
Genome-Wide Identification of DNA Methyltransferase and Demethylase in Populus sect. Turanga and Their Potential Roles in Heteromorphic Leaf Development in Populus euphratica

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
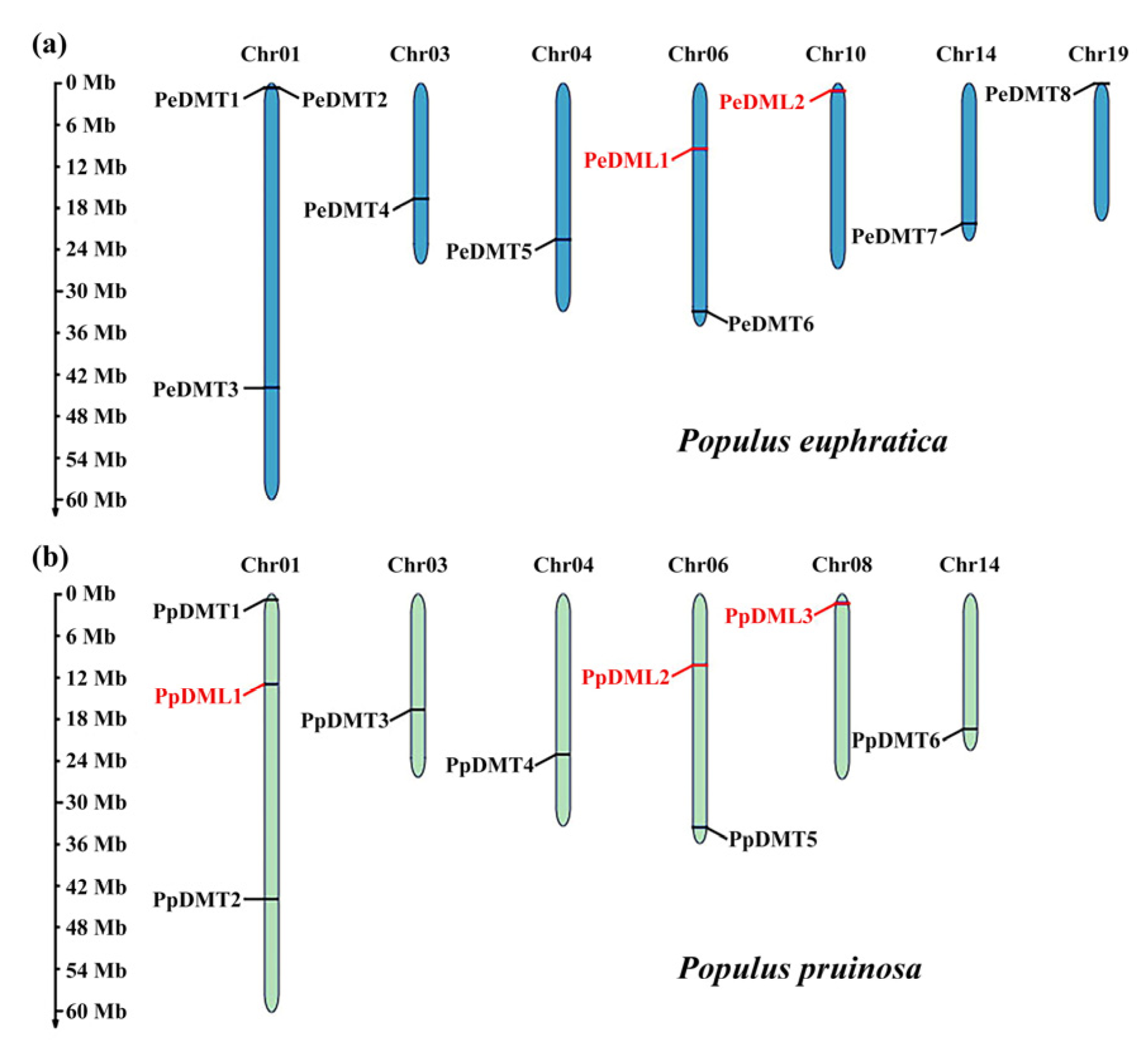
2.1. Identification and Chromosomal Classification of DMTs and DMLs
2.2. Protein Structure and Subcellular Localization Prediction
2.3. Phylogenetic Relationship of DMTs and DMLs
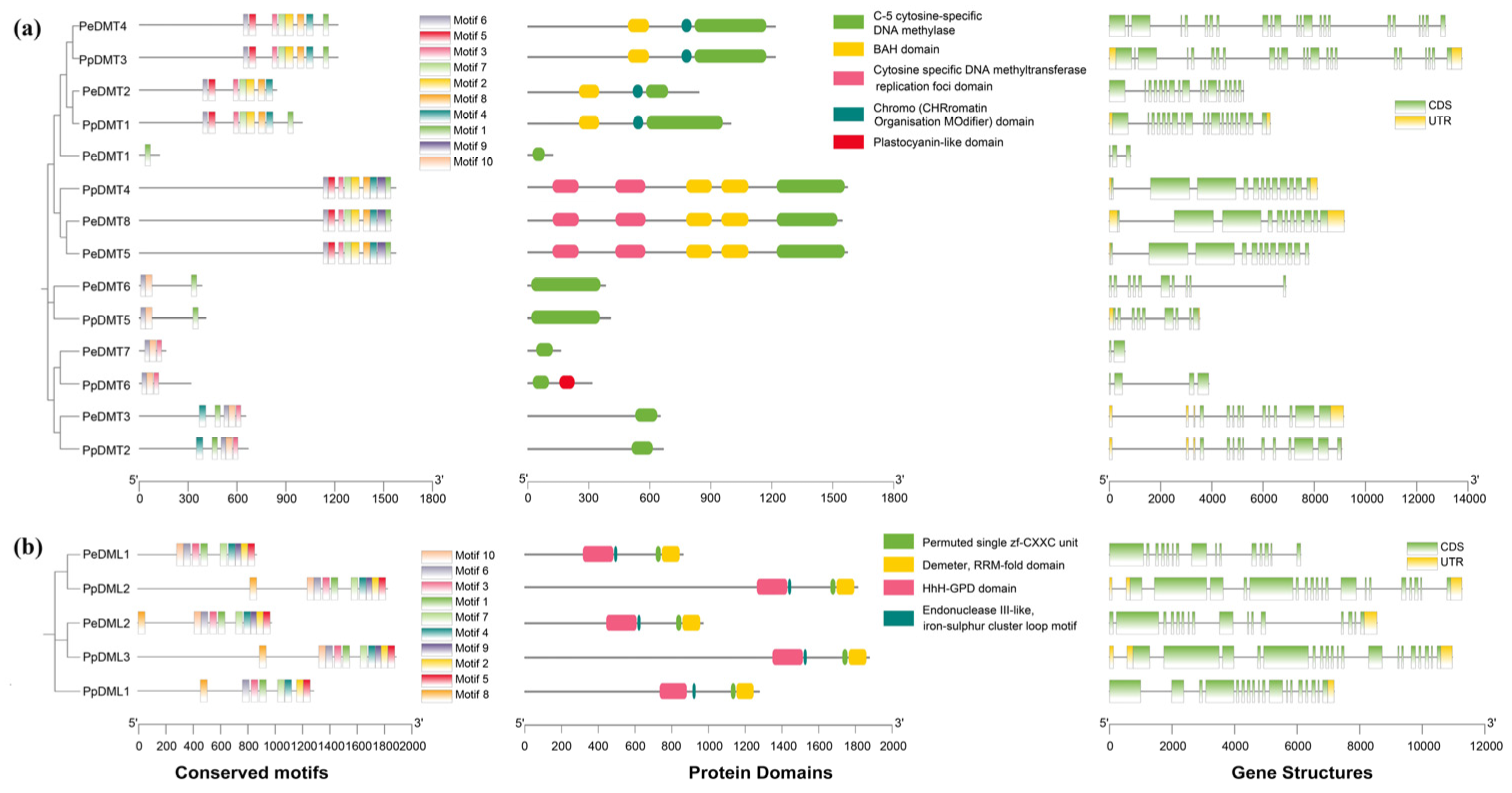
2.4. Conserved Motifs, Protein Domains, and Gene Structures of DMTs and DMLs
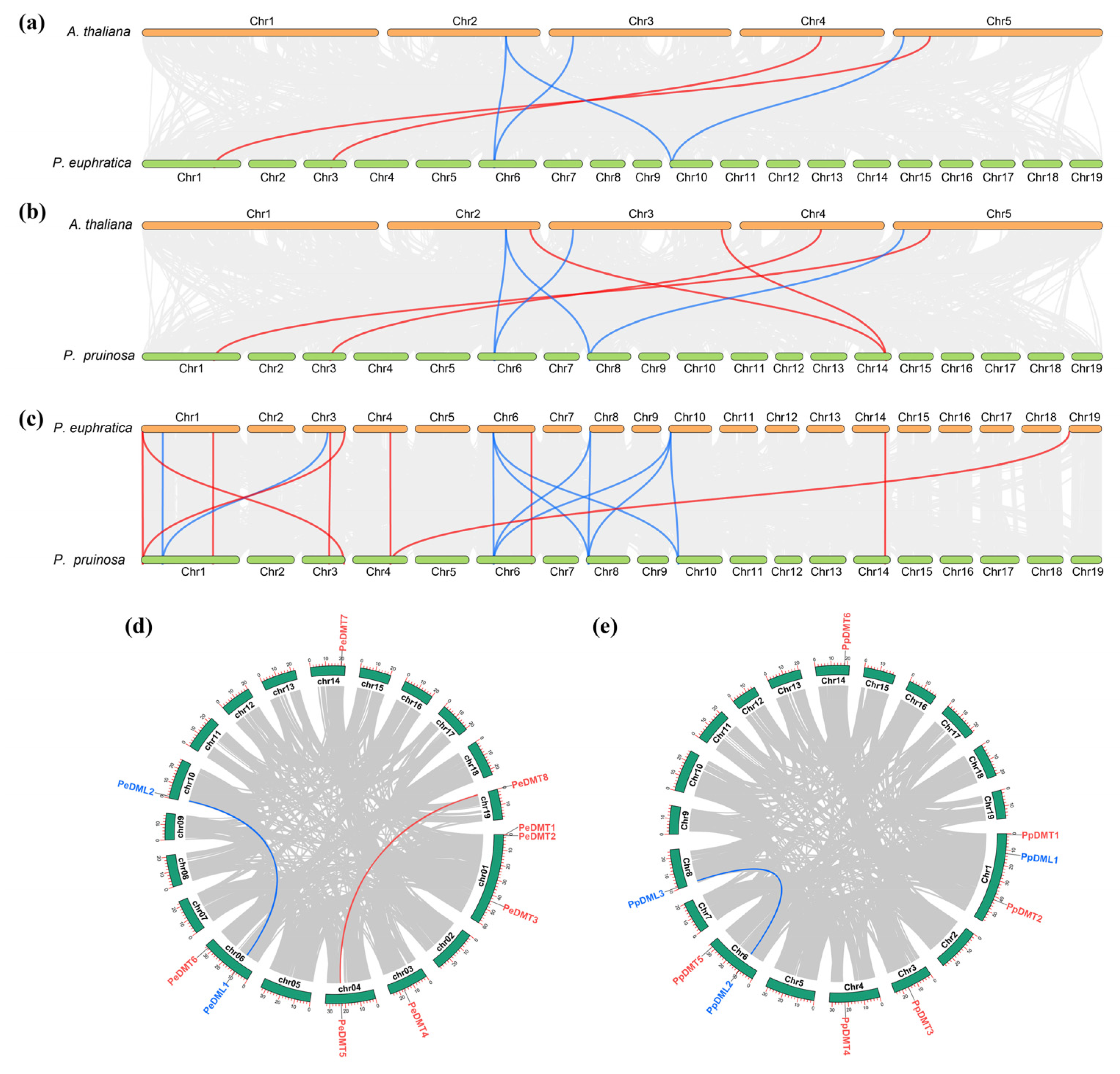
2.5. Interspecific and Intraspecific Collinearity Analysis of DMTs and DMLs Among Three Species
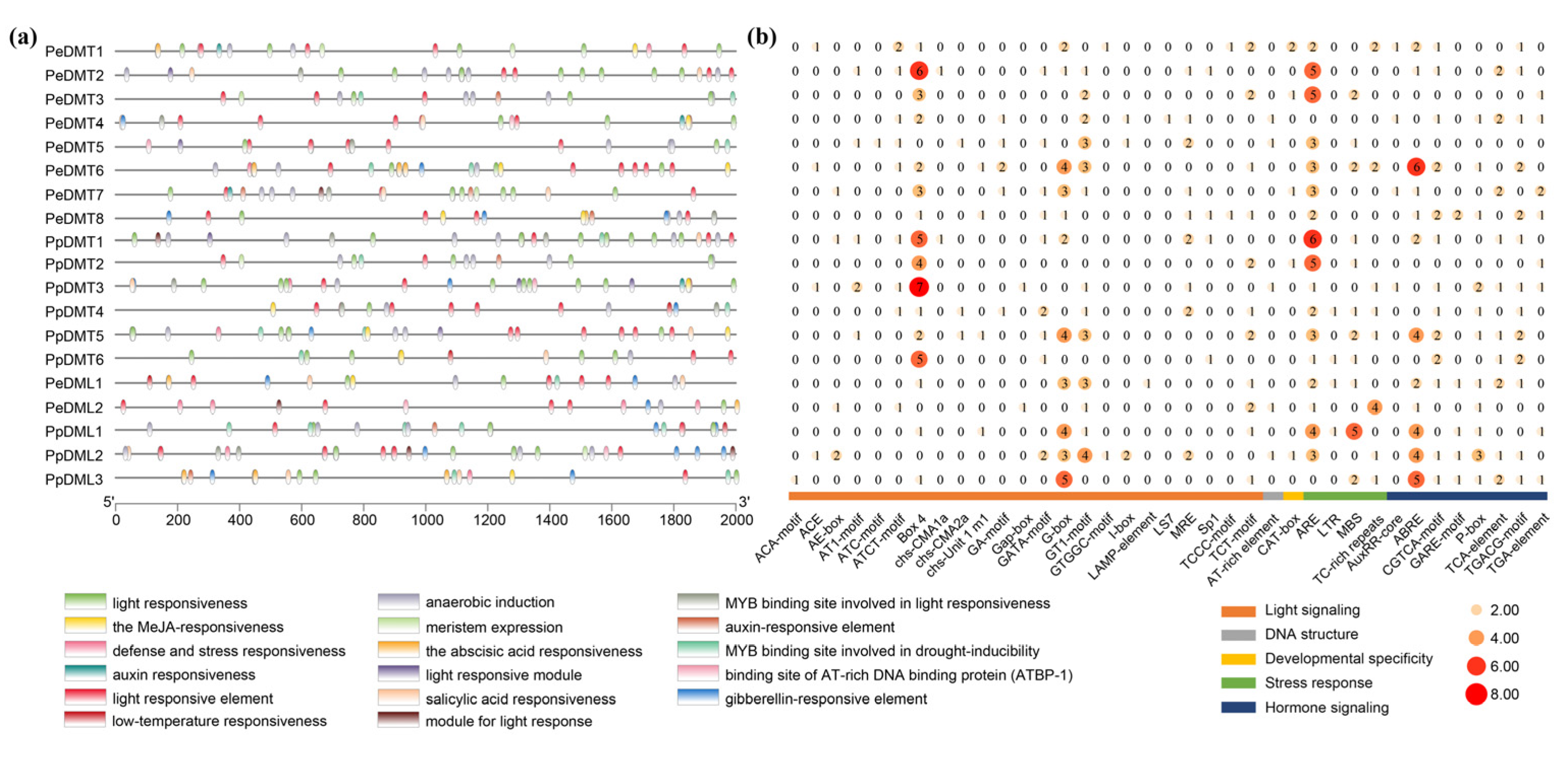
2.6. Analysis of the Cis-Acting Elements in the Promoter Region of DMTs and DMLs
2.7. Expression Pattern of PeDMTs and PeDMLs During the Development of Heteromorphic Leaves of P. euphratica
2.8. The Correlation Between DNA Methylation and Expression of PeDMTs/DMLs During the Development of Heteromorphic Leaves of P. euphratica
3. Discussion
4. Materials and Methods
4.1. Identification of the DMT and DML Gene Families
4.2. Protein Tertiary Structure Prediction and Subcellular Localization Analysis
4.3. Construction and Analysis of the Evolutionary Tree
4.4. Analysis of Gene Structure and Conserved Motif
4.5. Analysis of Cis-Acting Elements and Collinearity
4.6. Analysis of RNA-Seq and WGBS Data
4.7. qRT-PCR Validation
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DMT | DNA methyltransferase |
| DML | DNA demethylase |
| MET | methyltransferase |
| CMT | chromomethylase |
| DRM | domains rearranged methylase |
| DNMT2 | DNA methyltransferase 2 |
| ROS1 | repressor of silence 1 |
| DME | transcriptional activator demeter |
| DML2 | demeter-like protein 2 |
| DML3 | demeter-like protein 3 |
