
Genome-Wide Identification, Evolution and Expression Analysis of GRAS Transcription Factor Gene Family Under Viral Stress in Nicotiana benthamiana


Abstract
1. Introduction
2. Results
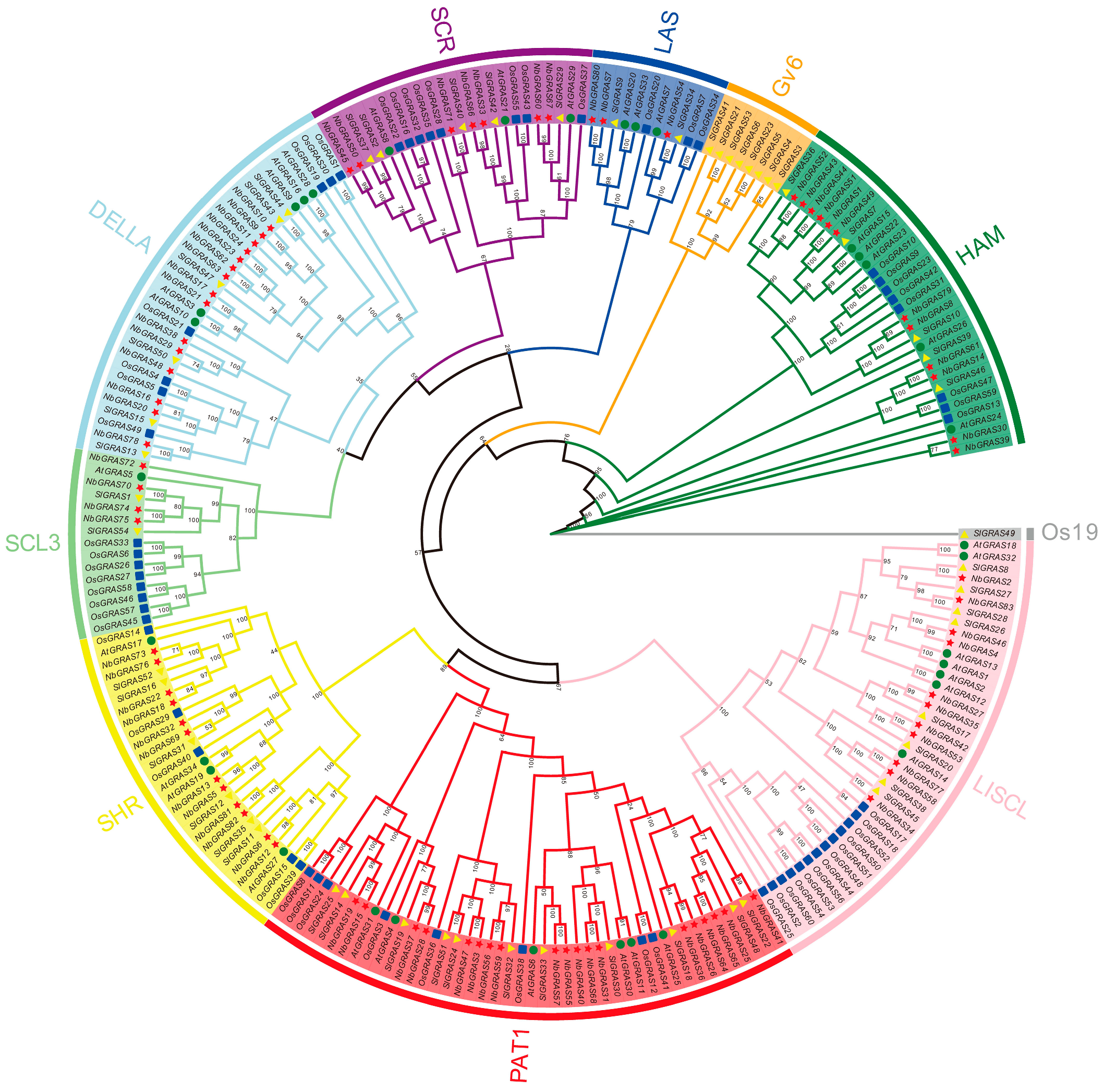
2.1. Identification and Characterization of the GRAS Gene Family in Nicotiana benthamiana
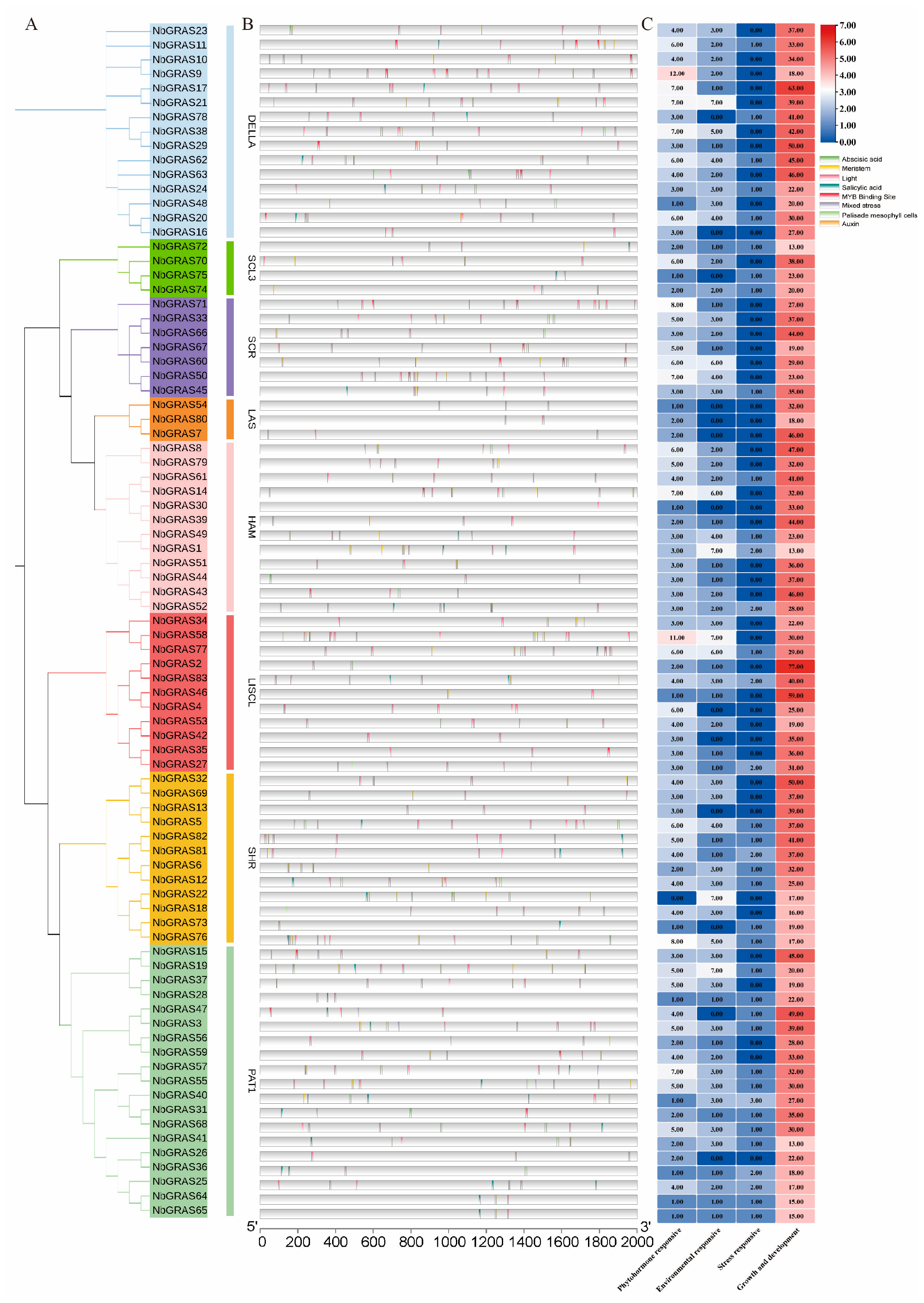
2.2. Analysis of Conserved Motifs and Gene Structures of the NbGRASs
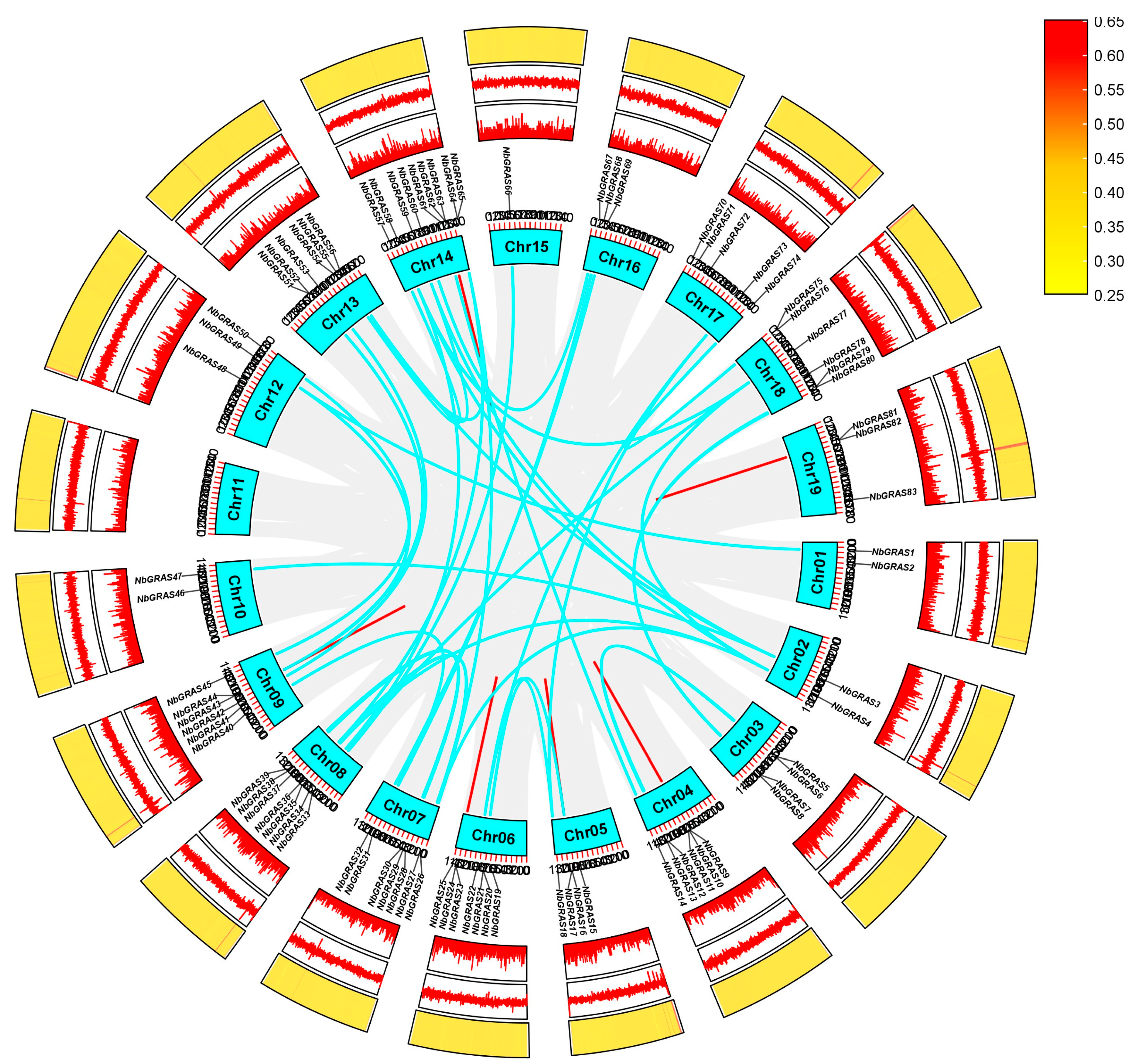
2.3. Chromosomal Distribution and Gene Duplication of the NbGRAS Family
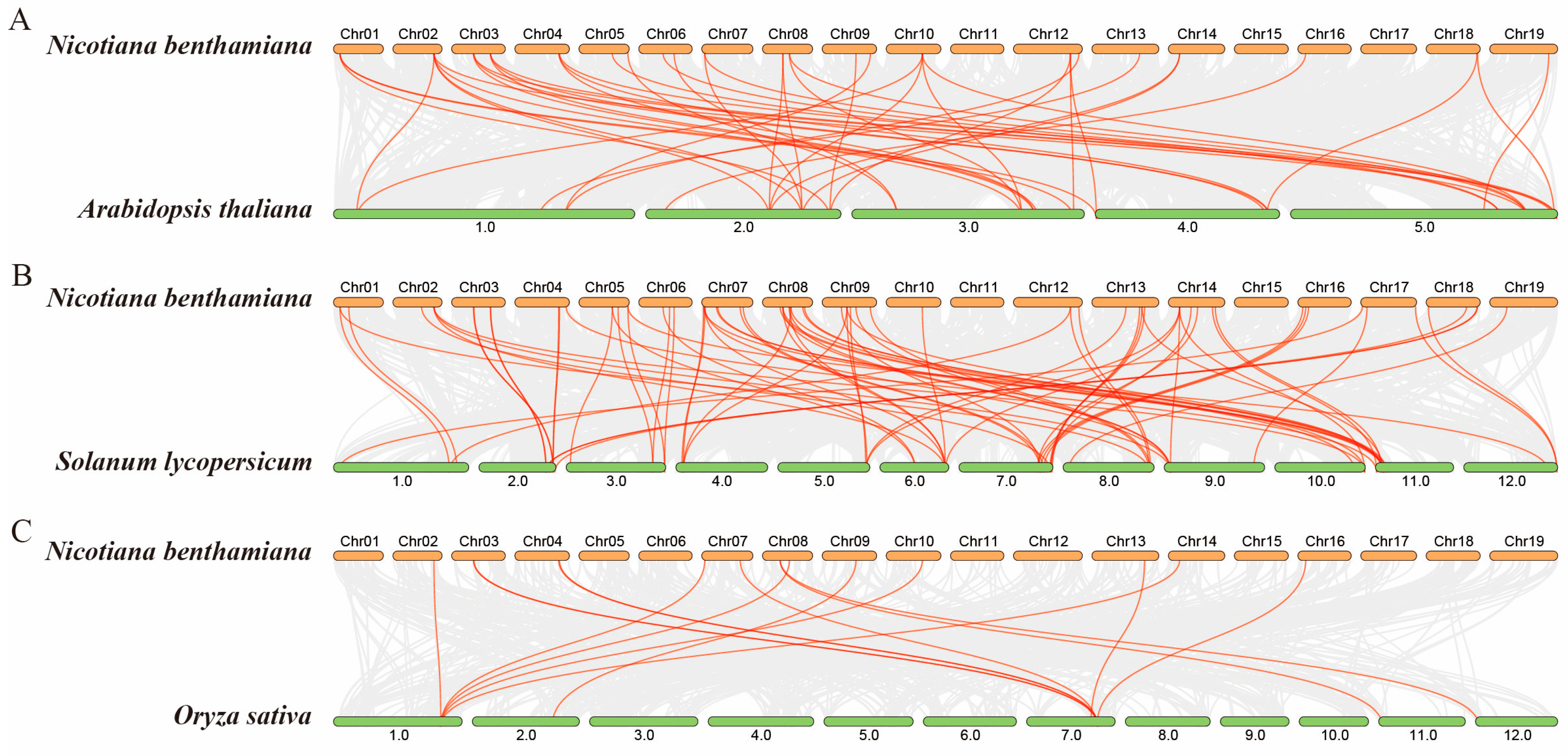
2.4. Synteny Analysis of GRAS Genes
2.5. Prediction of Cis-Acting Elements in the NbGRASs Promoters
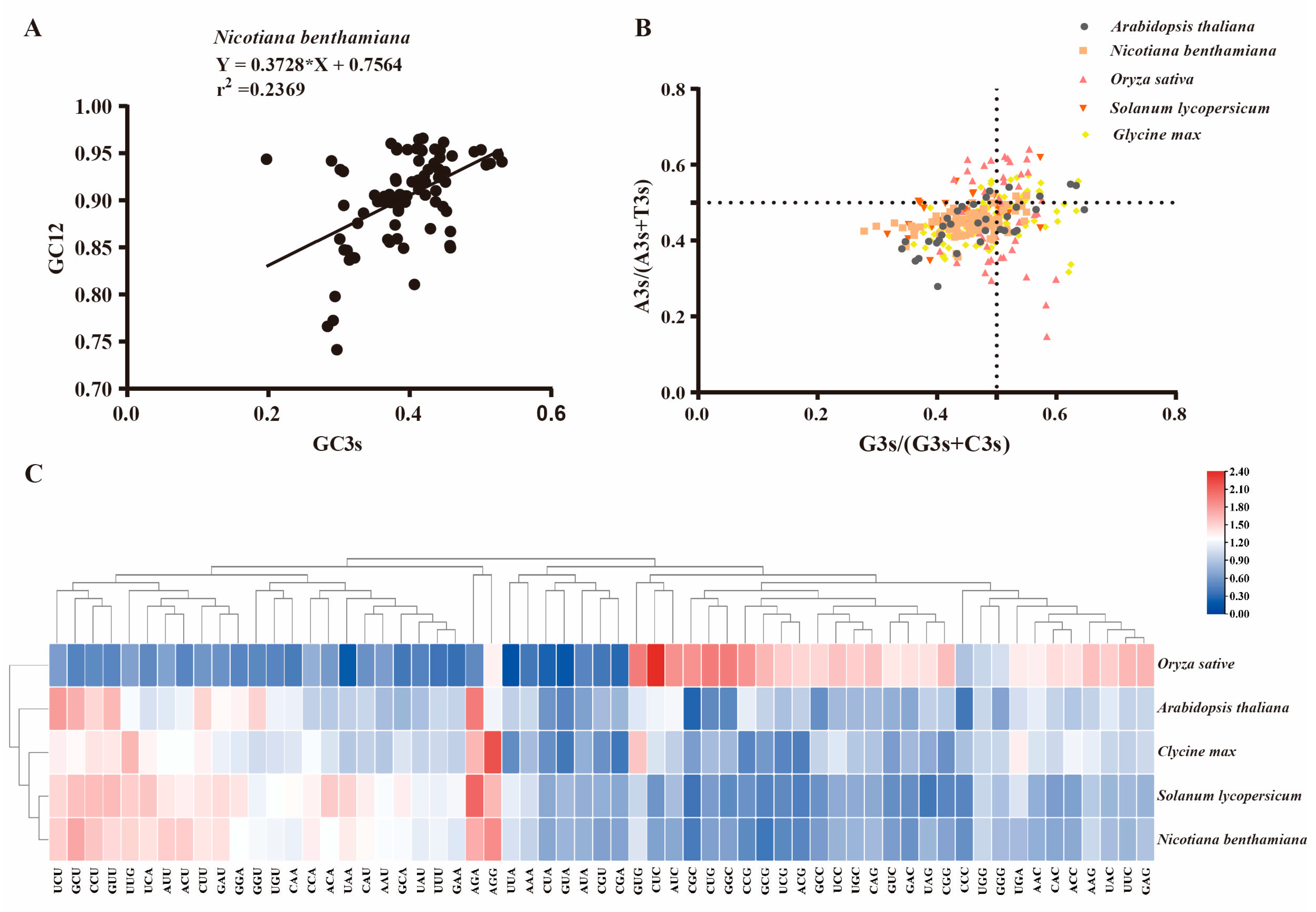
2.6. Analysis of Codon Usage Patterns for GRAS Genes Across Different Species
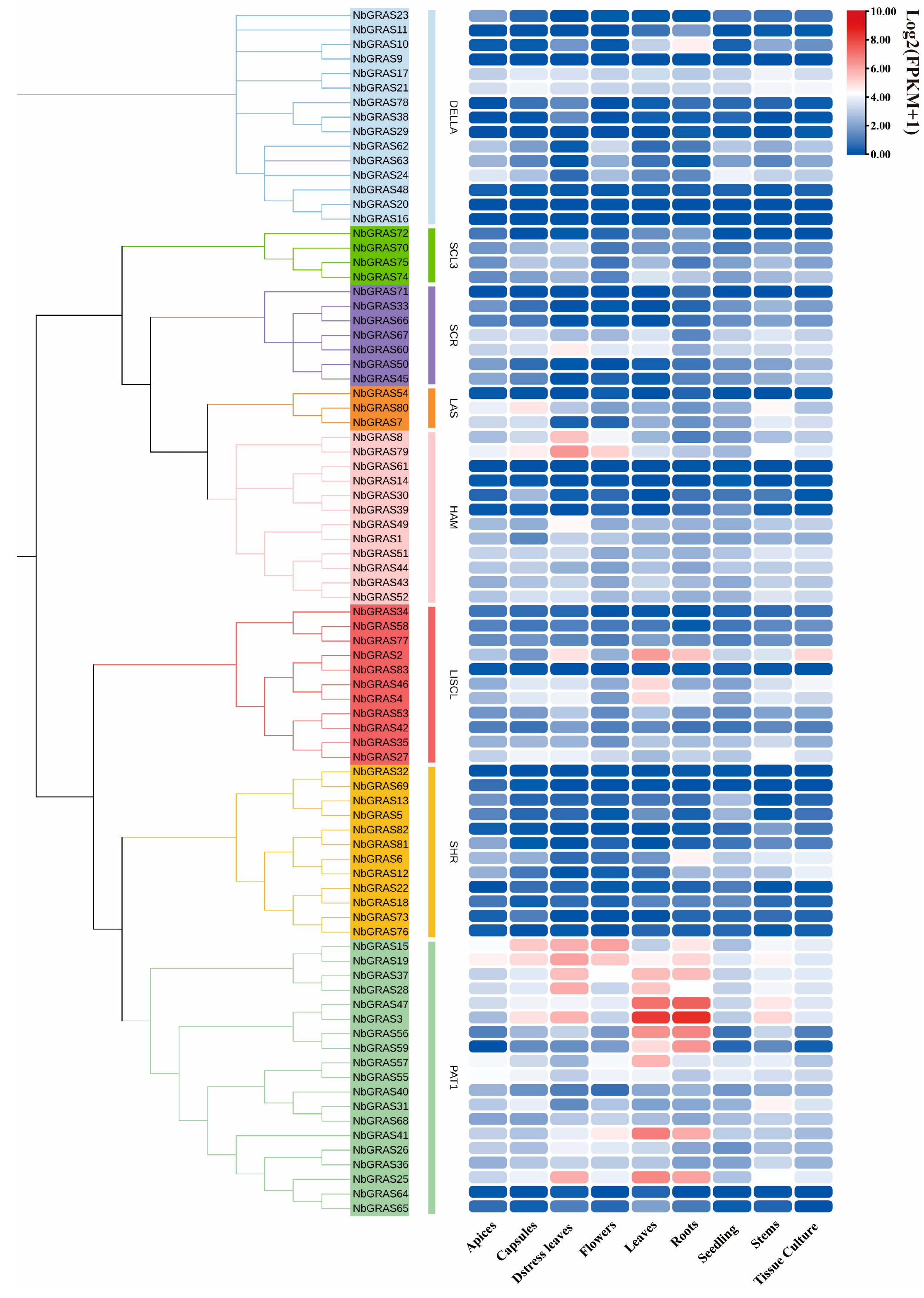
2.7. Expression Analysis of NbGRASs in Different Tissues
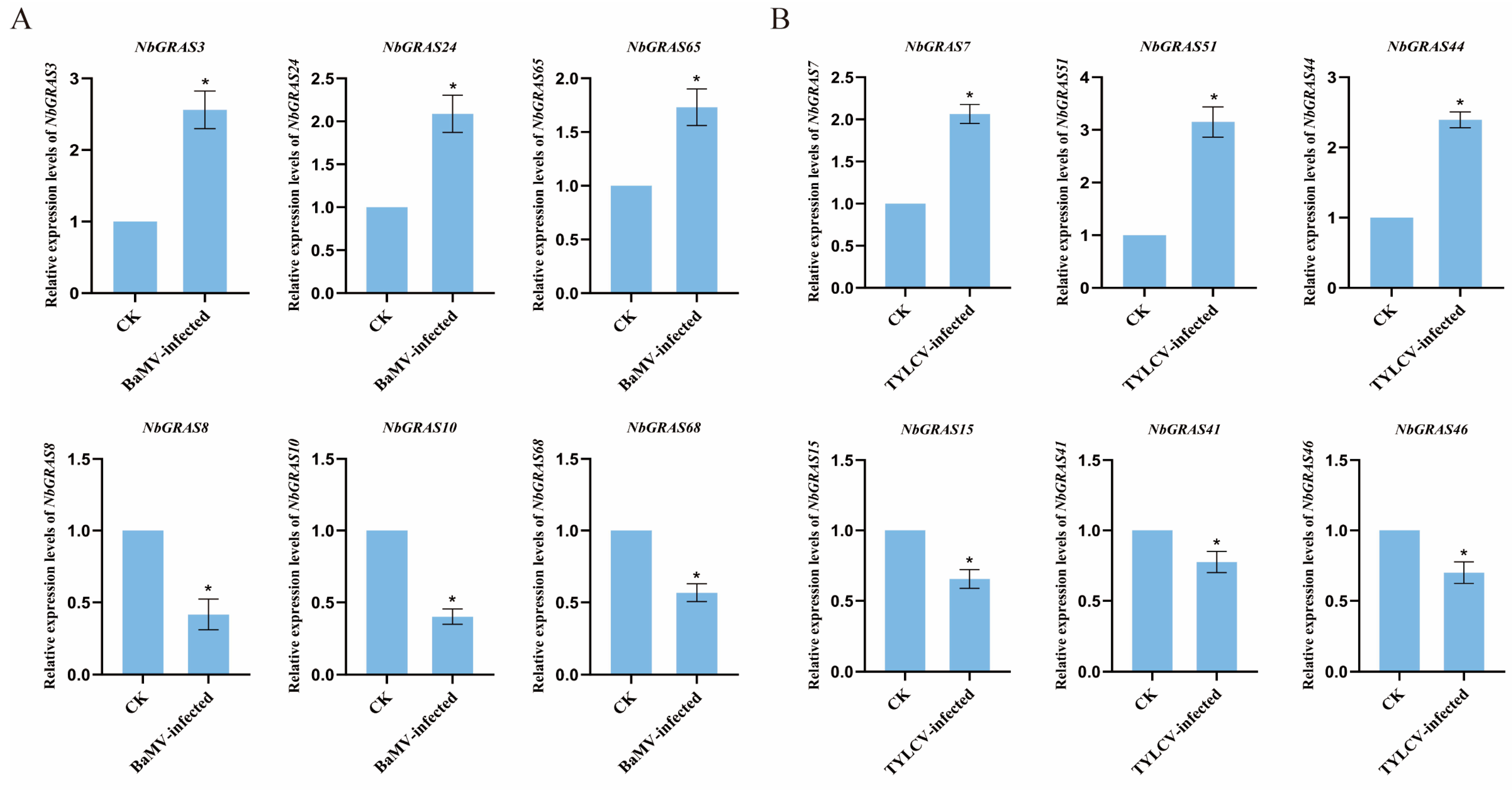
2.8. Expression Analysis of NbGRASs Under Viral Stress
2.9. Expression Analysis of NbGRASs Under Viral Stress
3. Discussion
4. Materials and Methods
4.1. Identification of the GRAS Family in Nicotiana benthamiana
4.2. Phylogenetic Analysis of GRAS Genes
4.3. Chromosome Location and Collinearity Analysis
4.4. Promoter Element Analysis
4.5. Motif and Gene Structure Analysis
4.6. RNA-Seq Analysis of Expression Patterns
4.7. Plant Growth and Viral Stress
4.8. Codon Usage Bias Analysis
4.9. RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TYLCV | tomato yellow leaf curl virus |
| BaMV | bamboo mosaic virus |
| TFs | transcription factors |
| MWs | molecular weights |
| PIs | isoelectric points |
| chr | chromosome |
| RSCU | relative synonymous codon usage |
| ENC | effective number of codons |
| RT-qPCR | real-time quantitative PCR |
References
- Wang, B.; Sun, Z.; Liu, Y.; Xu, B.; Li, J.; Chi, M.; Xing, Y.; Yang, B.; Li, J.; Liu, J.; et al. A pervasive phosphorylation cascade modulation of plant transcription factors in response to abiotic stress. Planta 2023, 258, 73. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Carol, P.; Richards, D.E.; King, K.E.; Cowling, R.J.; Murphy, G.P.; Harberd, N.P. The Arabidopsis GAI gene defines a signaling pathway that negatively regulates gibberellin responses. Genes Dev. 1997, 11, 3194–3205. [Google Scholar] [CrossRef] [PubMed]
