
Transcriptomic Profiling of Buds Unveils Insights into Floral Initiation in Tea-Oil Tree (Camellia oleifera ‘changlin53’)

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
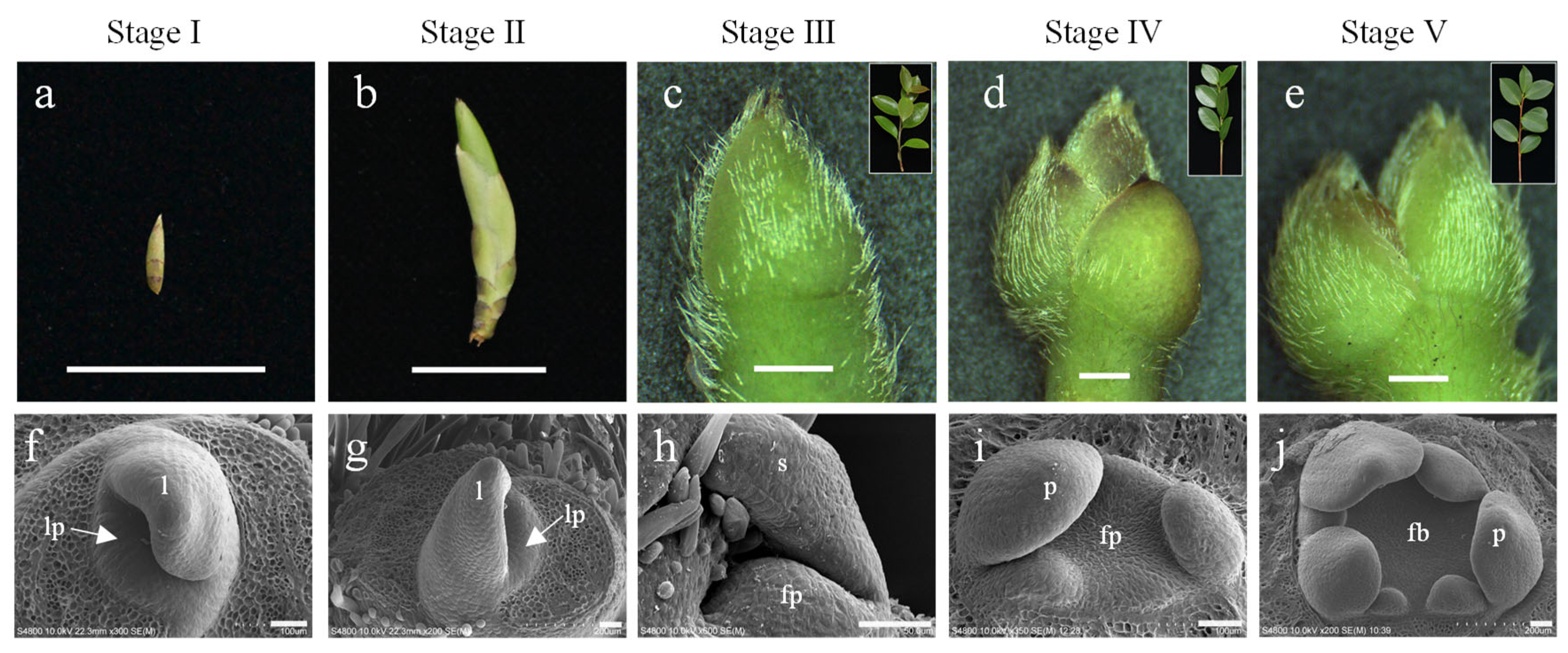
2.1. Morphological Characteristics of Floral Initiation in C. oleifera at Different Developmental Stages
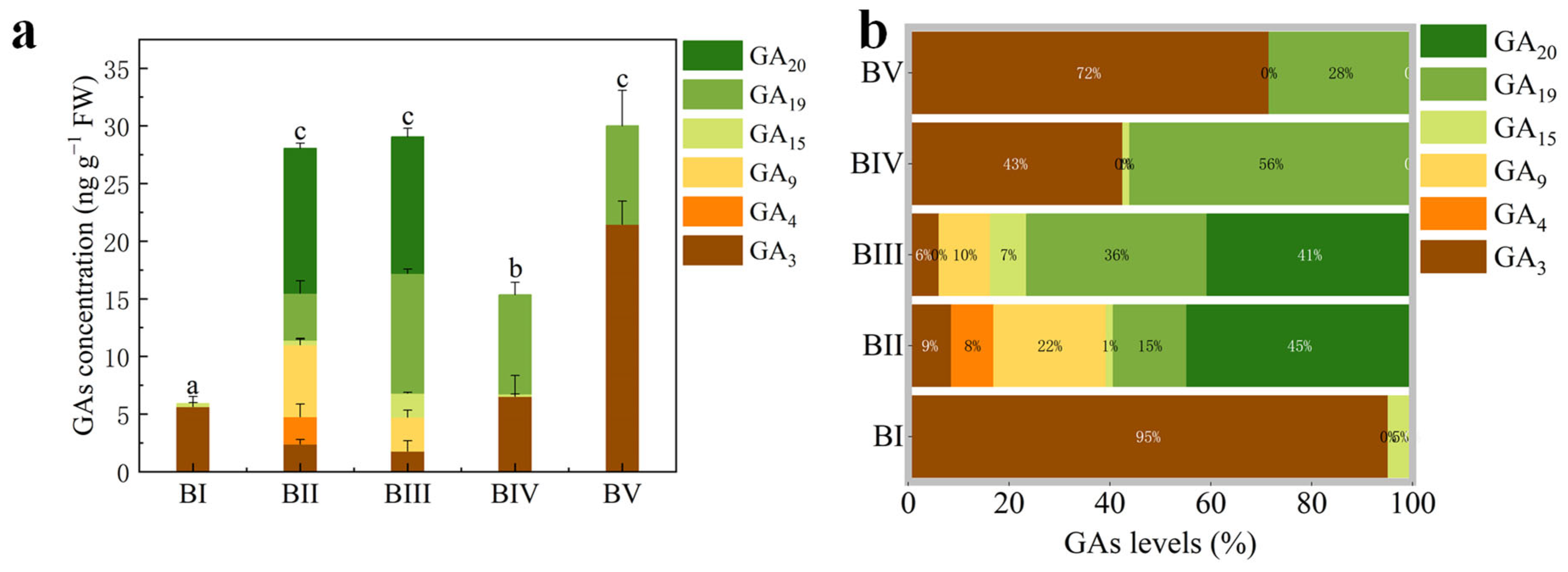
2.2. Quantitative Analysis of Endogenous Gibberellins in Buds at Different Developmental Stages
2.3. Transcriptome Assembly and Annotation
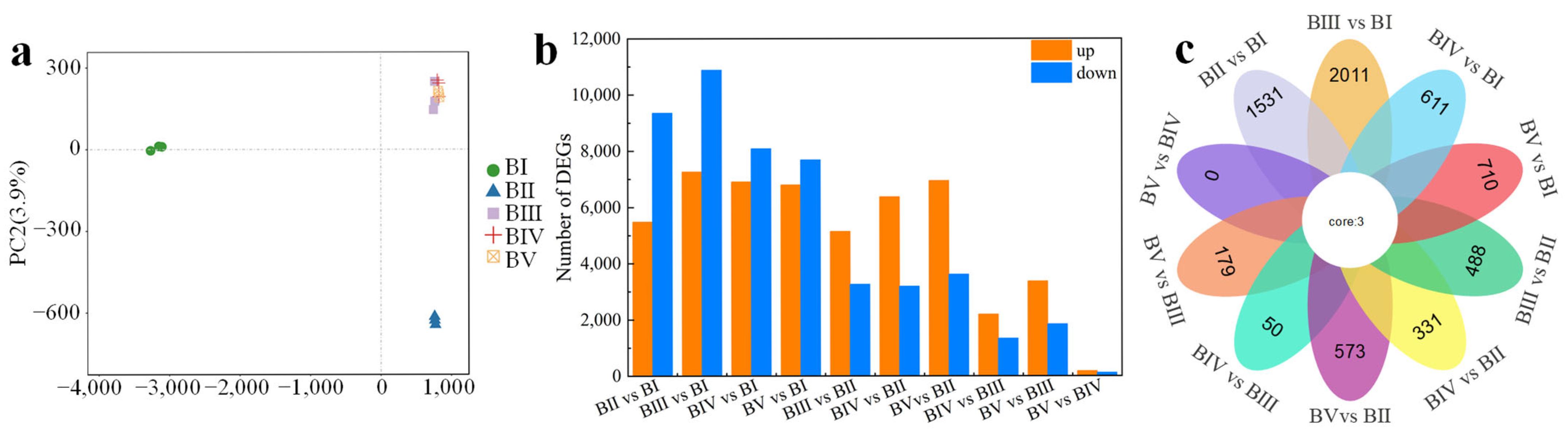
2.4. Differentially Expressed Genes at Different Developmental Stages
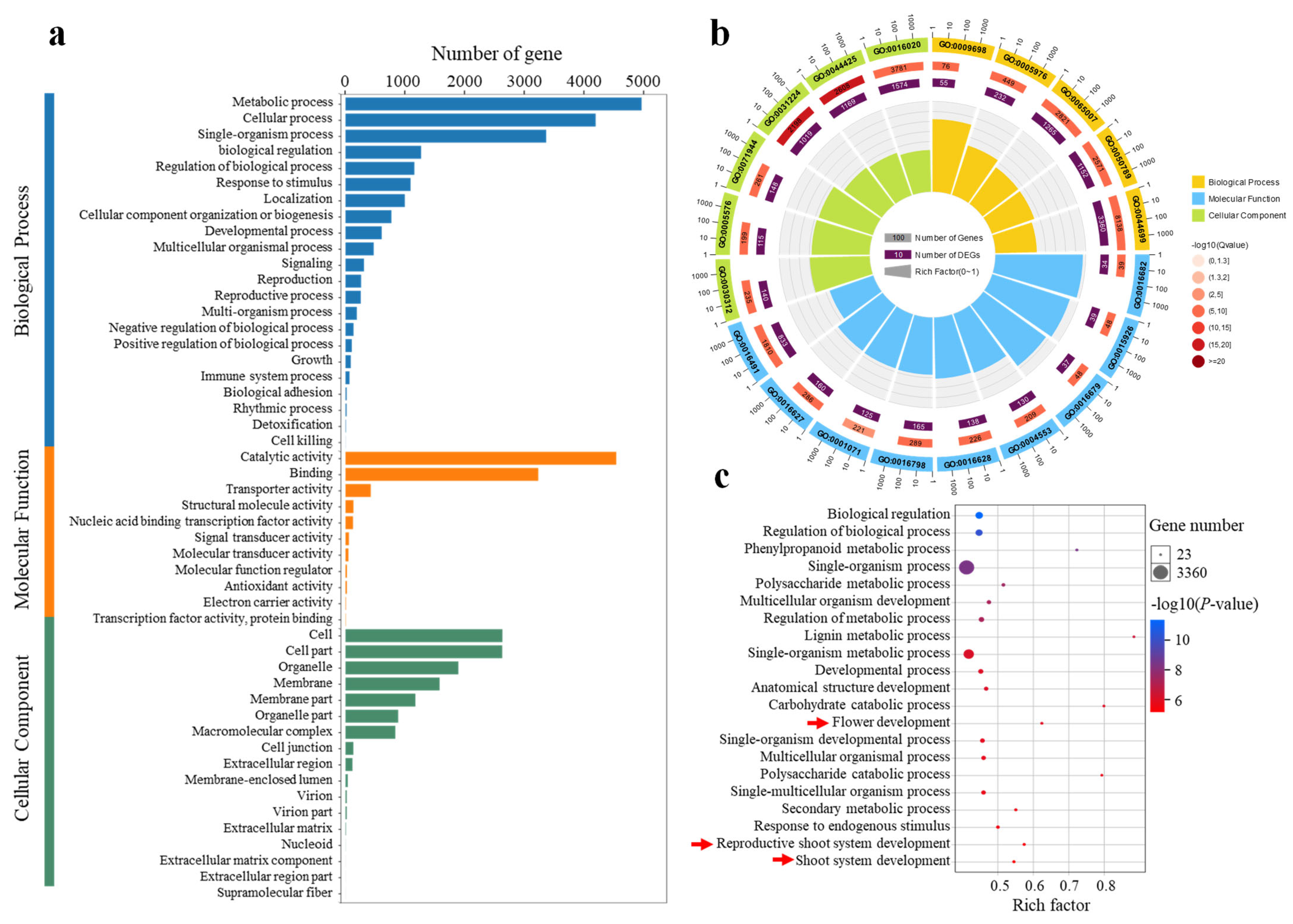
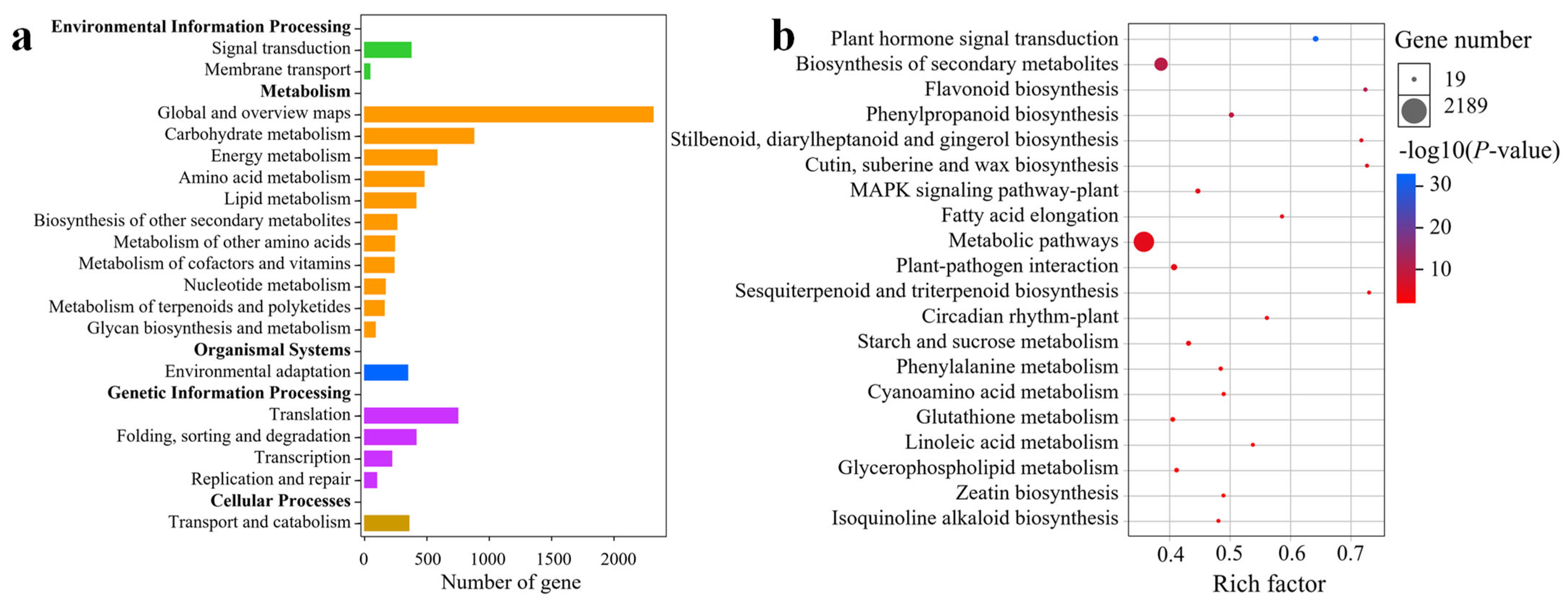
2.5. GO Enrichment Analysis of DEGs
2.6. KEGG Enrichment Analysis of DEGs
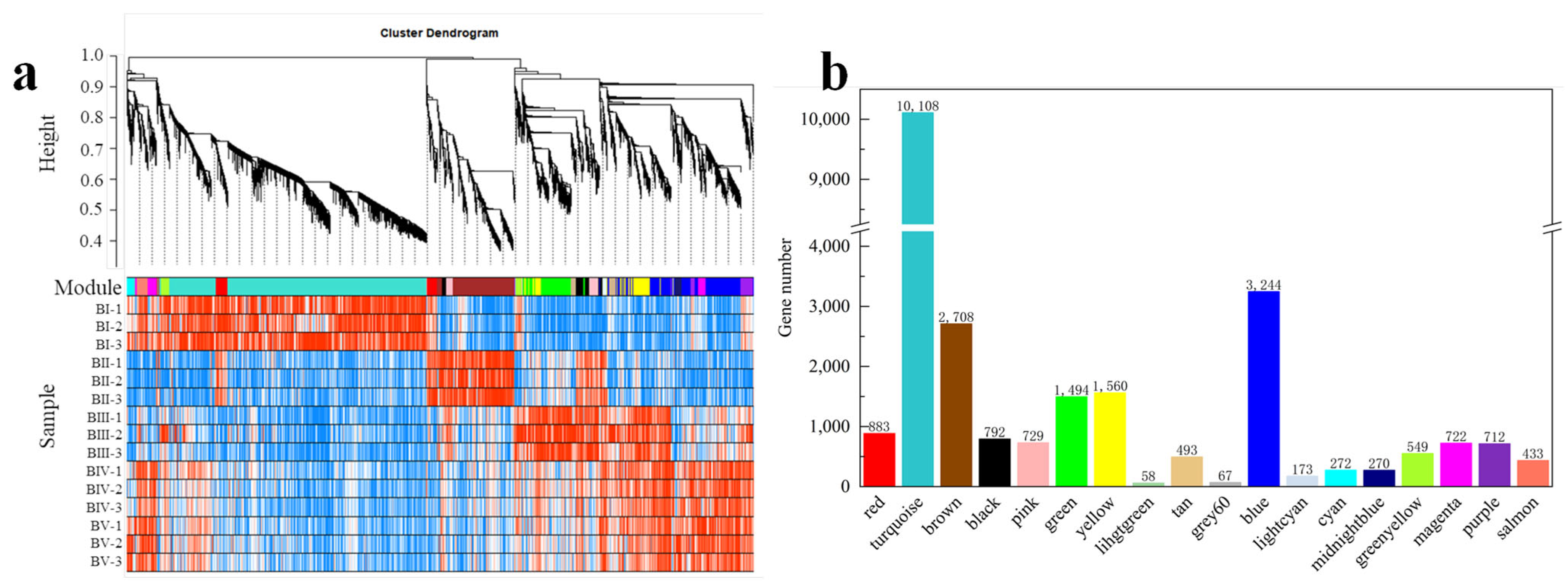
2.7. WGCNA Analysis of DEGs
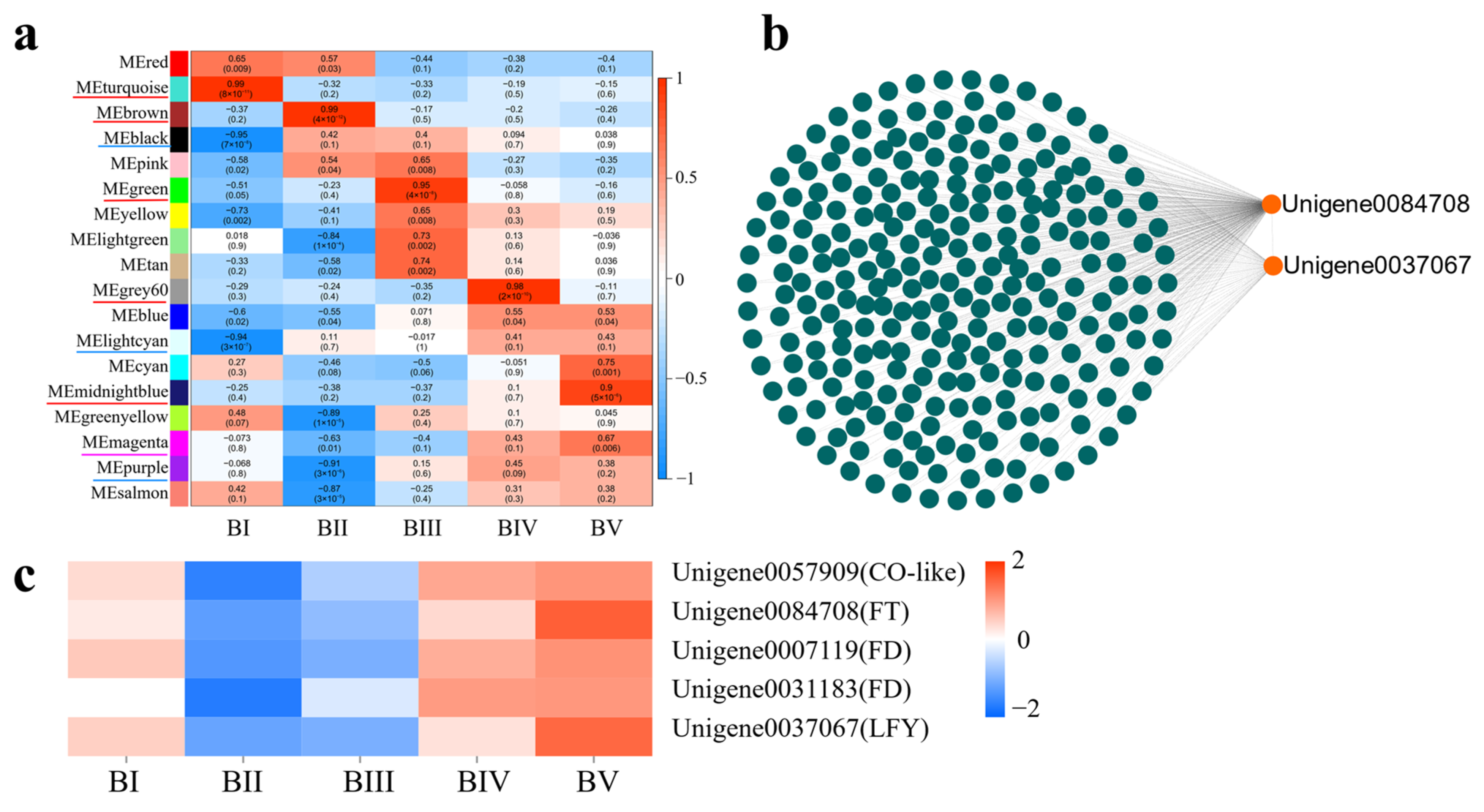
2.8. Identification of Core DEGs at Each Developmental Stage
2.9. Identification of Core DEGs Related to Floral Initiation During Whole Floral Initiation
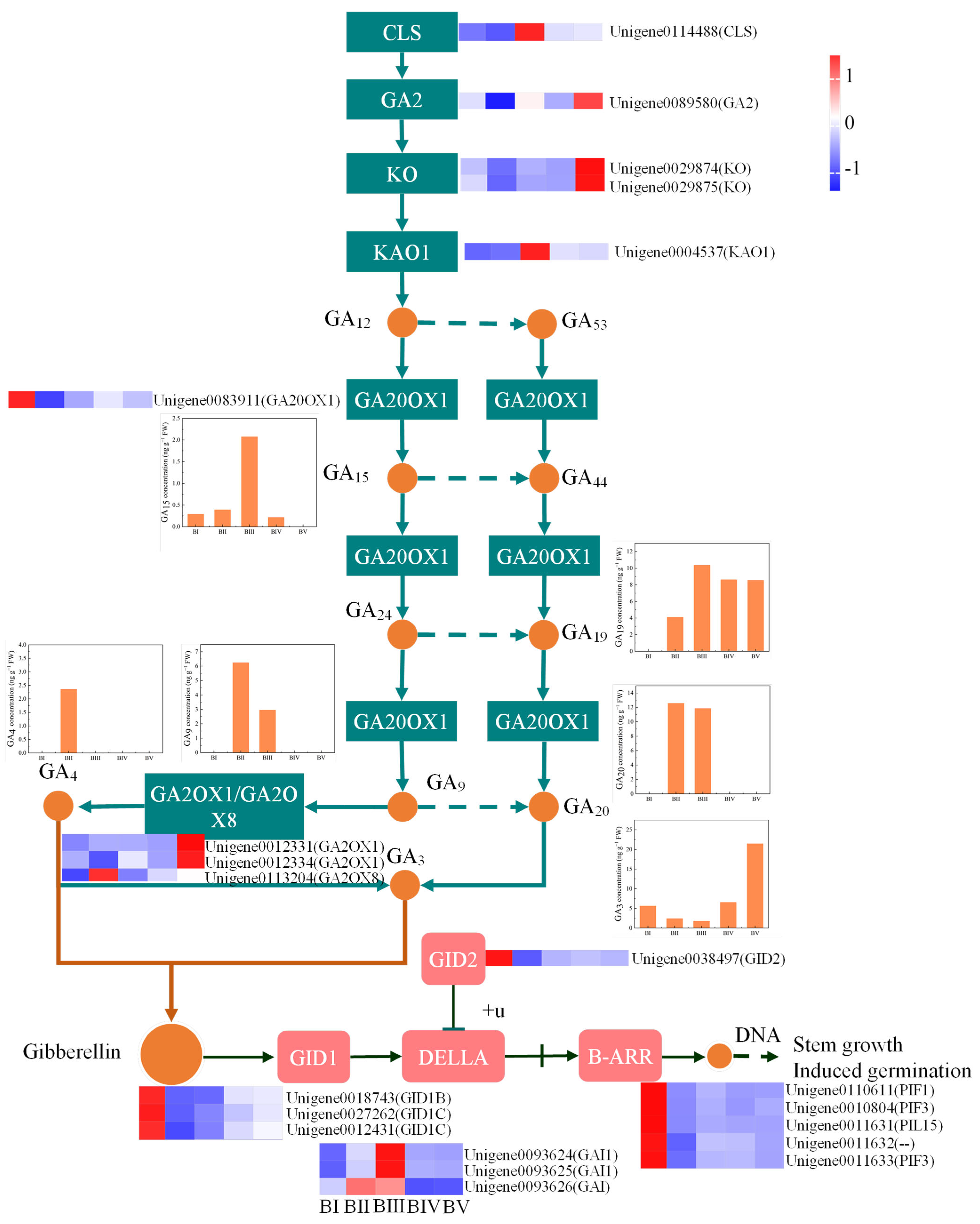
2.10. Identification of the DEGs Involved in Gibberellin Biosynthesis and Signal Transduction
2.11. Quantitative Real-Time PCR Analysis
3. Discussion
3.1. Reproductive Shoot Development Is a Key Component of the Reproductive Growth Initiation in the Annual Growth Cycle of Adult C. oleifera
3.2. The Photoperiod Pathway Involving CoCO-like, CoFT, and CoLFY Promotes Stem Apex Meristem Differentiation and Floral Initiation in C. oleifera
3.3. GA4 and GA3 Might Play Different Roles in the Floral Initiation of C. oleifera
3.4. GA20OX1 and GA2OX8 Are the Key Candidate Genes in GA-Mediated Regulation of Floral Initiation by Gibberellin Pathway
4. Materials and Methods
4.1. Plant Growth, Sample Collection, and Morphological Characteristics Analysis
4.2. Quantitative Analysis of Endogenous Gibberellins
4.3. Transcriptome Sequencing
4.4. Functional Annotation of the Transcriptome
4.5. Differentially Expressed Genes
4.6. GO and KEGG Enrichment Analysis of DEGs and Construction of Coexpression Network
4.7. Quantitative Real-Time PCR (qRT-PCR) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.; Yang, X.; Huang, X.; Duan, S.; Long, C.; Chen, J.; Rong, J. Leaf transcriptome analysis of a subtropical evergreen broadleaf plant, wild oil-tea camellia (Camellia oleifera), revealing candidate genes for cold acclimation. BMC Genom. 2017, 18, 211. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, J.; Li, Z.; Zhang, F.; Tan, X. Transcriptomic Analyses of Camellia oleifera ‘Huaxin’ Leaf Reveal Candidate Genes Related to Long-Term Cold Stress. Int. J. Mol. Sci. 2020, 21, 846. [Google Scholar] [CrossRef]