| HMM | Hidden Markov Model |
| GRAVY | grand average of hydrophobicity |
| Li | linear |
| La | lanceolate |
| Ov | ovate |
| Bo | broad-ovate |
| TPM | transcripts per million |
| MW | molecular weight |
| pI | isoelectric point |
| CDS | coding sequence |
References
- Zhong, X.; Du, J.; Hale, C.J.; Gallego-Bartolome, J.; Feng, S.; Vashisht, A.A.; Chory, J.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 2014, 157, 1050–1060. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef]
- Xiao, K.; Chen, J.; He, Q.; Wang, Y.; Shen, H.; Sun, L. DNA methylation is involved in the regulation of pepper fruit ripening and interacts with phytohormones. J. Exp. Bot. 2020, 71, 1928–1942. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 201–220. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef]
- Liu, R.; Lang, Z. The mechanism and function of active DNA demethylation in plants. J. Integr. Plant Biol. 2020, 62, 148–159. [Google Scholar] [CrossRef]
- Pavlopoulou, A.; Kossida, S. Plant cytosine-5 DNA methyltransferases: Structure, function, and molecular evolution. Genomics 2007, 90, 530–541. [Google Scholar] [CrossRef]
- Greb-Markiewicz, B.; Orłowski, M.; Dobrucki, J.; Ożyhar, A. Sequences that direct subcellular traffic of the Drosophila methoprene-tolerant protein (MET) are located predominantly in the PAS domains. Mol. Cell Endocrinol. 2011, 345, 16–26. [Google Scholar] [CrossRef]
- Clark, T.A.; Murray, I.A.; Morgan, R.D.; Kislyuk, A.O.; Spittle, K.E.; Boitano, M.; Fomenkov, A.; Roberts, R.J.; Korlach, J. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 2012, 40, e29. [Google Scholar] [CrossRef]