- Silverstone, A.L.; Ciampaglio, C.N.; Sun, T. The Arabidopsis RGA gene encodes a transcriptional regulator repressing the gibberellin signal transduction pathway. Plant Cell 1998, 10, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Cenci, A.; Rouard, M. Evolutionary Analyses of GRAS Transcription Factors in Angiosperms. Front. Plant Sci. 2017, 8, 273. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Wan, P.; Sun, S.; Li, J.; Chen, M. Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant Mol. Biol. 2004, 54, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Bolle, C. The role of GRAS proteins in plant signal transduction and development. Planta 2004, 218, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xue, B.; Jones, W.T.; Rikkerink, E.; Dunker, A.K.; Uversky, V.N. A functionally required unfoldome from the plant kingdom: Intrinsically disordered N-terminal domains of GRAS proteins are involved in molecular recognition during plant development. Plant Mol. Biol. 2011, 77, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Nakasugi, K.; Crowhurst, R.N.; Bally, J.; Wood, C.C.; Hellens, R.P.; Waterhouse, P.M. De novo transcriptome sequence assembly and analysis of RNA silencing genes of Nicotiana benthamiana. PLoS ONE 2013, 8, e59534. [Google Scholar] [CrossRef] [PubMed]
- Brule, C.E.; Grayhack, E.J. Synonymous Codons: Choose Wisely for Expression. Trends Genet. 2017, 33, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, X.; Fan, S.; Zhou, L.; Wang, Y. Overexpression of HcSCL13, a Halostachys caspica GRAS transcription factor, enhances plant growth and salt stress tolerance in transgenic Arabidopsis. Plant Physiol. Biochem. PPB 2020, 151, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Lee, L.Y.; Xia, K.; Yan, Y.; Yu, H. DELLAs modulate jasmonate signaling via competitive binding to JAZs. Dev. Cell 2010, 19, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Fang, L.; Karungo, S.K.; Zhang, L.; Gao, Y.; Li, S.; Xin, H. Overexpression of VaPAT1, a GRAS transcription factor from Vitis amurensis, confers abiotic stress tolerance in Arabidopsis. Plant Cell Rep. 2016, 35, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Stuurman, J.; Jäggi, F.; Kuhlemeier, C. Shoot meristem maintenance is controlled by a GRAS-gene mediated signal from differentiating cells. Genes Dev. 2002, 16, 2213–2218. [Google Scholar] [CrossRef] [PubMed]