- Fang, H.; Bai, H. Cultural distribution and site classification for Camellia oleifera. Sci. Silvae Sin. 2002, 38, 64–72. [Google Scholar]
- Ma, J.; Ye, H.; Rui, Y.; Chen, G.; Zhang, N. Fatty acid composition of Camellia oleifera oil. J. Für Verbraucherschutz Und Leb. 2010, 6, 9–12. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Z.; Zhou, J.; Gu, Y.; Tan, X. Comparative study on fruit development and oil synthesis in two cultivars of Camellia oleifera. BMC Plant Biol. 2021, 21, 348. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Yang, Q.; Fang, F.; Li, Y. The camelliagenin from defatted seeds of Camellia oleifera as antibiotic substitute to treat chicken against infection of Escherichia coli and Staphylococcus aureus. BMC Vet. Res. 2015, 11, 214. [Google Scholar] [CrossRef]
- Wilkie, J.D.; Sedgley, M.; Olesen, T. Regulation of floral initiation in horticultural trees. J. Exp. Bot. 2008, 59, 3215–3228. [Google Scholar] [CrossRef]
- Bolouri Moghaddam, M.R.; Van den Ende, W. Sugars, the clock and transition to flowering. Front. Plant. Sci. 2013, 4, 22. [Google Scholar]
- Bendix, C.; Marshall, C.M.; Harmon, F.G. Circadian Clock Genes Universally Control Key Agricultural Traits. Mol. Plant 2015, 8, 1135–1152. [Google Scholar] [CrossRef]
- Blackman, B.K. Changing Responses to Changing Seasons: Natural Variation in the Plasticity of Flowering Time. Plant Physiol. 2017, 173, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Bouche, F.; Lobet, G.; Tocquin, P.; Perilleux, C. FLOR-ID: An interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Res. 2016, 44, D1167–D1171. [Google Scholar] [CrossRef]
- Domagalska, M.A.; Sarnowska, E.; Nagy, F.; Davis, S.J. Genetic analyses of interactions among gibberellin, abscisic acid, and brassinosteroids in the control of flowering time in Arabidopsis thaliana. PLoS ONE 2010, 5, e14012. [Google Scholar] [CrossRef]
- Cho, L.H.; Yoon, J.; An, G. The control of flowering time by environmental factors. Plant J. 2017, 90, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Conti, L. Hormonal control of the floral transition: Can one catch them all? Dev. Biol. 2017, 430, 288–301. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, A.; Richter, R. Genetic and molecular basis of floral induction in Arabidopsis thaliana. J. Exp. Bot. 2020, 71, 2490–2504. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, E.; Frommlet, F.; Nordborg, M. The genetic architecture of the network underlying flowering time variation in Arabidopsis thaliana. bioRxiv 2017. [Google Scholar] [CrossRef]