- Du, J.; Johnson, L.M.; Groth, M.; Feng, S.; Hale, C.J.; Li, S.; Vashisht, A.A.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Mechanism of DNA methylation-directed histone methylation by KRYPTONITE. Mol. Cell 2014, 55, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Hatlen, A.; Kelly, L.J.; Becher, H.; Wang, W.; Kovarik, A.; Leitch, I.J.; Leitch, A.R. Angiosperms Are Unique among Land Plant Lineages in the Occurrence of Key Genes in the RNA-Directed DNA Methylation (RdDM) Pathway. Genome Biol. Evol. 2015, 7, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Deleris, A.; Halter, T.; Navarro, L. DNA Methylation and Demethylation in Plant Immunity. Annu. Rev. Phytopathol. 2016, 54, 579–603. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, G.; Silva, J.A.T.d.; Li, M.; Zhao, C.; He, C.; Si, C.; Zhang, M.; Duan, J. Genome-wide identification and analysis of DNA methyltransferase and demethylase gene families in Dendrobium officinale reveal their potential functions in polysaccharide accumulation. BMC Plant Biol. 2021, 21, 21. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, S.; Zhou, C.; Chen, L.; Fu, H.; Li, X.; Lin, Y.; Lai, Z.; Guo, Y. Genome-wide investigation and transcriptional analysis of cytosine-5 DNA methyltransferase and DNA demethylase gene families in tea plant (Camellia sinensis) under abiotic stress and withering processing. PeerJ 2020, 8, e8432. [Google Scholar] [CrossRef]
- Malabarba, J.; Windels, D.; Xu, W.; Verdier, J. Regulation of DNA (de)Methylation Positively Impacts Seed Germination during Seed Development under Heat Stress. Genes 2021, 12, 457. [Google Scholar] [CrossRef]
- Bharti, P.; Mahajan, M.; Vishwakarma, A.K.; Bhardwaj, J.; Yadav, S.K. AtROS1 overexpression provides evidence for epigenetic regulation of genes encoding enzymes of flavonoid biosynthesis and antioxidant pathways during salt stress in transgenic tobacco. J. Exp. Bot. 2015, 66, 5959–5969. [Google Scholar] [CrossRef]