- Morohashi, K.; Minami, M.; Takase, H.; Hotta, Y.; Hiratsuka, K. Isolation and characterization of a novel GRAS gene that regulates meiosis-associated gene expression. J. Biol. Chem. 2003, 278, 20865–20873. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Chen, S.; Li, T.; Ma, X.; Liang, X.; Ding, X.; Liu, H.; Luo, L. OsGRAS23, a rice GRAS transcription factor gene, is involved in drought stress response through regulating expression of stress-responsive genes. BMC Plant Biol. 2015, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Cody, J.P.; Maher, M.F.; Nasti, R.A.; Starker, C.G.; Chamness, J.C.; Voytas, D.F. Direct delivery and fast-treated Agrobacterium co-culture (Fast-TrACC) plant transformation methods for Nicotiana benthamiana. Nat. Protoc. 2023, 18, 81–107. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Sivakrishna Rao, G.; Aman, R.; Butt, H.; Kamel, R.; Sedeek, K.; Mahfouz, M.M. High-efficiency retron-mediated single-stranded DNA production in plants. Synth. Biol. 2022, 7, ysac025. [Google Scholar] [CrossRef] [PubMed]
- Kwan, B.D.; Seligmann, B.; Nguyen, T.D.; Franke, J.; Dang, T.T. Leveraging synthetic biology and metabolic engineering to overcome obstacles in plant pathway elucidation. Curr. Opin. Plant Biol. 2023, 71, 102330. [Google Scholar] [CrossRef] [PubMed]
- Gülck, T.; Booth, J.K.; Carvalho, Â.; Khakimov, B.; Crocoll, C.; Motawia, M.S.; Møller, B.L.; Bohlmann, J.; Gallage, N.J. Synthetic Biology of Cannabinoids and Cannabinoid Glucosides in Nicotiana benthamiana and Saccharomyces cerevisiae. J. Nat. Prod. 2020, 83, 2877–2893. [Google Scholar] [CrossRef] [PubMed]
- Torres-Galea, P.; Hirtreiter, B.; Bolle, C. Two GRAS proteins, SCARECROW-LIKE21 and PHYTOCHROME A SIGNAL TRANSDUCTION1, function cooperatively in phytochrome A signal transduction. Plant Physiol. 2013, 161, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wu, H.; Li, X.; Li, Q.; Zhao, X.; Duan, X.; An, Y.; Lv, W.; An, H. Identification and expression of GRAS family genes in maize (Zea mays L.). PLoS ONE 2017, 12, e0185418. [Google Scholar] [CrossRef] [PubMed]