- An, H.; Roussot, C.; Suarez-Lopez, P.; Corbesier, L.; Vincent, C.; Pineiro, M.; Hepworth, S.; Mouradov, A.; Justin, S.; Turnbull, C.; et al. CONSTANS acts in the phloem to regulate a systemic signal that induces photoperiodic flowering of Arabidopsis. Development 2004, 131, 3615–3626. [Google Scholar] [CrossRef]
- Golembeski, G.S.; Imaizumi, T. Photoperiodic Regulation of Florigen Function in Arabidopsis thaliana. Arab. Book 2015, 13, e0178. [Google Scholar] [CrossRef]
- Kinmonth-Schultz, H.; Lewandowska-Sabat, A.; Imaizumi, T.; Ward, J.K.; Rognli, O.A.; Fjellheim, S. Flowering Times of Wild Arabidopsis Accessions From Across Norway Correlate With Expression Levels of FT, CO, and FLC Genes. Front. Plant Sci. 2021, 12, 747740. [Google Scholar] [CrossRef]
- Andres, F.; Coupland, G. The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012, 13, 627–639. [Google Scholar] [CrossRef]
- Hsu, C.Y.; Adams, J.P.; Kim, H.; No, K.; Ma, C.; Strauss, S.H.; Drnevich, J.; Vandervelde, L.; Ellis, J.D.; Rice, B.M.; et al. FLOWERING LOCUS T duplication coordinates reproductive and vegetative growth in perennial poplar. Proc. Natl. Acad. Sci. USA 2011, 108, 10756–10761. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Winter, C.M.; Wu, M.; Kanno, Y.; Yamaguchi, A.; Seo, M.; Wagner, D. Gibberellin Acts Positively Then Negatively to Control Onset of Flower Formation in Arabidopsis. Science 2014, 344, 638–641. [Google Scholar] [CrossRef]
- Goslin, K.; Zheng, B.; Serrano-Mislata, A.; Rae, L.; Ryan, P.T.; Kwasniewska, K.; Thomson, B.; O’Maoileidigh, D.S.; Madueno, F.; Wellmer, F.; et al. Transcription Factor Interplay between LEAFY and APETALA1/CAULIFLOWER during Floral Initiation. Plant Physiol. 2017, 174, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.M.; Yamaguchi, N.; Wu, M.F.; Wagner, D. Transcriptional programs regulated by both LEAFY and APETALA1 at the time of flower formation. Physiol Plant 2015, 155, 55–73. [Google Scholar] [CrossRef]
- Jue, D.; Sang, X.; Liu, L.; Shu, B.; Wang, Y.; Liu, C.; Wang, Y.; Xie, J.; Shi, S. Comprehensive analysis of the longan transcriptome reveals distinct regulatory programs during the floral transition. BMC Genom. 2019, 20, 126. [Google Scholar] [CrossRef]