- Yang, X.; Lu, X.; Chen, X.; Wang, D.; Wang, J.; Wang, S.; Guo, L.; Chen, C.; Wang, X.; Wang, X.; et al. Genome-wide identification and expression analysis of DNA demethylase family in cotton. J. Cotton Res. 2019, 2, 16. [Google Scholar] [CrossRef]
- Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J.-K.; Lang, Z. Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. USA 2019, 116, 1430–1436. [Google Scholar] [CrossRef]
- Liu, R.; How-Kit, A.; Stammitti, L.; Teyssier, E.; Rolin, D.; Mortain-Bertrand, A.; Halle, S.; Liu, M.; Kong, J.; Wu, C.; et al. A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc. Natl. Acad. Sci. USA 2015, 112, 10804–10809. [Google Scholar] [CrossRef]
- Liu, J.; Wu, X.; Yao, X.; Yu, R.; Larkin, P.J.; Liu, C.-M. Mutations in the DNA demethylase OsROS1 result in a thickened aleurone and improved nutritional value in rice grains. Proc. Natl. Acad. Sci. USA 2018, 115, 11327–11332. [Google Scholar] [CrossRef]
- Moritoh, S.; Eun, C.-H.; Ono, A.; Asao, H.; Okano, Y.; Yamaguchi, K.; Shimatani, Z.; Koizumi, A.; Terada, R. Targeted disruption of an orthologue of DOMAINS REARRANGED METHYLASE 2, OsDRM2, impairs the growth of rice plants by abnormal DNA methylation. Plant J. 2012, 71, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Tarutani, Y.; Miyao, A.; Ito, T.; Yamazaki, M.; Sakai, H.; Fukai, E.; Hirochika, H. Loss of function mutations in the rice chromomethylase OsCMT3a cause a burst of transposition. Plant J. 2015, 83, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Jacobsen, S.E. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl. Acad. Sci. USA 2002, 9 (Suppl. 4), 16491–16498. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Gao, C.; Bian, X.; Zhao, S.; Zhao, C.; Xia, H.; Song, H.; Hou, L.; Wan, S.; Wang, X. Genome-Wide Identification and Comparative Analysis of Cytosine-5 DNA Methyltransferase and Demethylase Families in Wild and Cultivated Peanut. Front. Plant Sci. 2016, 7, 7. [Google Scholar] [CrossRef]
