
Bioinformatics and Functional Validation of CqPRX9L1 in Chenopodium quinoa


Abstract
1. Introduction
2. Results and Discussion
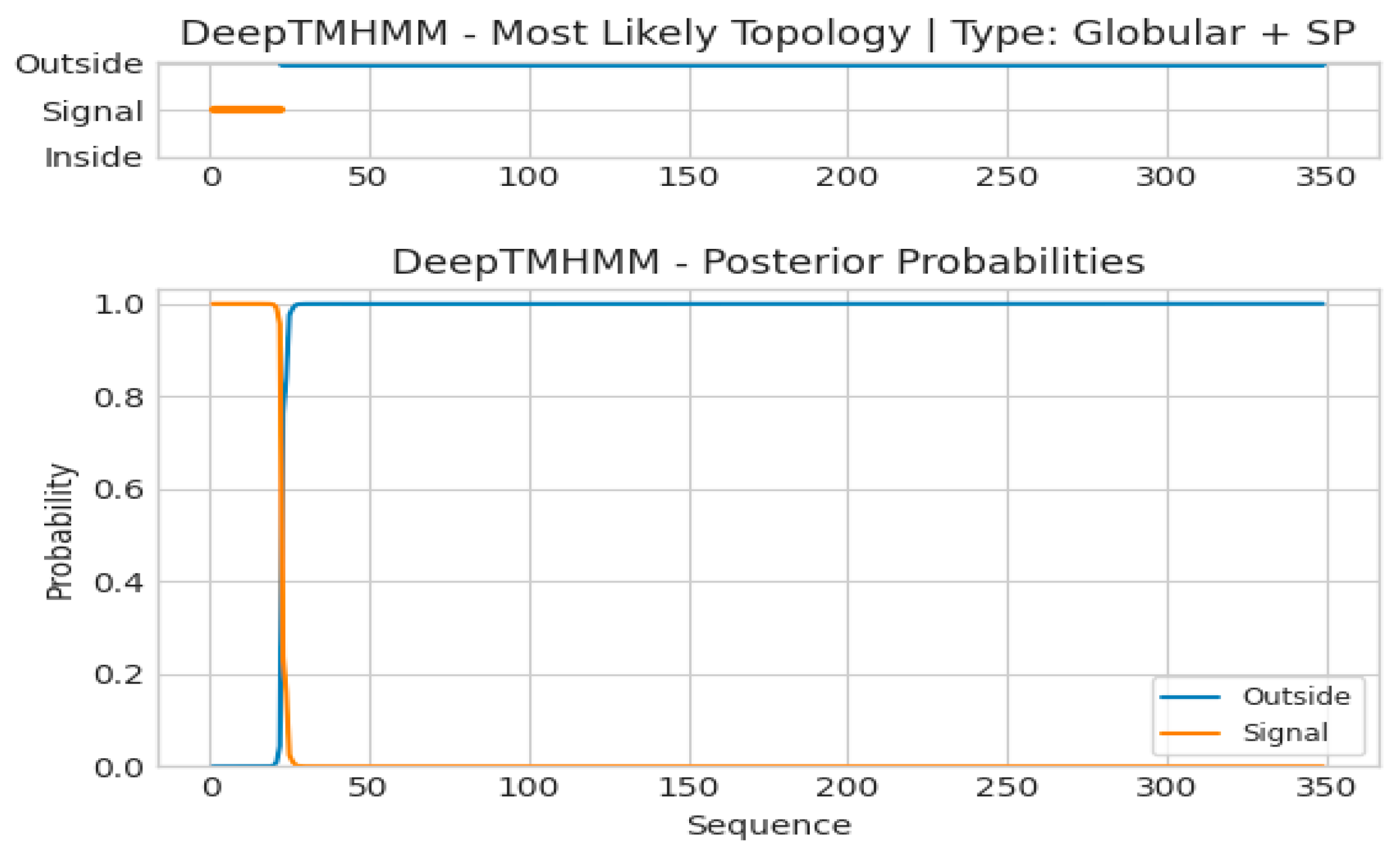
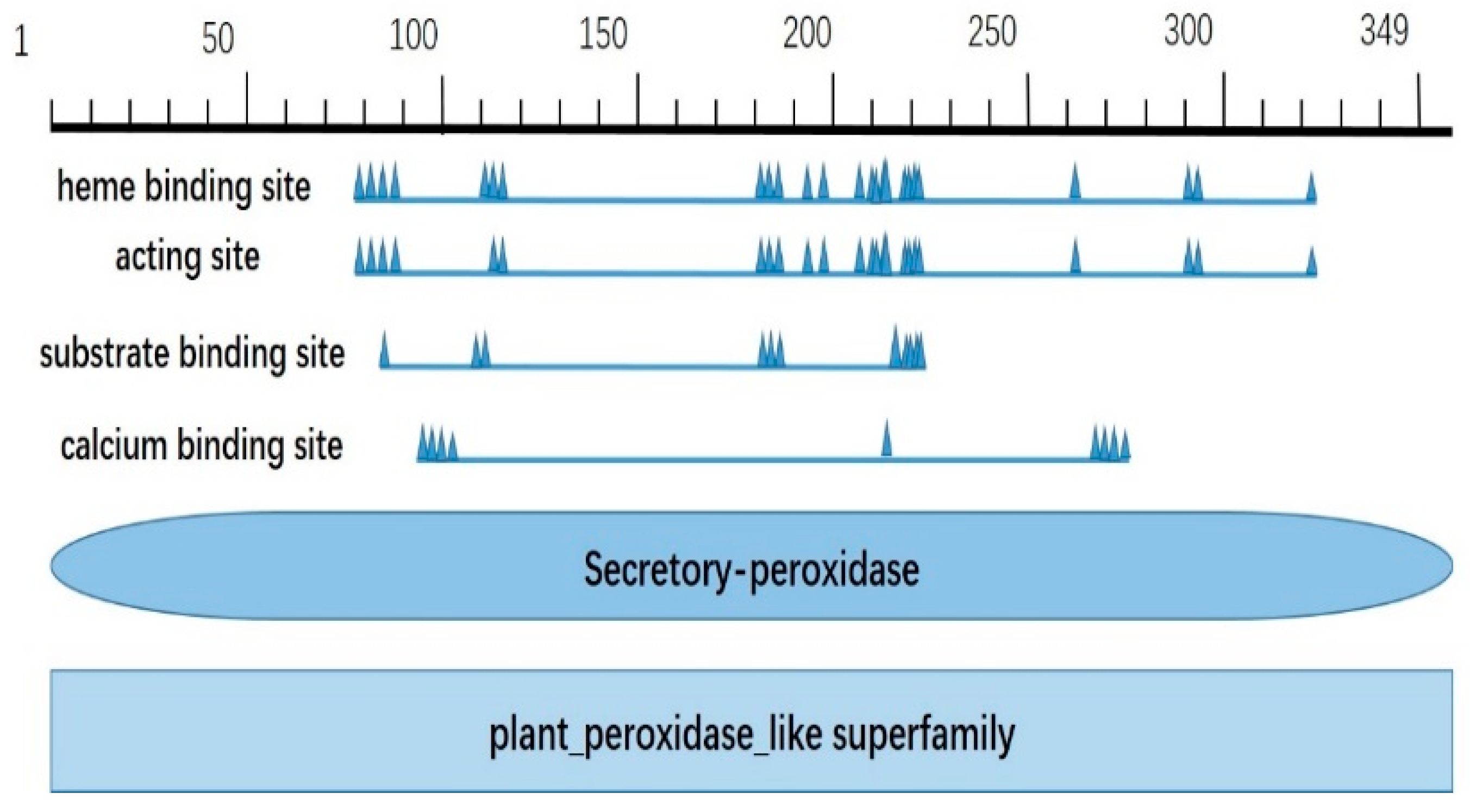
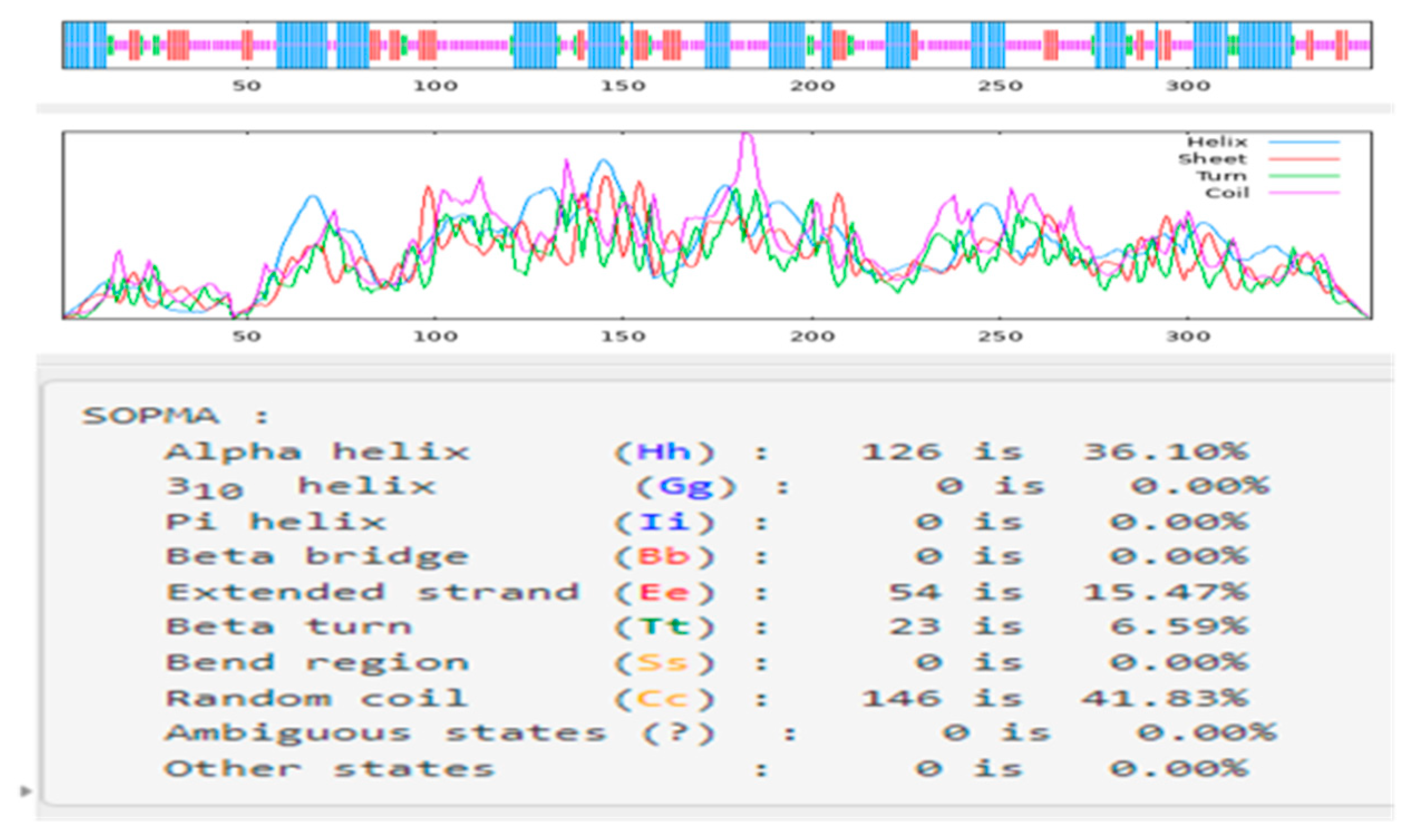
2.1. Bioinformatic Analysis of CqPRX9L1
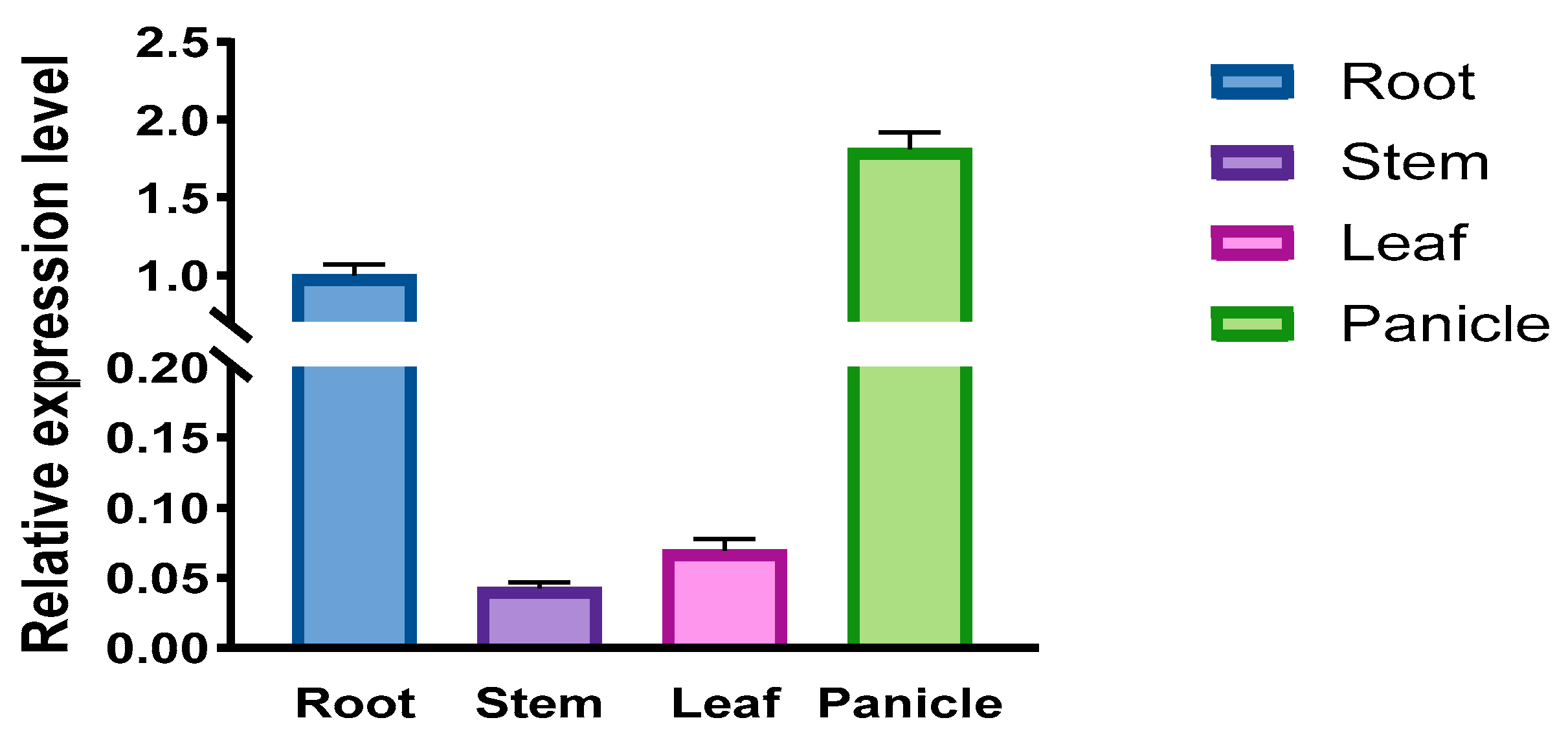