- Song, X.M.; Liu, T.K.; Duan, W.K.; Ma, Q.H.; Ren, J.; Wang, Z.; Li, Y.; Hou, X.L. Genome-wide analysis of the GRAS gene family in Chinese cabbage (Brassica rapa ssp. pekinensis). Genomics 2014, 103, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Xian, Z.; Kang, X.; Tang, N.; Li, Z. Genome-wide identification, phylogeny and expression analysis of GRAS gene family in tomato. BMC Plant Biol. 2015, 15, 209. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Chen, Z.; Ahmed, N.; Han, B.; Cui, Q.; Liu, A. Genome-Wide Identification, Evolutionary Analysis, and Stress Responses of the GRAS Gene Family in Castor Beans. Int. J. Mol. Sci. 2016, 17, 1004. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yan, M.; Chen, S.; Sun, J.; Wang, J.; Meng, D.; Li, J.; Zhang, L.; Guo, L. The complete genome assembly of Nicotiana benthamiana reveals the genetic and epigenetic landscape of centromeres. Nat. Plants 2024, 10, 1928–1943. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Q.; Tung, J.; Zhang, X.; Liu, D.; Deng, Y.; Tian, Z.; Chen, H.; Wang, T.; Yin, W.; et al. High-quality assembled and annotated genomes of Nicotiana tabacum and Nicotiana benthamiana reveal chromosome evolution and changes in defense arsenals. Mol. Plant 2024, 17, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, W. Characterization of the GRAS gene family reveals their contribution to the high adaptability of wheat. PeerJ 2021, 9, e10811. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wan, X.; Zhang, X.; Zhang, J.; Zheng, C.; Yang, Q.; Yang, L.; Li, X.; Feng, L.; Zou, L.; et al. GRAS gene family in rye (Secale cereale L.): Genome-wide identification, phylogeny, evolutionary expansion and expression analyses. BMC Plant Biol. 2024, 24, 46. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Biłas, R.; Szafran, K.; Hnatuszko-Konka, K.; Kononowicz, A.K. Cis-regulatory elements used to control gene expression in plants. Plant Cell Tissue Organ Cult. (PCTOC) 2016, 127, 269–287. [Google Scholar] [CrossRef]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant Sci. Int. J. Exp. Plant Biol. 2014, 217–218, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.N.; Chiang-Hsieh, Y.F.; Chien, C.H.; Zheng, H.Q.; Lee, T.Y.; Wu, N.Y.; Tseng, K.C.; Hou, P.F.; Chang, W.C. Delineation of condition specific Cis- and Trans-acting elements in plant promoters under various Endo- and exogenous stimuli. BMC Genom. 2018, 19 (Suppl. S2), 85. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Arce, A.L.; Schmitz, G.; Koornneef, M.; Novikova, P.; Beyer, A.; de Meaux, J. The Footprint of Polygenic Adaptation on Stress-Responsive Cis-Regulatory Divergence in the Arabidopsis Genus. Mol. Biol. Evol. 2016, 33, 2088–2101. [Google Scholar] [CrossRef] [PubMed]