- Yang, S.; Qiao, S.; Yang, Y.; Wang, F.; Song, W.; Tan, W.; Li, Y.; Zhu, Y. Genome-Wide Identification and Analysis of DNA Methyltransferase and Demethylase Gene Families in Sweet Potato and Its Diploid Relative. Plants 2025, 14, 1735. [Google Scholar] [CrossRef]
- Conde, D.; Moreno-Cortés, A.; Dervinis, C.; Ramos-Sánchez, J.M.; Kirst, M.; Perales, M.; González-Melendi, P.; Allona, I. Overexpression of DEMETER, a DNA demethylase, promotes early apical bud maturation in poplar. Plant Cell Environ. 2017, 40, 2806–2819. [Google Scholar] [CrossRef]
- Wang, Q.; Qu, Y.; Yu, Y.; Mao, X.; Fu, X. Genome-wide identification and comparative analysis of DNA methyltransferase and demethylase gene families in two ploidy Cyclocarya paliurus and their potential function in heterodichogamy. BMC Genom. 2023, 24, 287. [Google Scholar] [CrossRef]
- Wu, Z.; Jiang, Z.; Li, Z.; Jiao, P.; Zhai, J.; Liu, S.; Han, X.; Zhang, S.; Sun, J.; Gai, Z.; et al. Multi-omics analysis reveals spatiotemporal regulation and function of heteromorphic leaves in Populus. Plant Physiol. 2023, 192, 188–204. [Google Scholar] [CrossRef]
- Wang, J.; Xu, J.; Sun, J.; Qiu, C.; Wu, Z.; Li, Z. Genome-wide identification of TCP transcription factors family in Populus sect. Turanga (Populus pruinosa Schrenk and Populus euphratica Olive) reveal the roles of TCPS in leaf morphology. Appl. Ecol. Environ. Res. 2023, 21, 1665–1696. [Google Scholar]
- Sun, J.; Xu, J.; Qiu, C.; Zhai, J.; Zhang, S.; Zhang, X.; Wu, Z.; Li, Z. The chromosome-scale genome and population genomics reveal the adaptative evolution of Populus pruinosa to desertification environment. Hortic. Res. 2024, 11, uhae034. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, X.; Chen, G.; Li, M.; Liu, M.; Liu, D. Epidermal Micromorphology and Mesophyll Structure of Populus euphratica Heteromorphic Leaves at Different Development Stages. PLoS ONE 2015, 10, e0137701. [Google Scholar]