2.2. Expression Analysis of CqPRX9L1 in Quinoa
2.2.1. Spatiotemporal Expression Pattern Analysis of CqPRX9L1
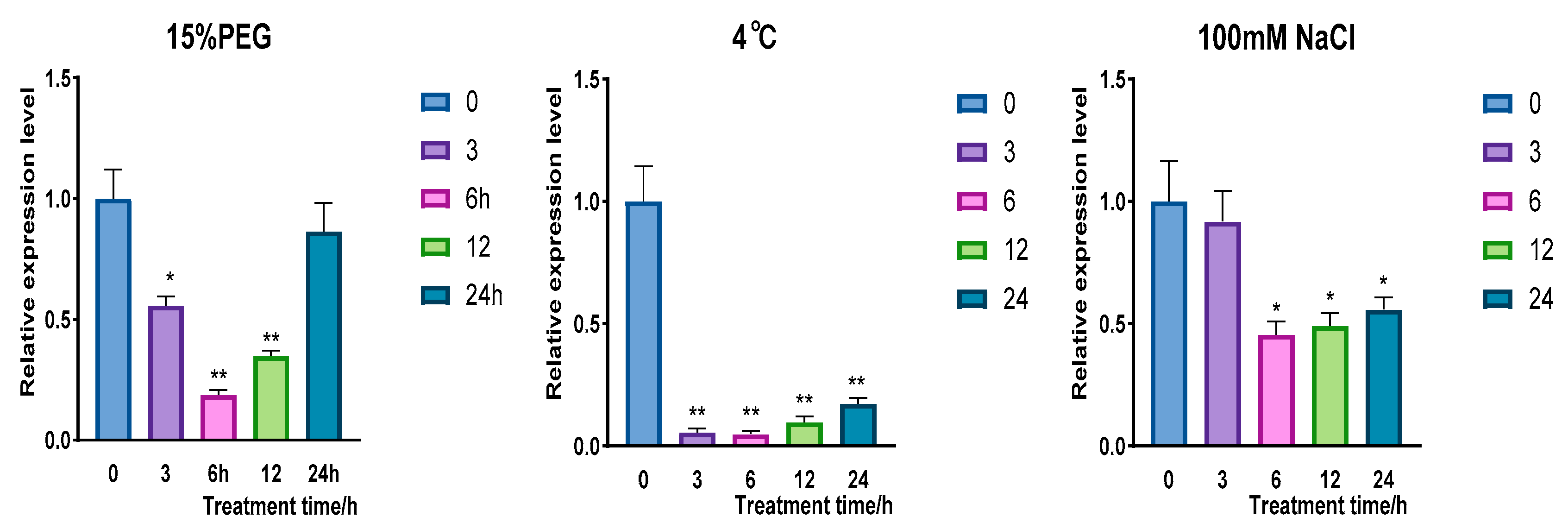
2.2.2. Expression Analysis of CqPRX9L1 Under Different Stress Treatments
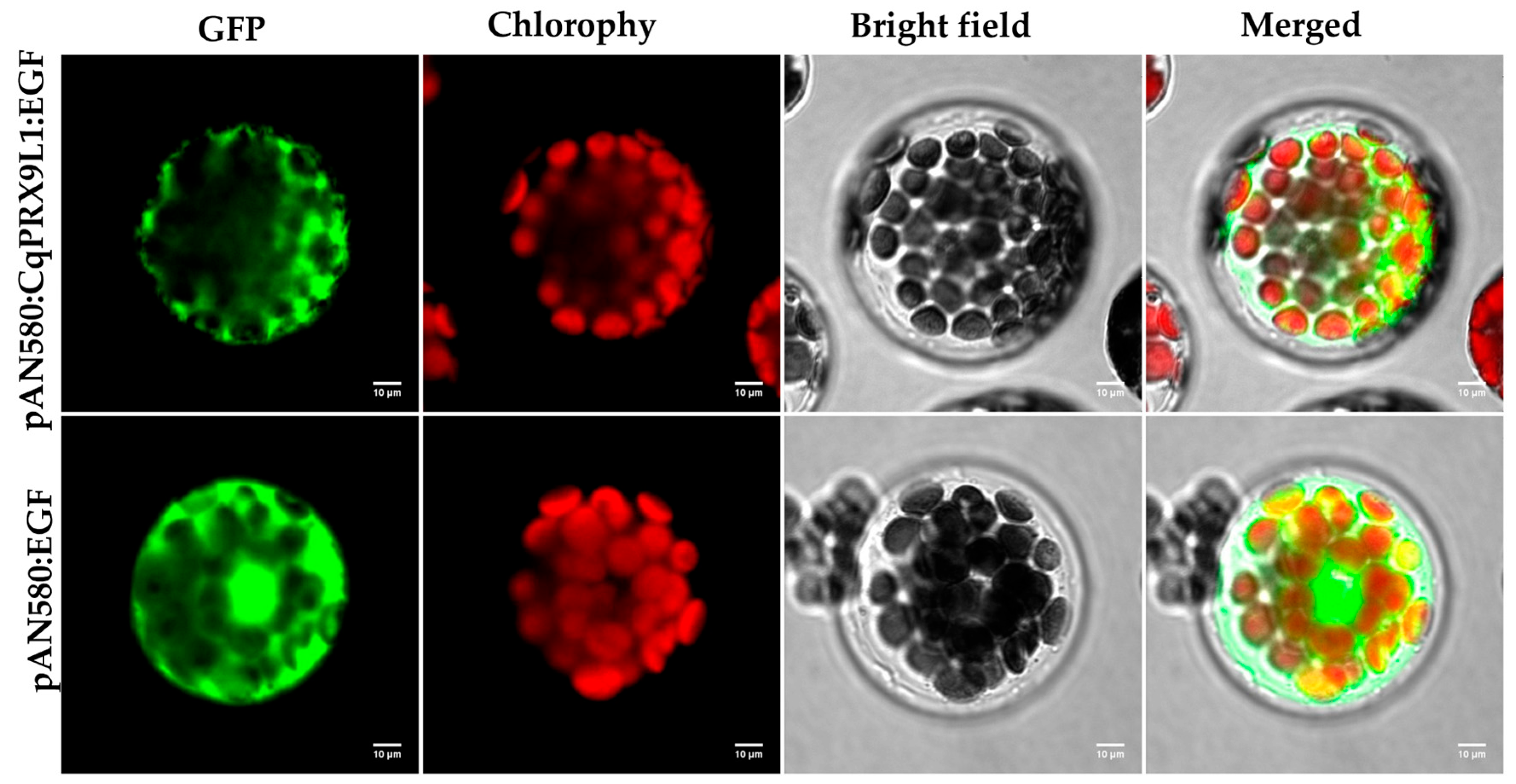
2.3. Subcellular Localization of CqPRX9L1 Protein
2.4. Expression Pattern of CqPRX9L1
