonoids in plant–environment interactions and stress responses. Discov. Plants 2024, 1, 68. [Google Scholar] [CrossRef]
- Shomali, A.; Das, S.; Arif, N.; Sarraf, M.; Zahra, N.; Yadav, V.; Aliniaeifard, S.; Chauhan, D.K.; Hasanuzzaman, M. Diverse physiological roles of flavonoids in plant environmental stress responses and tolerance. Plants 2022, 11, 3158. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, R.; Mori, T.; Saito, K. Alternation of flavonoid accumulation under drought stress in Arabidopsis thaliana. Plant Signal. Behav. 2014, 9, e29518. [Google Scholar] [CrossRef]
- Rabeh, K.; Hnini, M.; Oubohssaine, M. A comprehensive review of transcription factor-mediated regulation of secondary metabolites in plants under environmental stress. Stress Biol. 2025, 5, 15. [Google Scholar] [CrossRef]
- Yan, M.; Nie, H.; Wang, Y.; Wang, X.; Jarret, R.; Zhao, J.; Wang, H.; Yang, J. Exploring and exploiting genetics and genomics for sweetpotato improvement: Status and perspectives. Plant Commun. 2022, 3, 100338. [Google Scholar] [CrossRef]
- Johnson, M.; Pace, R.D. Sweet potato leaves: Properties and synergistic interactions that promote health and prevent disease. Nutr. Rev. 2010, 68, 604–615. [Google Scholar] [CrossRef]
- Ojong, P.B.; Njiti, V.; Guo, Z.; Gao, M.; Besong, S.; Barnes, S.L. Variation of flavonoid content among sweetpotato accessions. J. Am. Soc. Hortic. Sci. 2008, 133, 819–824. [Google Scholar] [CrossRef]
- Cheng, P.; Kong, F.; Han, Y.; Liu, X.; Xia, J. Transcriptome analysis reveals genes and pathways associated with drought tolerance of early stages in sweet potato (Ipomoea batatas (L.) Lam.). Genes 2024, 15, 948. [Google Scholar] [CrossRef]
- Gao, H.; Zhang, Y.; Duan, Q.; Ren, Q.; Deng, L.; Huo, Y.; Zhang, B.; Zhen, X. Widely targeted metabolomics analyses clarify the biosynthetic pathways responsible for flavonoids in sweet potato (Ipomoea batatas (L.) Lam.) storage roots. Agriculture 2023, 13, 1955. [Google Scholar] [CrossRef]
- Yang, X.L.; Chen, Y.Q.; Steenhuis, T.S.; Pacenka, S.; Gao, W.S.; Ma, L.; Zhang, M.; Sui, P. Mitigating groundwater depletion in North China Plain with cropping system that alternates deep and shallow rooted crops. Front. Plant Sci. 2017, 8, 980. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Sun, Y.; Wang, S.; Liu, Q.; Li, H. Photosynthesis product allocation and yield in sweet potato in response to different late-season irrigation levels. Plants 2023, 12, 1780. [Google Scholar] [CrossRef]
- Wu, Y.; Jin, X.; Wang, L.; Lei, J.; Chai, S.; Wang, C.; Zhang, W.; Yang, X. Integrated transcriptional and metabolomic analysis of factors influencing root tuber enlargement during early sweet potato development. Genes 2024, 15, 1319. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Fu, L.; Tie, W.; Yan, Y.; Wu, C.; Dai, J.; Zhang, J.; Hu, W. Highly dynamic, coordinated, and stage-specific profiles are revealed by a multi-omics integrative analysis during tuberous root development in cassava. J. Exp. Bot. 2020, 71, 7003–7017. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Li, R.; Ren, L.; Gao, X.; Zhang, Y.; Ma, Z.; Ma, D.; Luo, Y. A comparative metabolomics study of flavonoids in sweet potato with different flesh colors (Ipomoea batatas (L.) Lam). Food Chem. 2018, 260, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Wu, J.; Wang, X.; Cui, L.; Xiao, Q. Accumulation patterns of flavonoids and phenolic acids in different colored sweet potato flesh revealed based on untargeted metabolomics. Food Chem. X 2024, 23, 101551. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, J.; Su, Z.; Ma, H. Developmentally dependent reprogramming of the Arabidopsis floral transcriptome under sufficient and limited water availability. BMC Plant Biol. 2024, 24, 273. [Google Scholar] [CrossRef]