- Hao, J.; Yue, N.; Zheng, C. Analysis of changes in anatomical characteristics and physiologic features of heteromorphic leaves in a desert tree, Populus euphratica. Acta Physiol. Plant 2017, 39, 160. [Google Scholar] [CrossRef]
- Zhai, J.T.; Li, Y.L.; Han, Z.J.; Li, Z.J. Morphological, structural and physiological differences in heteromorphic leaves of Euphrates poplar during development stages and at crown scales. Plant Biol. 2020, 22, 366–375. [Google Scholar] [CrossRef]
- Li, Z.; Jiao, P.; Wu, Z.; Zhai, J.; Zhang, X.; Zhang, S.; Gai, Z.; Guo, X. Heterophylly and Growth Adaptation Strategies of Populus euphratica and Populus pruinosa; Science Press: Beijing, China, 2021. [Google Scholar]
- Ma, T.; Wang, K.; Hu, Q.; Xi, Z.; Wan, D.; Wang, Q.; Feng, J.; Jiang, D.; Ahani, H.; Abbott, R.J.; et al. Ancient polymorphisms and divergence hitchhiking contribute to genomic islands of divergence within a poplar species complex. Proc. Natl. Acad. Sci. USA 2018, 115, E236–E243. [Google Scholar] [CrossRef]
- Jia, H.; Liu, G.; Li, J.; Zhang, J.; Sun, P.; Zhao, S.; Zhou, X.; Lu, M.; Hu, J. Genome resequencing reveals demographic history and genetic architecture of seed salinity tolerance in Populus euphratica. J. Exp. Bot. 2020, 71, 4308–4320. [Google Scholar] [CrossRef]
- Gai, Z.; Zhai, J.; Chen, X.; Jiao, P.; Zhang, S.; Sun, J.; Qin, R.; Liu, H.; Wu, Z.; Li, Z. Phylogeography Reveals Geographic and Environmental Factors Driving Genetic Differentiation of Populus sect. Turanga in Northwest China. Front. Plant Sci. 2021, 12, 705083. [Google Scholar] [CrossRef]
- Ogneva, Z.V.; Dubrovina, A.S.; Kiselev, K.V. Age-associated alterations in DNA methylation and expression of methyltransferase and demethylase genes in Arabidopsis thaliana. Biol. Plant 2016, 60, 628–634. [Google Scholar] [CrossRef]