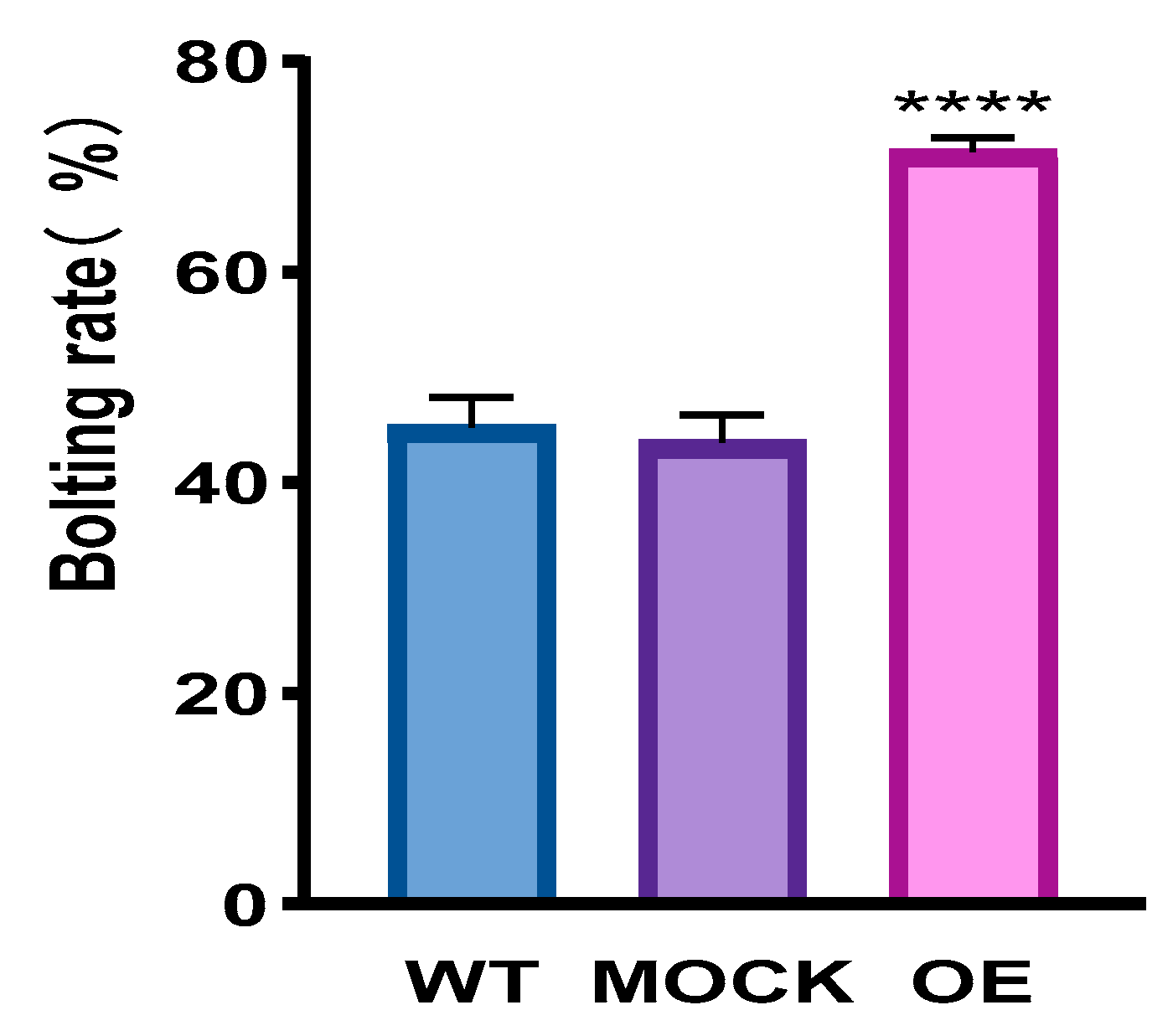
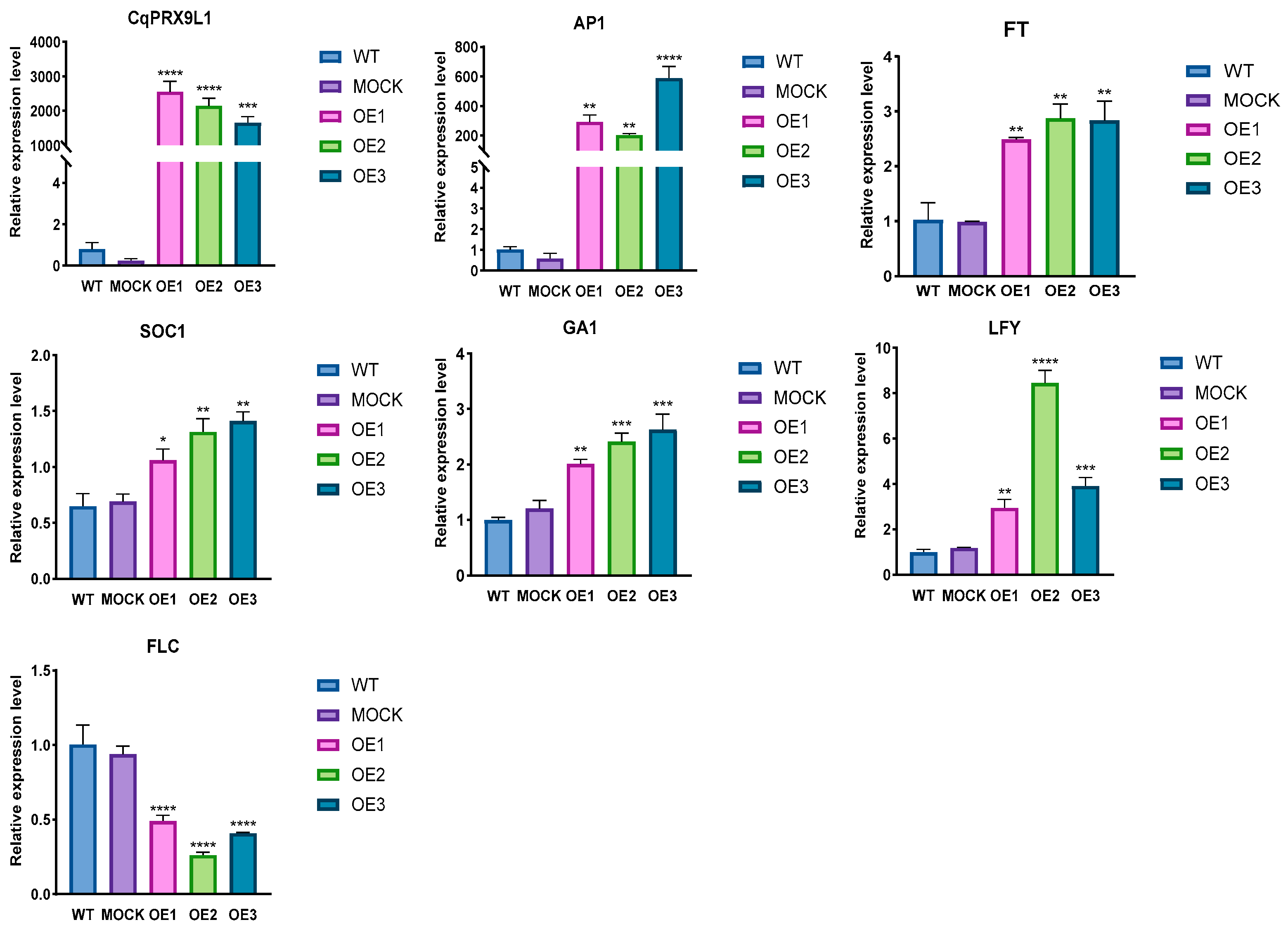
2.4.1. Overexpression of CqPRX9L1 Promotes Arabidopsis Plants to Bolt and Flower by Improving the Expression of Flowering Marker Genes
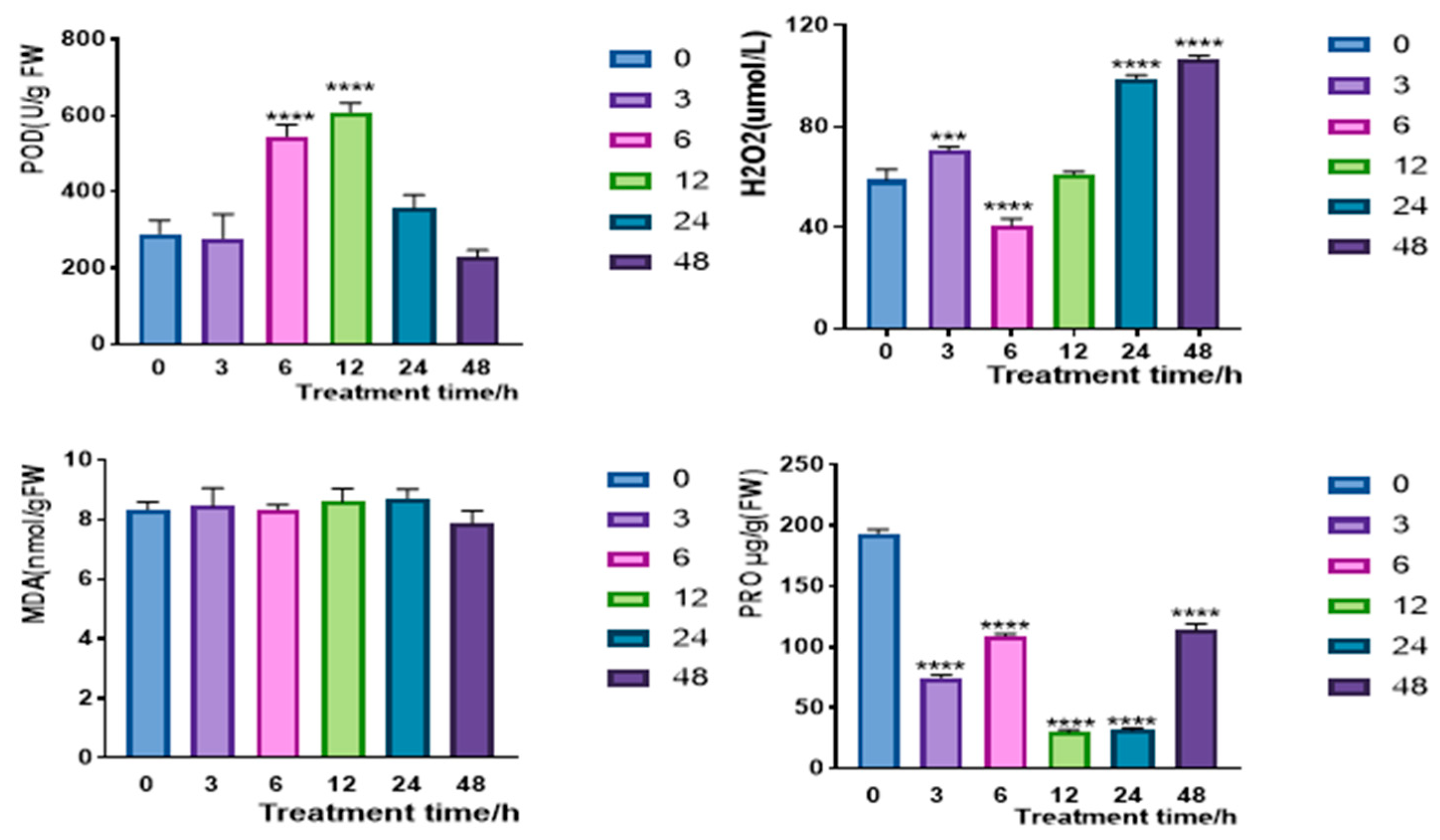
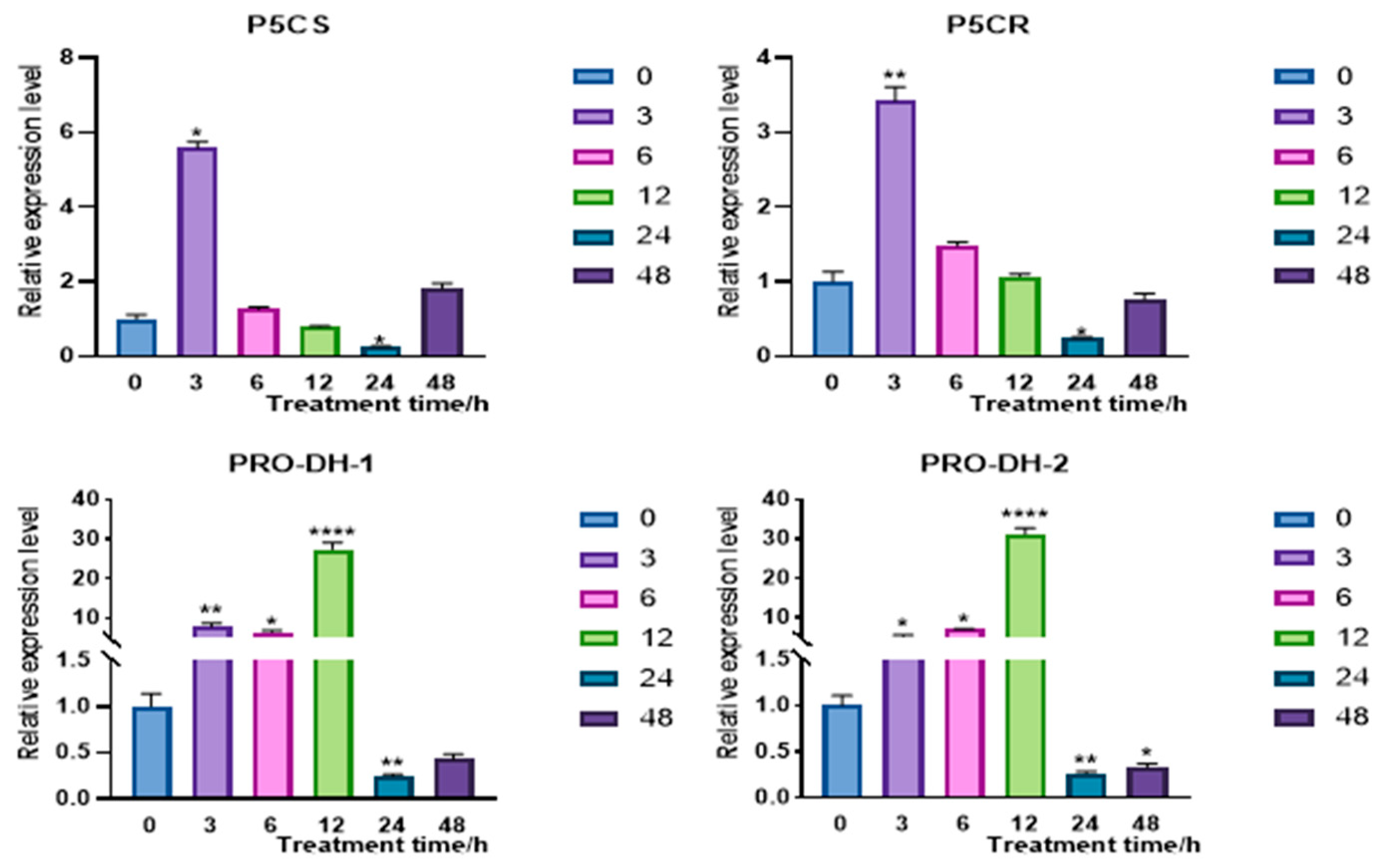
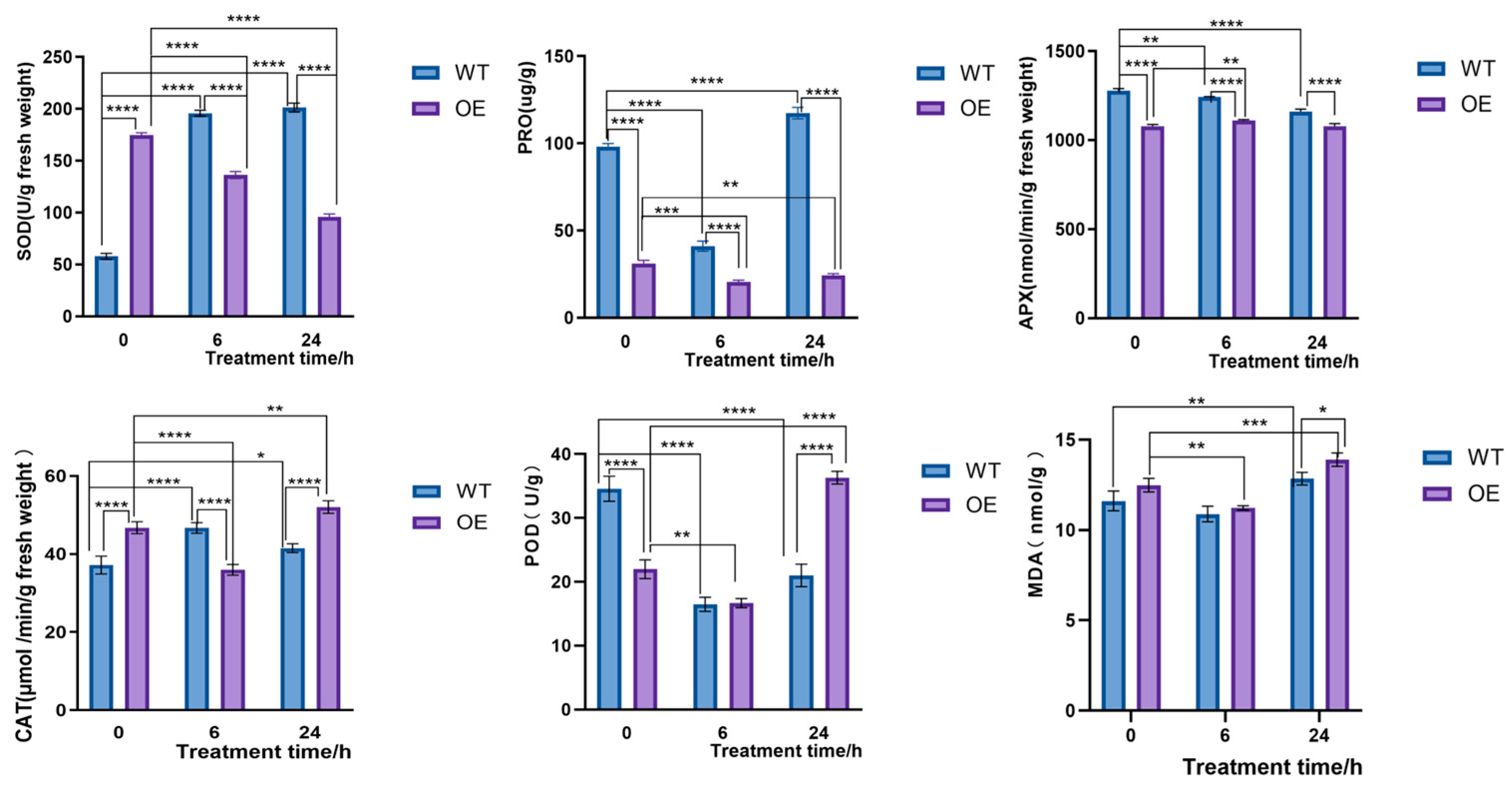