- Das, A.; Geetha, G.A.; Ravishankar, K.V.; Shivashankara, K.S.; Roy, T.K.; Dinesh, M.R. Interrelations of growth regulators, carbohydrates and expression of flowering genes (FT, LFY, AP1) in leaf and shoot apex of regular and alternate bearing mango (Mangifera indica L.) cultivars during flowering. Sci. Hortic. 2019, 253, 263–269. [Google Scholar] [CrossRef]
- Song, G.-Q.; Liu, Z.; Zhong, G.-Y. Regulatory frameworks involved in the floral induction, formation and developmental programming of woody horticultural plants: A case study on blueberries. Front. Plant Sci. 2024, 15, 1336892. [Google Scholar] [CrossRef]
- Hong-yan, G.; Xin-jian, T.; Feng, T.; Qiu-ping, Z. The Relationship between Floral Initiation and Spring-shoot Growth in Camellia oleifera. For. Res. 2022, 35, 123–130. [Google Scholar]
- Colasanti, J.; Sundaresan, V. Control of the transition to flowering. Plant Biotechnol. 1996, 7, 145–169. [Google Scholar] [CrossRef]
- Xing, L.B.; Zhang, D.; Li, Y.M.; Shen, Y.W.; Zhao, C.P.; Ma, J.J.; An, N.; Han, M.Y. Transcription Profiles Reveal Sugar and Hormone Signaling Pathways Mediating Flower Induction in Apple (Malus domestica Borkh.). Plant Cell Physiol. 2015, 56, 2052–2068. [Google Scholar] [CrossRef]
- Jiang, Z.; Sun, L.; Wei, Q.; Ju, Y.; Zou, X.; Wan, X.; Liu, X.; Yin, Z. A New Insight into Flowering Regulation: Molecular Basis of Flowering Initiation in Magnolia × soulangeana ‘Changchun’. Genes 2019, 11, 15. [Google Scholar] [CrossRef]
- Agusti, M.; Reig, C.; Martinez-Fuentes, A.; Mesejo, C. Advances in Citrus Flowering: A Review. Front. Plant Sci. 2022, 13, 868831. [Google Scholar] [CrossRef]
- Munoz-Fambuena, N.; Nicolas-Almansa, M.; Martinez-Fuentes, A.; Reig, C.; Iglesias, D.J.; Primo-Millo, E.; Mesejo, C.; Agusti, M. Genetic inhibition of flowering differs between juvenile and adult Citrus trees. Ann. Bot. 2019, 123, 483–490. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Niu, Q.; He, L.; Teng, Y.; Bai, S. Abscisic Acid (ABA) Promotes the Induction and Maintenance of Pear (Pyrus pyrifolia White Pear Group) Flower Bud Endodormancy. Int. J. Mol. Sci. 2018, 19, 310. [Google Scholar] [CrossRef]
- Winterhagen, P.; Hegele, M.; Tiyayon, P.; Wünsche, J.N. Cytokinin accumulation and flowering gene expression are orchestrated for floral meristem development in longan (Dimocarpus longan Lour.) after chemical flower induction. Sci. Hortic. 2020, 270, 109467. [Google Scholar] [CrossRef]