- Gu, T.; Ren, S.; Wang, Y.; Han, Y.; Li, Y. Characterization of DNA methyltransferase and demethylase genes in Fragaria vesca. Mol. Genet. Genom. 2016, 291, 1333–1345. [Google Scholar] [CrossRef]
- Peng, Z.; Xu, K.; Zhou, K.; Tang, D. Identification and Biological Information Analysis of Phyllostachys edulis DNMT Gene Family. Mol. Plant Breed. 2022, 23, 4630–4636. [Google Scholar]
- Wei, X.; Sun, Q.; Xia, W.; Chen, R.; Xiao, Y. Bioinformatics Analysis of Genes Associated with Coconut DNMT Family. Mol. Plant Breed. 2022, 21, 5967–5973. [Google Scholar]
- Gahlaut, V.; Samtani, H.; Gautam, T.; Khurana, P. Identification and Characterization of DNA Demethylase Genes and Their Association With Thermal Stress in Wheat (Triticum aestivum L.). Front. Genet. 2022, 13, 894020. [Google Scholar] [CrossRef] [PubMed]
- SWL, C.; IR, H.; SE, J. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [PubMed]
- Silva, H.G.; Sobral, R.S.; Magalhães, A.P.; Morais-Cecílio, L.; Costa, M.M.R. Genome-Wide Identification of Epigenetic Regulators in Quercus suber L. Int. J. Mol. Sci. 2020, 21, 3783. [Google Scholar] [CrossRef]
- Qiu, C.; Liu, S.; Sun, J.; Gai, Z.; Han, X.; Jiao, P.; Zhai, J.; Yang, Y.; Jiang, Z.; Liu, H.; et al. DNA methylation profile revealed the dynamically epigenetic regulation of the distinct heteromorphic leaf development in Populus euphratica. Ind. Crop. Prod. 2024, 216, 118688. [Google Scholar] [CrossRef]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.-K. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, Z.; Ma, D.; Zhai, J.; Han, X.; Jiang, Z.; Liu, S.; Xu, J.; Jiao, P.; Li, Z. Chromosome-scale assemblies of the male and female Populus euphratica genomes reveal the molecular basis of sex determination and sexual dimorphism. Commun. Biol. 2022, 5, 1186. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.e.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005. [Google Scholar]