- Felipez, W.; de Freitas, K.E.J.; Dos Santos, R.S.; Yamamoto, R.R.; Costa de Oliveira, A. The roles of WRKY transcription factors in Malus spp. and Pyrus spp. Funct. Integr. Genom. 2022, 22, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Cao, Q.; Li, Y.; He, M.; Liu, X. Advances in cis-element- and natural variation-mediated transcriptional regulation and applications in gene editing of major crops. J. Exp. Bot. 2023, 74, 5441–5457. [Google Scholar] [CrossRef] [PubMed]
- Marand, A.P.; Eveland, A.L.; Kaufmann, K.; Springer, N.M. cis-Regulatory Elements in Plant Development, Adaptation, and Evolution. Annu. Rev. Plant Biol. 2023, 74, 111–137. [Google Scholar] [CrossRef] [PubMed]
- Behura, S.K.; Severson, D.W. Comparative analysis of codon usage bias and codon context patterns between dipteran and hymenopteran sequenced genomes. PLoS ONE 2012, 7, e43111. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Pan, Z.; Gao, S.; He, Y.; Xia, Q.; Jin, Y.; Yao, H. Analysis of synonymous codon usage of chloroplast genome in Porphyra umbilicalis. Genes Genom. 2019, 41, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.D.; Hsu, Y.H.; Lee, Y.S.; Lin, N.S. Importin α2 participates in RNA interference against bamboo mosaic virus accumulation in Nicotiana benthamiana via NbAGO10a-mediated small RNA clearance. Mol. Plant Pathol. 2024, 25, e13422. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Ding, X.; Fu, X.; Lozano-Duran, R. Transcriptional reprogramming caused by the geminivirus Tomato yellow leaf curl virus in local or systemic infections in Nicotiana benthamiana. BMC Genom. 2019, 20, 542. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Fujiwara, A.; Kosetsu, K.; Horiuchi, Y.; Kamamoto, N.; Umakawa, N.; Tamada, Y.; Zhang, L.; Matsushita, K.; Palfalvi, G.; et al. GRAS transcription factors regulate cell division planes in moss overriding the default rule. Proc. Natl. Acad. Sci. USA 2023, 120, e2210632120. [Google Scholar] [CrossRef] [PubMed]