- Liang, Q.; Song, K.; Lu, M.; Dai, T.; Yang, J.; Wan, J.; Li, L.; Chen, J.; Zhan, R.; Wang, S. Transcriptome and Metabolome Analyses Reveal the Involvement of Multiple Pathways in Flowering Intensity in Mango. Front. Plant Sci. 2022, 13, 933923. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Hernandez, E.C.; Quiroz, S.; Garcia-Ponce, B.; Alvarez-Buylla, E.R. The flowering transition pathways converge into a complex gene regulatory network that underlies the phase changes of the shoot apical meristem in Arabidopsis thaliana. Front. Plant Sci. 2022, 13, 852047. [Google Scholar] [CrossRef]
- Jin, R.; Klasfeld, S.; Zhu, Y.; Fernandez Garcia, M.; Xiao, J.; Han, S.-K.; Konkol, A.; Wagner, D. LEAFY is a pioneer transcription factor and licenses cell reprogramming to floral fate. Nat. Commun. 2021, 12, 626. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Klasfeld, S.; Jeong, C.W.; Jin, R.; Goto, K.; Yamaguchi, N.; Wagner, D. TERMINAL FLOWER 1-FD complex target genes and competition with FLOWERING LOCUS T. Nat. Commun. 2020, 11, 5118. [Google Scholar] [CrossRef]
- Lei, H.; Su, S.; Ma, L.; Wen, Y.; Wang, X. Molecular cloning and functional characterization of CoFT1, a homolog of FLOWERING LOCUS T (FT) from Camellia oleifera. Gene 2017, 626, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; He, J.; Li, J.; Ren, S.; Wang, Y.; Zhou, J.; Tan, X. Analysis of Camellia oleifera transcriptome reveals key pathways and hub genes involved during different photoperiods. BMC Plant Biol. 2022, 22, 435. [Google Scholar] [CrossRef]
- Yan, J.; Liu, Q.; Guo, P.; Wang, Y.; Sheng, S.; Liu, X.; Zhang, R.; Li, J.; Tan, X. Time-Course Transcriptome Analysis Unveils the CoFKF1-CoMYB4-CoFT1 Regulatory Module in Flowering Control of Camellia oleifera Abel. Plant Cell Environ. 2025, 48, 6153–6169. [Google Scholar] [CrossRef]
- Wu, T.; Liu, Z.; Yu, T.; Zhou, R.; Yang, Q.; Cao, R.; Nie, F.; Ma, X.; Bai, Y.; Song, X. Flowering genes identification, network analysis, and database construction for 837 plants. Hortic. Res. 2024, 11, uhae013. [Google Scholar] [CrossRef]
- Davis, S.J. Integrating hormones into the floral-transition pathway of Arabidopsis thaliana. Plant Cell Environ. 2009, 32, 1201–1210. [Google Scholar] [CrossRef]
- Hauvermale, A.L.; Ariizumi, T.; Steber, C.M. Gibberellin signaling: A theme and variations on DELLA repression. Plant Physiol. 2012, 160, 83–92. [Google Scholar] [CrossRef]