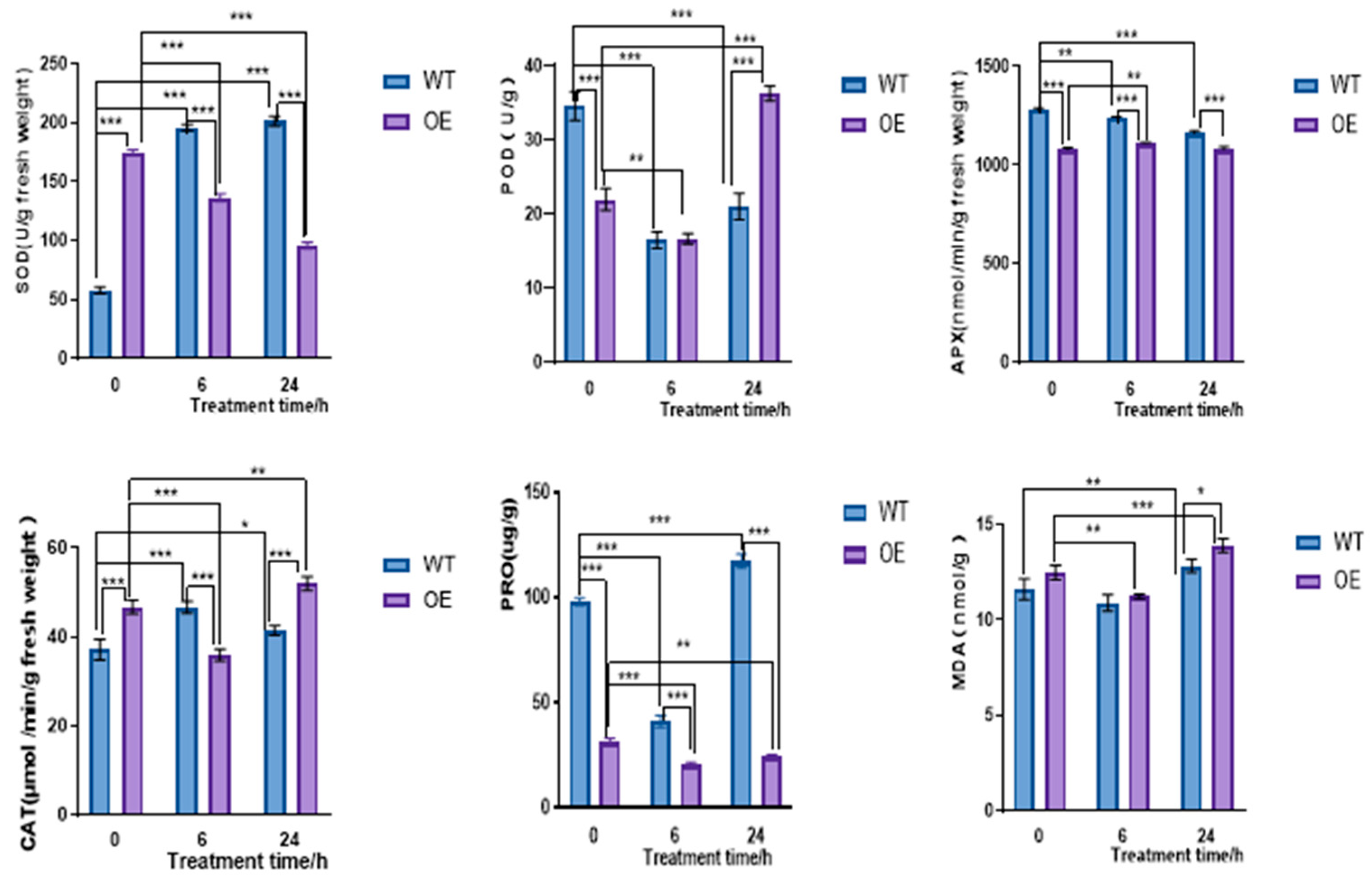
2.4.2. Overexpression of CqPRX9L1 Reduces Abiotic Stress Tolerance
3. Materials and Methods
3.1. Test Materials
3.2. Bioinformatics Analysis of CqPRX9L1
3.3. Subcellular Localization of CqPRX9L1
3.3.1. Construction of Expression Vectors
3.3.2. Transformation of Arabidopsis Protoplasts
3.4. Expression Pattern of CqPRX9L1 in Quinoa
3.4.1. Spatiotemporal Expression Pattern of CqPRX9L1
3.4.2. Expression Patterns and Functional Analysis of CqPRX9L1 Under Different Stress Treatments
3.5. Construction of Plant Heterologous Overexpression Vector CAM-CqPRX9L1-GFP and Genetic Transformation of Arabidopsis thaliana
3.6. Physiological Analysis of Drought Stress Responses
3.6.1. Peroxidase (POD) Activity Assay
- (1)
- Sample pretreatment: Fresh leaf tissue (0.1 g) was homogenized in 1 mL ice-cold extraction buffer using a pre-chilled mortar and pestle. The homogenate was centrifuged at 12,000 rpm for 10 min at 4 °C, and the resulting supernatant was immediately placed on ice for subsequent analysis.