- Achard, P.; Gong, F.; Cheminant, S.; Alioua, M.; Hedden, P.; Genschik, P. The cold-inducible CBF1 factor-dependent signaling pathway modulates the accumulation of the growth-repressing DELLA proteins via its effect on gibberellin metabolism. Plant Cell 2008, 20, 2117–2129. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Baloch, A.M. Overview of Sustainable Plant Growth and Differentiation and the Role of Hormones in Controlling Growth and Development of Plants Under Various Stresses. Recent Pat. Food Nutr. Agric. 2020, 11, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Luo, X.; Wu, M.; Wei, L.; Fan, Z.; Zhu, Y. Genome-wide identification and expression of GRAS gene family members in cassava. BMC Plant Biol. 2020, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cao, Y.; Shang, C.; Li, J.; Wang, J.; Wu, Z.; Ma, L.; Qi, T.; Fu, C.; Bai, Z.; et al. Genome-wide characterization of GRAS family genes in Medicago truncatula reveals their evolutionary dynamics and functional diversification. PLoS ONE 2017, 12, e0185439. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wang, T.; Xu, Z.; Sun, L.; Zhang, Q. Genome-wide analysis of the GRAS gene family in Prunus mume. Mol. Genet. Genom. 2015, 290, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Zhao, T.; Xu, X.; Li, J. Genome-wide identification and characterization of GRAS transcription factors in tomato (Solanum lycopersicum). PeerJ 2017, 5, e3955. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Kim, B.; Song, S.K.; Heo, J.O.; Yu, N.I.; Lee, S.A.; Kim, M.; Kim, D.G.; Sohn, S.O.; Lim, C.E.; et al. Large-scale analysis of the GRAS gene family in Arabidopsis thaliana. Plant Mol. Biol. 2008, 67, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Dassanayake, M.; Oh, D.H.; Haas, J.S.; Hernandez, A.; Hong, H.; Ali, S.; Yun, D.J.; Bressan, R.A.; Zhu, J.K.; Bohnert, H.J.; et al. The genome of the extremophile crucifer Thellungiella parvula. Nat. Genet. 2011, 43, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Magadum, S.; Banerjee, U.; Murugan, P.; Gangapur, D.; Ravikesavan, R. Gene duplication as a major force in evolution. J. Genet. 2013, 92, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Waseem, M.; Nkurikiyimfura, O.; Niyitanga, S.; Jakada, B.H.; Shaheen, I.; Aslam, M.M. GRAS transcription factors emerging regulator in plants growth, development, and multiple stresses. Mol. Biol. Rep. 2022, 49, 9673–9685. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni-Putman, S.M.; Brumos, J.; Zhao, C.; Alonso, J.M.; Stepanova, A.N. Auxin Interactions with Other Hormones in Plant Development. Cold Spring Harb. Perspect. Biol. 2021, 13, a039990. [Google Scholar] [CrossRef] [PubMed]