- Shi, B.; Felipo-Benavent, A.; Cerutti, G.; Galvan-Ampudia, C.; Jilli, L.; Brunoud, G.; Mutterer, J.; Vallet, E.; Sakvarelidze-Achard, L.; Davière, J.-M.; et al. A quantitative gibberellin signaling biosensor reveals a role for gibberellins in internode specification at the shoot apical meristem. Nat. Commun. 2024, 15, 3895. [Google Scholar] [CrossRef]
- Nemoto, K.; Ramadan, A.; Arimura, G.-I.; Imai, K.; Tomii, K.; Shinozaki, K.; Sawasaki, T. Tyrosine phosphorylation of the GARU E3 ubiquitin ligase promotes gibberellin signalling by preventing GID1 degradation. Nat. Commun. 2017, 8, 1004. [Google Scholar] [CrossRef]
- de Lucas, M.; Davière, J.-M.; Rodríguez-Falcón, M.; Pontin, M.; Iglesias-Pedraz, J.M.; Lorrain, S.; Fankhauser, C.; Blázquez, M.A.; Titarenko, E.; Prat, S. A molecular framework for light and gibberellin control of cell elongation. Nature 2008, 451, 480–484. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, W.; Aiwaili, P.; Zhang, H.; Xu, Y.; Gu, Z.; Gao, J.; Hong, B. PHOTOLYASE/BLUE LIGHT RECEPTOR2 regulates chrysanthemum flowering by compensating for gibberellin perception. Plant Physiol. 2023, 193, 2848–2864. [Google Scholar] [CrossRef]
- Girardi, F.; Canton, M.; Populin, F.; Tijero, V.; Bettio, G.; Munné-Bosch, S.; Rasori, A.; Cardillo, V.; Costa, G.; Botton, A. A gibberellin-assisted study of the transcriptional and hormonal changes occurring at floral transition in peach buds (Prunus persica L. Batsch). BMC Plant Biol. 2024, 24, 643. [Google Scholar] [CrossRef]
- Yuxi, Z.; Yanchao, Y.; Zejun, L.; Tao, Z.; Feng, L.; Chunying, L.; Shupeng, G. GA3 is superior to GA4 in promoting bud endodormancy release in tree peony (Paeonia suffruticosa) and their potential working mechanism. BMC Plant Biol. 2021, 21, 323. [Google Scholar] [CrossRef]
- Zhuang, W.; Gao, Z.; Wen, L.; Huo, X.; Cai, B.; Zhang, Z. Metabolic changes upon flower bud break in Japanese apricot are enhanced by exogenous GA4. Hortic. Res. 2015, 2, 15046. [Google Scholar] [CrossRef]
- Nakagawa, M.; Honsho, C.; Kanzaki, S.; Shimizu, K.; Utsunomiya, N. Isolation and expression analysis of FLOWERING LOCUS T-like and gibberellin metabolism genes in biennial-bearing mango trees. Sci. Hortic. 2012, 139, 108–117. [Google Scholar] [CrossRef]
- Susawaengsup, C.; Rayanakorn, M.; Wongpornchai, S.; Wangkarn, S. Investigation of plant hormone level changes in shoot tips of longan (Dimocarpus longan Lour.) treated with potassium chlorate by liquid chromatography-electrospray ionization mass spectrometry. Talanta 2011, 85, 897–905. [Google Scholar] [CrossRef]