- (2)
- In this study, 10 µL of supernatant and 190 µL of working solution were mixed well, and the absorbance A1 at 470 nm for 10 s and the absorbance A2 at 470 nm for 1 min and 10 s were recorded; ΔA = A2 − A1 is calculated; If the absorbance value at 1 min and 10 s is greater than 1.5 or ΔA is greater than 0.5, the sample can be diluted with extraction solution for detection; If ΔA is less than 0.005, the reaction time can be extended to 5 min.
- (3)
- Quantitative unit definition:
3.6.2. Superoxide Dismutase (SOD) Activity Assay
- (1)
- Sample pretreatment: Fresh leaf tissue (0.1 g) was homogenized in 1 mL ice-cold extraction buffer using a pre-chilled mortar and pestle. The homogenate was centrifuged at 12,000 rpm for 10 min at 4° C, and the resulting supernatant was immediately placed on ice for subsequent analysis.
- (2)
- In this study, 10 µL of supernatant (distilled water was taken as a control), 10 µL of reagent 3, 20 µL of reagent 4, and 160 µL of working solution were mixed well and incubated at room temperature (25 °C) for 30 min, and the absorbance value A of each tube at 450 nm was recorded. Cautionary notes: Reagent 3 contains thermolabile enzymes and must not be frozen. Keep on ice during use.
- (3)
- Quantitative results
3.6.3. Ascorbate Peroxidase (APX) Activity Assay
- (1)
- Sample pretreatment: Fresh leaf tissue (0.5 g) was homogenized in 1 mL ice-cold extraction buffer using a pre-chilled mortar and pestle. The homogenate was centrifuged at 12,000 rpm for 10 min at 4 °C, and the resulting supernatant was immediately placed on ice for subsequent analysis.
- (2)
- In this study, 10 µL of supernatant, 140 µL of reagent 1, 20 µL of reagent 2, and 30 µL of diluted reagent 3 were mixed well, and the absorbance values A1 and A2 at 290 nm for 10 s and 130 s were recorded; ΔA = A2 − A1 is calculated. If a large number of bubbles are generated during the reaction process, it indicates high enzyme activity. The supernatant can be diluted before measurement. If ΔA is negative and the value is small, the reaction time can be extended.
- (3)
- Quantitative definition of active unit:
- ε is the molar absorptivity of AsA at 290 nm, 2.8 × 103 L/mol/cm;
- d is the 96-well plate optical diameter (cm), 0.5 cm;
- Vtr is the total volume of the reaction system (L);
- 200 μL = 2 × 10−4 L;
- 109: 1 mol = 1 × 109 nmol;
- W is the sample quality;
- Vs is the volume of supernatant (mL) added to the reaction system;
- 20 μL = 0.02 mL;
- Vts is the extraction volume, 1 mL;
- T is the catalytic reaction time (min), 2 min.
3.6.4. Catalase (CAT) Activity Assay
- (1)
- Sample pretreatment: Fresh leaf tissue (0.5 g) was homogenized in 1 mL ice-cold extraction buffer using a pre-chilled mortar and pestle. The homogenate was centrifuged at 12,000 rpm for 10 min at 4 °C, and the resulting supernatant was immediately placed on ice for subsequent analysis.
- (2)
- In this study, 10 µL of supernatant and 190 µL of the working solution were mixed well, and the absorbance values A1 at 240 nm for 10 s and A2 for 1 min and 10 s were recorded; and ΔA = A2 − A1 is calculated; Note: If ΔA is negative during detection, a kinetic cycle can be set (To measure absorbance every 30 s or 1 min for 5–10 min), and the absorbance difference within a 1-minute interval in the decreasing range should be taken. If visible bubbles are observed after the reaction, it indicates high sample activity, and the sample can be diluted before detection. For a large number of samples, a multichannel pipette can be used to add the working solution.
- (3)
- Quantitative results
- Vtr is the total volume of the reaction system, 2 × 10−4 L;
- Ε is the molar extinction coefficient of H2O2, 43.6 L/mol/cm;
- D is the 96-well plate optical diameter, 0.5 cm;
- Vs is the added sample volume, 0.01 mL;
- Vts is the added extraction solution volume, 1 mL;
- T is the reaction time, 1 min;
- W is the sample quality, g.
3.6.5. Determination of Proline Content
- (1)
- Sample pretreatment: Fresh leaf tissue (0.05 g) was homogenized in 0.5 mL extraction buffer in a 90 °C water bath for 10 min. The homogenate was centrifuged at 10,000 rpm for 10 min at 25 °C, and the resulting supernatant was immediately placed on ice for subsequent analysis.
- (2)
- In this study, 250 µL of supernatant (or 250 µL of extract as control), 250 µL of reagent 1, and 250 µL of reagent 2 were mixed well and placed in a 92 °C water bath for 30 min (covered tightly to prevent water loss), cooled down, 500 µL of reagent 3 was added, and shaken for 30 s, and then the pigment was transferred to reagent 3. Then, 200 μL of the upper solution was sucked into a 96-well plate, A520 was measured, and ΔA = Am − Ab was calculated. If ΔA is greater than 1, the supernatant was diluted with the extraction solution for measurement and multiplied by the dilution factor when calculating. If ΔA is less than 0.0047, it is recommended to weigh 0.2 g of sample and add 1 mL of extraction solution for concentration treatment.
- (3)
- Quantitative results
- V1 is the sample volume added to the reaction system, 0.25 mL;
- V2 is the extract volume added, 0.5 mL;
- W is the sample quality, g;
- Am is the A measure;
- Ab is the A blank.
3.6.6. Determination of MDA Content
- (1)
- Sample pretreatment: Fresh leaf tissue (0.1 g) was homogenized in 1 mL extraction buffer, the mixture was homogenized at room temperature and centrifuged at a speed of 10,000 rpm for 10 min at room temperature, and the supernatant was taken for testing.