- Chao, J.; Li, Z.; Sun, Y.; Aluko, O.O.; Wu, X.; Wang, Q.; Liu, G. MG2C: A user-friendly online tool for drawing genetic maps. Mol. Hortic. 2021, 1, 16. [Google Scholar] [CrossRef]
- Geourjon, C.; Deléage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.d.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024, 52, W78–W82. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Peer, Y.V.d.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, C.; Sun, J.; Jia, M.; Han, X.; Song, J.; Gai, Z.; Li, Z. Genome-Wide Identification of DNA Methyltransferase and Demethylase in Populus sect. Turanga and Their Potential Roles in Heteromorphic Leaf Development in Populus euphratica. Plants 2025, 14, 2370. https://doi.org/10.3390/plants14152370
Qiu C, Sun J, Jia M, Han X, Song J, Gai Z, Li Z. Genome-Wide Identification of DNA Methyltransferase and Demethylase in Populus sect. Turanga and Their Potential Roles in Heteromorphic Leaf Development in Populus euphratica. Plants. 2025; 14(15):2370. https://doi.org/10.3390/plants14152370
Chicago/Turabian StyleQiu, Chen, Jianhao Sun, Mingyu Jia, Xiaoli Han, Jia Song, Zhongshuai Gai, and Zhijun Li. 2025. "Genome-Wide Identification of DNA Methyltransferase and Demethylase in Populus sect. Turanga and Their Potential Roles in Heteromorphic Leaf Development in Populus euphratica" Plants 14, no. 15: 2370. https://doi.org/10.3390/plants14152370
APA StyleQiu, C., Sun, J., Jia, M., Han, X., Song, J., Gai, Z., & Li, Z. (2025). Genome-Wide Identification of DNA Methyltransferase and Demethylase in Populus sect. Turanga and Their Potential Roles in Heteromorphic Leaf Development in Populus euphratica. Plants, 14(15), 2370. https://doi.org/10.3390/plants14152370