- Bao, S.; Hua, C.; Shen, L.; Yu, H. New insights into gibberellin signaling in regulating flowering in Arabidopsis. J. Integr. Plant Biol. 2020, 62, 118–131. [Google Scholar] [CrossRef]
- Teotia, S.; Tang, G. To Bloom or Not to Bloom: Role of MicroRNAs in Plant Flowering. Mol. Plant 2015, 8, 359–377. [Google Scholar] [CrossRef]
- Li, C.; Zheng, L.; Wang, X.; Hu, Z.; Zheng, Y.; Chen, Q.; Hao, X.; Xiao, X.; Wang, X.; Wang, G.; et al. Comprehensive expression analysis of Arabidopsis GA2-oxidase genes and their functional insights. Plant Sci. 2019, 285, 1–13. [Google Scholar] [CrossRef]
- Wu, W.; Zhu, L.; Wang, P.; Liao, Y.; Duan, L.; Lin, K.; Chen, X.; Li, L.; Xu, J.; Hu, H.; et al. Transcriptome-Based Construction of the Gibberellin Metabolism and Signaling Pathways in Eucalyptus grandis × E. urophylla, and Functional Characterization of GA20ox and GA2ox in Regulating Plant Development and Abiotic Stress Adaptations. Int. J. Mol. Sci. 2023, 24, 7051. [Google Scholar] [CrossRef]
- Guo, H.; Zhong, Q.; Tian, F.; Zhou, X.; Tan, X.; Luo, Z. Transcriptome Analysis Reveals Putative Induction of Floral Initiation by Old Leaves in Tea-Oil Tree (Camellia oleifera ‘changlin53’). Int. J. Mol. Sci. 2022, 23, 13021. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 19, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Developmental Stages | Sample Name | Number of Clean Reads | Number of Clean Data | Q20 (%) | Q30 (%) | GC (%) | Total Mapped (%) |
|---|---|---|---|---|---|---|---|
| I | BI-1 | 56,907,348 | 8,497,755,633 | 8,338,100,557 (98.12%) | 8,015,960,394 (94.33%) | 3,900,291,117 (45.90%) | 44,985,690 (79.05%) |
| BI-2 | 48,431,060 | 7,215,452,943 | 7,085,854,082 (98.20%) | 6,822,179,906 (94.55%) | 3,339,480,736 (46.28%) | 38,526,147 (79.55%) | |
| BI-3 | 58,652,190 | 8,740,998,410 | 8,563,905,357 (97.97%) | 8,212,926,590 (93.96%) | 4,031,458,880 (46.12%) | 45,998,276 (78.43%) | |
| II | BII-1 | 54,553,160 | 8,112,660,091 | 7,889,813,970 (97.25%) | 7,477,825,967 (92.17%) | 3,768,480,169 (46.45%) | 44,160,960 (80.95%) |
| BII-2 | 44,576,422 | 6,624,937,074 | 6,457,818,501 (97.48%) | 6,135,698,351 (92.62%) | 3,151,122,729 (47.56%) | 37,181,979 (83.41%) | |
| BII-3 | 46,778,886 | 6,962,446,522 | 6,759,631,130 (97.09%) | 6,392,012,670 (91.81%) | 3,234,932,717 (46.46%) | 37,839,837 (80.89%) | |
| III | BIII-1 | 50,985,660 | 7,586,849,110 | 7,365,728,863 (97.09%) | 6,964,564,010 (91.80%) | 3,511,913,268 (46.29%) | 41,408,399 (81.22%) |