- Li, H.; Duijts, K.; Pasini, C.; van Santen, J.E.; Lamers, J.; de Zeeuw, T.; Verstappen, F.; Wang, N.; Zeeman, S.C.; Santelia, D.; et al. Effective root responses to salinity stress include maintained cell expansion and carbon allocation. New Phytol. 2023, 238, 1942–1956. [Google Scholar] [CrossRef]
- Li, S.; Zhao, L.; Sun, N.; Liu, Q.; Li, H. Photosynthesis product allocation and yield in sweetpotato with different irrigation levels at mid-season. Agric. Water Manag. 2021, 246, 106708. [Google Scholar] [CrossRef]
- Daryanto, S.; Wang, L.; Jacinthe, P.A. Drought effects on root and tuber production: A meta-analysis. Agric. Water Manag. 2016, 176, 122–131. [Google Scholar] [CrossRef]
- Yu, J.; Su, D.; Yang, D.; Dong, T.; Tang, Z.; Li, H.; Han, Y.; Li, Z.; Zhang, B. Chilling and heat stress-induced physiological changes and microRNA-related mechanism in sweetpotato (Ipomoea batatas L.). Front. Plant Sci. 2020, 11, 687. [Google Scholar] [CrossRef]
- Ravi, V.; Chakrabarti, S.K.; Makeshkumar, T.; Saravanan, R. Molecular regulation of storage root formation and development in sweet potato. In Horticultural Reviews; Janick, J., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014; Volume 42, pp. 157–208. [Google Scholar]
- Pietta, P.G. Flavonoids as antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Nahar, K.; Gill, S.S.; Fujita, M. Drought stress responses in plants, oxidative stress, and antioxidant defense. In Climate Change and Plant Abiotic Stress Tolerance; Tuteja, N., Gill, S.S., Eds.; Wiley: Weinheim, Germany, 2014; pp. 209–250. [Google Scholar]
- Abhishek, K.; Sardar, A.H.; Das, S.; Kumar, A.; Ghosh, A.K.; Singh, R.; Saini, S.; Mandal, A.; Verma, S.; Kumar, A.; et al. Phosphorylation of translation initiation factor 2-alpha in Leishmania donovani under stress is necessary for parasite survival. Mol. Cell Biol. 2017, 37, e00344-16. [Google Scholar] [CrossRef]
- Nicolas-Espinosa, J.; Garcia-Ibañez, P.; Lopez-Zaplana, A.; Yepes-Molina, L.; Albaladejo-Marico, L.; Carvajal, M. Confronting secondary metabolites with water uptake and transport in plants under abiotic stress. Int. J. Mol. Sci. 2023, 24, 2826. [Google Scholar] [CrossRef]
- Araguirang, G.E.; Venn, B.; Kelber, N.M.; Feil, R.; Lunn, J.; Kleine, T.; Leister, D.; Mühlhaus, T.; Richter, A.S. Spliceosomal complex components are critical for adjusting the C:N balance during high-light acclimation. Plant J. 2024, 119, 153–175. [Google Scholar] [CrossRef]
- Solis, J.; Villordon, A.; Baisakh, N.; LaBonte, D.; Firon, N. Effect of drought on storage root development and gene expression profile of sweetpotato under greenhouse and field conditions. J. Am. Soc. Hortic. Sci. 2014, 139, 317–324. [Google Scholar] [CrossRef]
- Makhubu, F.N.; Laurie, S.M.; Rauwane, M.E.; Figlan, S. Trends and gaps in sweet potato (Ipomoea batatas L.) improvement in sub-Saharan Africa: Drought tolerance breeding strategies. Food Energy Secur. 2024, 13, e545. [Google Scholar] [CrossRef]
- Mo, X.; Wang, L.; Yu, C.; Kou, C. Combined metabolomics and transcriptomics analysis of the distribution of flavonoids in the fibrous root and taproot of Polygonatum kingianum Coll. et Hemsl. Genes 2024, 15, 828. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.; Wilkinson, M.; Lowe, C.; Xu, J.; Hughes, D.; Hassall, K.L.; Hassani-Pak, K.; Amberkar, S.; Noleto-Dias, C.; Ward, J.; et al. Novel molecules and target genes for vegetative heat tolerance in wheat. Plant Environ. Interact. 2022, 3, 264–289. [Google Scholar] [CrossRef]
- Sarker, U.; Oba, S. Drought stress enhances nutritional and bioactive compounds, phenolic acids and antioxidant capacity of Amaranthus leafy vegetable. BMC Plant Biol. 2018, 18, 258. [Google Scholar] [CrossRef]