- Kieber, J.J.; Schaller, G.E. Cytokinin signaling in plant development. Development 2018, 145, dev149344. [Google Scholar] [CrossRef] [PubMed]
- Jodder, J. miRNA-mediated regulation of auxin signaling pathway during plant development and stress responses. J. Biosci. 2020, 45, 91. [Google Scholar] [CrossRef]
- Woodward, A.W.; Bartel, B. Auxin: Regulation, action, and interaction. Ann. Bot. 2005, 95, 707–735. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Liu, W.; Xiang, C.; Li, X.; Wang, Q.; Wang, T.; Liu, Z.; Zhang, J.; Gao, L.; Zhang, W. Genome-Wide Characterization of GRAS Family and Their Potential Roles in Cold Tolerance of Cucumber (Cucumis sativus L.). Int. J. Mol. Sci. 2020, 21, 3857. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.T.; Yu, T.F.; Fu, J.D.; Su, H.G.; Chen, J.; Zhou, Y.B.; Chen, M.; Guo, J.; Ma, Y.Z.; Wei, W.L.; et al. Genome-Wide Analysis of the GRAS Gene Family and Functional Identification of GmGRAS37 in Drought and Salt Tolerance. Front. Plant Sci. 2020, 11, 604690. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, Y.; Li, F.; Zhang, X.; Ye, J.; Wei, T.; Li, Z.; Tao, X.; Cui, F.; Wang, X.; et al. Plant virology in the 21st century in China: Recent advances and future directions. J. Integr. Plant Biol. 2024, 66, 579–622. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Mei, Y.; Ye, R.; Deng, Y.; Zhang, X.; Hu, Z.; Zhou, X.; Zhang, M.; Yang, J. Tomato leaf curl New Delhi virus: An emerging plant begomovirus threatening cucurbit production. aBIOTECH 2023, 4, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Huang, M.; Wang, S.; Li, T.; Huang, Y. Tomato yellow leaf curl virus: Characteristics, influence, and regulation mechanism. Plant Physiol. Biochem. PPB 2024, 213, 108812. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Sharma, N.; Hari-Gowthem, G.; Muthamilarasan, M.; Prasad, M. Tomato Yellow Leaf Curl Virus: Impact, Challenges, and Management. Trends Plant Sci. 2020, 25, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Lee, C.C. Function and Structural Organization of the Replication Protein of Bamboo mosaic virus. Front. Microbiol. 2017, 8, 522. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.P.; Chen, I.H.; Tsai, C.H. Host Factors in the Infection Cycle of Bamboo mosaic virus. Front. Microbiol. 2017, 8, 437. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.Y.; Fan, H.Y.; Zeng, Z.; Zhou, Y.H.; Hu, S.Y.; Li, S.X.; Cheng, Y.J.; Meng, X.R.; Chen, F.; Shao, Z.Q.; et al. Comprehensive regulatory networks for tomato organ development based on the genome and RNAome of MicroTom tomato. Hortic. Res. 2023, 10, uhad147. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Itaya, H.; Oshita, K.; Arakawa, K.; Tomita, M. GEMBASSY: An EMBOSS associated software package for comprehensive genome analyses. Source Code Biol. Med. 2013, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Name | CAI | CBI | Fop | ENC | GC3s | GC Content |
|---|---|---|---|---|---|---|
| Arabidopsis thaliana | 0.207 | 0.013 | 0.426 | 55.41 | 0.438 | 0.467 |
| Nicotiana benthamiana | 0.188 | −0.08 | 0.373 | 53.92 | 0.375 | 0.435 |
| Solanum lycopersicum | 0.187 | −0.082 | 0.372 | 52.98 | 0.356 | 0.421 |
| Oryza sativa | 0.231 | 0.14 | 0.498 | 45.6 | 0.769 | 0.634 |
| Glycine max | 0.201 | −0.024 | 0.406 | 55.24 | 0.476 | 0.471 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, K.; Cui, S.; Zhang, S.; Cao, H.; He, L.; Chen, J. Genome-Wide Identification, Evolution and Expression Analysis of GRAS Transcription Factor Gene Family Under Viral Stress in Nicotiana benthamiana. Plants 2025, 14, 2295. https://doi.org/10.3390/plants14152295
Yao K, Cui S, Zhang S, Cao H, He L, Chen J. Genome-Wide Identification, Evolution and Expression Analysis of GRAS Transcription Factor Gene Family Under Viral Stress in Nicotiana benthamiana. Plants. 2025; 14(15):2295. https://doi.org/10.3390/plants14152295
Chicago/Turabian StyleYao, Keyan, Shuhao Cui, Songbai Zhang, Hao Cao, Long He, and Jie Chen. 2025. "Genome-Wide Identification, Evolution and Expression Analysis of GRAS Transcription Factor Gene Family Under Viral Stress in Nicotiana benthamiana" Plants 14, no. 15: 2295. https://doi.org/10.3390/plants14152295
APA StyleYao, K., Cui, S., Zhang, S., Cao, H., He, L., & Chen, J. (2025). Genome-Wide Identification, Evolution and Expression Analysis of GRAS Transcription Factor Gene Family Under Viral Stress in Nicotiana benthamiana. Plants, 14(15), 2295. https://doi.org/10.3390/plants14152295