| BIII-2 | 50,562,454 | 7,545,683,270 | 7,317,183,863 (96.97%) | 6,915,262,966 (91.65%) | 3,461,771,628 (45.88%) | 40,696,612 (80.49%) | |
| BIII-3 | 46,393,600 | 6,891,860,986 | 6,708,819,561 (97.34%) | 6367327747 (92.39%) | 3,208,481,881 (46.55%) | 38,154,413 (82.24%) | |
| IV | BIV-1 | 41,886,036 | 6,228,273,002 | 6,066,396,478 (97.40%) | 5,763,909,687 (92.54%) | 28,57,941,556 (45.89%) | 33,925,334 (80.99%) |
| BIV-2 | 51,563,192 | 7,699,619,603 | 7485225086 (97.22%) | 7,085,303,270 (92.02%) | 3,537,890,895 (45.95%) | 42,049,101 (81.55%) | |
| BIV-3 | 52,264,300 | 7,811,770,783 | 7614802319 (97.48%) | 7,238,347,545 (92.66%) | 3,592,384,741 (45.99%) | 42,578,151 (81.47%) | |
| V | BV-1 | 49,788,996 | 7,439,686,499 | 7,227,532,557 (97.15%) | 6,835,950,341 (91.88%) | 3,417,730,361 (45.94%) | 40,375,415 (81.09%) |
| BV-2 | 48,638,636 | 7,269,786,502 | 7,078,040,967 (97.36%) | 6,716,162,908 (92.38%) | 3,349,964,297 (46.08%) | 39,285,275 (80.77%) | |
| BV-3 | 57,903,406 | 8,647,231,084 | 8,394,242,589 (97.07%) | 7,932,070,431 (91.73%) | 3,978,878,410 (46.01%) | 46,499,461 (80.31%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Zhou, Z.; Zhou, J.; Yan, C.; Zhong, W.; Li, C.; Jiang, Y.; Yuan, Y.; Cao, L.; Pan, W.; et al. Transcriptomic Profiling of Buds Unveils Insights into Floral Initiation in Tea-Oil Tree (Camellia oleifera ‘changlin53’). Plants 2025, 14, 2348. https://doi.org/10.3390/plants14152348
Guo H, Zhou Z, Zhou J, Yan C, Zhong W, Li C, Jiang Y, Yuan Y, Cao L, Pan W, et al. Transcriptomic Profiling of Buds Unveils Insights into Floral Initiation in Tea-Oil Tree (Camellia oleifera ‘changlin53’). Plants. 2025; 14(15):2348. https://doi.org/10.3390/plants14152348
Chicago/Turabian StyleGuo, Hongyan, Zongshun Zhou, Jian Zhou, Chao Yan, Wenbin Zhong, Chang Li, Ying Jiang, Yaqi Yuan, Linqing Cao, Wenting Pan, and et al. 2025. "Transcriptomic Profiling of Buds Unveils Insights into Floral Initiation in Tea-Oil Tree (Camellia oleifera ‘changlin53’)" Plants 14, no. 15: 2348. https://doi.org/10.3390/plants14152348
APA StyleGuo, H., Zhou, Z., Zhou, J., Yan, C., Zhong, W., Li, C., Jiang, Y., Yuan, Y., Cao, L., Pan, W., Wang, J., Wang, J., He, T., Hua, Y., Liu, Y., Cao, L., & Chen, C. (2025). Transcriptomic Profiling of Buds Unveils Insights into Floral Initiation in Tea-Oil Tree (Camellia oleifera ‘changlin53’). Plants, 14(15), 2348. https://doi.org/10.3390/plants14152348