- Escobar-Puentes, A.A.; Palomo, I.; Rodríguez, L.; Fuentes, E.; Villegas-Ochoa, M.A.; González-Aguilar, G.A.; Olivas-Aguirre, F.J.; Wall-Medrano, A. Sweet potato (Ipomoea batatas L.) phenotypes: From agroindustry to health effects. Foods 2022, 11, 1058. [Google Scholar] [CrossRef]
- Huang, X.; Chu, G.; Wang, J.; Luo, H.; Yang, Z.; Sun, L.; Rong, W.; Wang, M. Integrated metabolomic and transcriptomic analysis of specialized metabolites and isoflavonoid biosynthesis in Sophora alopecuroides L. under different degrees of drought stress. Ind. Crops Prod. 2023, 197, 116595. [Google Scholar] [CrossRef]
- Fiehn, O.; Wohlgemuth, G.; Scholz, M.; Kind, T.; Lee, D.Y.; Lu, Y.; Moon, S.; Nikolau, B. Quality control for plant metabolomics: Reporting MSI-compliant studies. Plant J. 2008, 53, 691–704. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, D.; Li, D.; Ji, Q.; Gao, J.; Hou, F. Metagenomic and metabolite analysis reveals microbial community and metabolite dynamics in fermented Indigo naturalis. Heliyon 2024, 10, 17. [Google Scholar] [CrossRef]
- Wang, M.; Wang, C.; Han, X. Selection of internal standards for accurate quantification of complex lipid species in biological extracts by electrospray ionization mass spectrometry—What, how and why? Mass Spectrom. Rev. 2017, 36, 693–714. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, J.; Zhu, Y.; Zhang, Y.; Zhu, J.; Ming, Y. Metabolomics analysis of patients with Schistosoma japonicum infection based on UPLC-MS method. Parasites Vectors 2024, 17, 350. [Google Scholar] [CrossRef]
- Marco-Ramell, A.; Palau-Rodriguez, M.; Alay, A.; Tulipani, S.; Urpi-Sarda, M.; Sanchez-Pla, A.; Andres-Lacueva, C. Evaluation and comparison of bioinformatic tools for the enrichment analysis of metabolomics data. BMC Bioinform. 2018, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Ichihashi, Y.; Kimura, S.; Chitwood, D.H.; Headland, L.R.; Peng, J.; Maloof, J.N.; Sinha, N.R. A high-throughput method for Illumina RNA-Seq library preparation. Front. Plant Sci. 2012, 3, 202. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Internet]. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 November 2024).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Jiang, M.; Li, H. High-throughput sequencing analysis of Euphorbia fischeriana Steud provides insights into the molecular mechanism of pharmaceutical ingredient biosynthesis. 3 Biotech 2018, 8, 449. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, T.; Song, C.; Li, H.; Wang, S.; Wei, W.; Meng, J.; Liu, Q. Unveiling Stage-Specific Flavonoid Dynamics Underlying Drought Tolerance in Sweet Potato (Ipomoea batatas L.) via Integrative Transcriptomic and Metabolomic Analyses. Plants 2025, 14, 2383. https://doi.org/10.3390/plants14152383
Yin T, Song C, Li H, Wang S, Wei W, Meng J, Liu Q. Unveiling Stage-Specific Flavonoid Dynamics Underlying Drought Tolerance in Sweet Potato (Ipomoea batatas L.) via Integrative Transcriptomic and Metabolomic Analyses. Plants. 2025; 14(15):2383. https://doi.org/10.3390/plants14152383
Chicago/Turabian StyleYin, Tao, Chaoyu Song, Huan Li, Shaoxia Wang, Wenliang Wei, Jie Meng, and Qing Liu. 2025. "Unveiling Stage-Specific Flavonoid Dynamics Underlying Drought Tolerance in Sweet Potato (Ipomoea batatas L.) via Integrative Transcriptomic and Metabolomic Analyses" Plants 14, no. 15: 2383. https://doi.org/10.3390/plants14152383
APA StyleYin, T., Song, C., Li, H., Wang, S., Wei, W., Meng, J., & Liu, Q. (2025). Unveiling Stage-Specific Flavonoid Dynamics Underlying Drought Tolerance in Sweet Potato (Ipomoea batatas L.) via Integrative Transcriptomic and Metabolomic Analyses. Plants, 14(15), 2383. https://doi.org/10.3390/plants14152383