- (2)
- In this study, 100 µL of supernatant and 300 µL of reagent 2 were mixed well and water-bathed at 92 °C for 30 min (covered tightly to prevent water loss); after cooling, they were centrifuged at a speed of 12,000 rpm at 25 °C for 10 min. Then, 200 μL of supernatant was taken, and the absorbance values A at 532 nm and 600 nm were measured in a 96-well plate, denoted as A532 and A600, with ΔA = A532 − A600. If ΔA ≤ 0, it indicates that the MDA content of the sample is low, and the sampling quality can be increased, such as 0.2 g plus 1 mL of extraction solution.
- (3)
- Quantitative results
3.6.7. Determination of H2O2 Content
- (1)
- Sample pretreatment: Fresh leaf tissue (0.1 g) was homogenized in 1 mL TCA, the mixture was homogenized at 4 °C, and centrifuged at a speed of 12,000 rpm for 15 min.
- (2)
- In this study, 200 µL of supernatant, 200 µL of phosphate buffer, and 400 µL of KI were mixed, vortexed thoroughly, and incubated for 10 min, and the OD value at 390 nm was measured.
- (3)
- Quantitative results
3.7. Statistical Analyses
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dunford, H.B.; Stillman, J.S. On the function and mechanism of action of peroxidases. Coord. Chem. Rev. 1976, 19, 187–251. [Google Scholar] [CrossRef]
- Bhatt, I.; Tripathi, B.N. Plant peroxiredoxins: Catalytic mechanisms, functional signifificance and future perspectives. Biotechnol. Adv. 2011, 29, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Feng, J.J.; Qu, Y.F.; Zhang, J.L.; Zhang, L.; Liang, D.; Yang, Y.J.; Huang, J.L. Genome-wide identifification and functional analysis of class III peroxidases in Gossypium hirsutum. PeerJ 2022, 10, e13635. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.L.; Xu, R.Q.; Suo, X.M.; Li, J.S.; Gu, P.P.; Yao, R.; Lin, X.H.; Gao, H. Genome-wide identification of the Class III PRX gene family in foxtail millet (Setaria italica L.) and expression analysis under drought stress. Acta Agron. Sin. 2022, 48, 2517–2532. [Google Scholar]
- Cosio, C.; Ranocha, P.; Francoz, E.; Burlat, V.; Zheng, Y.; Perry, S.E.; Ripoll, J.J.; Yanofsky, M.; Dunand, C. The class III peroxidase PRX17 is a direct target of the MADS-box transcription factor Agamous-like15 (AGL15) and participates in lignified tissue formation. New Phytol. 2017, 213, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Tognoli, M.; Penel, C.; Greppin, H.; Simon, P. Analysis and expression of the class III peroxidase large gene family in Arabidopsis thaliana. Gene 2002, 288, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Shen, X.L.; Sun, X.X.; Wang, J.; Yuan, Q.P. Research progress in recombinant expression and application of peroxidases. Synth. Biol. J. 2024, 5, 1498–1517. [Google Scholar] [CrossRef]
- Hiraga, S.; Sasaki, K.; Ito, H.; Ohashi, Y.; Matsui, H. A large family of class III plant peroxidases. Plant Cell Physiol. 2001, 42, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Passardi, F.; Longet, D.; Penel, C.; Dunand, C. The Class III peroxidase Multigenic Family in Rice and Its Evolution in Land Plants. Phytochemistry 2004, 65, 1879–1893. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Liu, Y.; Yang, J.; Ma, H.L.; Qin, C.; Wang, N.; Shen, W.Q. The Class III Peroxidase Gene IPH1 Regulates Plant Height in Rice. J. Southwest Univ. Nat. Sci. Ed. 2024, 46, 24–33. [Google Scholar]
- Joseph, R.J.; William, C.D.; Weng, J.K. PRX9 and PRX40 are extensin peroxidases Essential for maintaining tapetum and microscope cell wall integrity during Arabidopsis anther development. Plant Cell 2019, 31, 848–861. [Google Scholar] [CrossRef]
- Luthie, S.; Martinez, C.T. Membrane-bound class III peroxidases: Unexpected enzymes with exciting functions. Int. J. Mol. Sci. 2018, 19, 2876. [Google Scholar] [CrossRef] [PubMed]
- Passardi, F.; Penel, C.; Dunand, C. Performing the paradoxical:how plant peroxidases modify the cell wall. Trends Plant Sci. 2004, 9, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rubio, M.C.; Alassimone, J.; Geldner, N. A mechanism for localized lignin deposition in the endodermis. Cell 2013, 153, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Kang, C.H.; Park, J.H.; Lee, S.Y. Physiological significance of plant peroxiredoxins and the structure related and multifunctional biochemistry of peroxiredoxin. Antioxid. Redox Signal. 2018, 28, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Devireddy, A.R.; Zandalinas, S.I.; Fichman, Y. Integration of reactive oxygen species and hormone signaling during abiotic stress. Plant J. 2021, 105, 459–476. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.; Edouard, P. Phenoloxidases in Plants—How Structural Diversity Enables Functional Specificity. Front Plant Sci. 2021, 12, 754601. [Google Scholar] [CrossRef]
- Shigeto, J.; Tsutsumi, Y. Diverse functions and reactions of class III peroxides. New Phytol. 2016, 209, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Liu, X.X.; Wang, C.H.; Zhang, J.G.; Rao, D.L. Evolution and expression analysis of the class III peroxidase family in olive. J. Beijing For. Univ. 2023, 45, 36–49. [Google Scholar] [CrossRef]
- Meng, Y.Y.; Fan, S.L.; Song, M.Z.; Pang, C.Y.; Yu, S.X. Advance in Research on Class III Peroxidases and Its Function in Plants. Acta Bot Boreali-Occident. Sin. 2011, 31, 1908–1916. [Google Scholar]
- Liu, H.; Dong, S.Y.; Li, M.; Gu, F.W.; Yang, G.L.; Guo, T.; Chen, Z.Q.; Wang, J.F. The Class III peroxidase gene OsPrx30, transcriptionally modulated by the AT-hook protein OsATH1, mediates rice bacterial blight-induced ROS accumulation. J. Integr. Plant Biol. 2021, 63, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Mei, W.Q.; Qin, Y.M.; Song, W.Q.; Li, J. Cotton GhPOX1encoding plant class III peroxidase may be responsible for the high level of reactive oxygen species production that is related to cotton fiber elongation. J. Genet. Genom. 2009, 36, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Q.Q.; Zhao, Y.; Han, G.M.; Zhu, S.W. Systematic analysis of maize class III peroxidase gene family reveals a conserved subfamily involved in abiotic stress response. Gene 2015, 566, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yuan, J.; Luo, W.; Qin, M.; Yang, J.; Wu, W.; Xie, X. Genome-wide identification and expression analysis of the class III peroxidase gene family in potato (Solanum tuberosum L.). Front. Genet. 2020, 11, 593577. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.M.; Ranocha, P.; Kasulin, L.; Fusari, C.M.; Servi, L.; Aptekmann, A.A.; Gabarain, V.B.; Peralta, J.M.; Borassi, C.; Marzol, E.; et al. Apoplastic class III peroxidases PRX62 and PRX69 promote Arabidopsis root hair growth at low temperature. Nat. Commun. 2022, 13, 1310. [Google Scholar] [CrossRef] [PubMed]
- Llorente, F.; López-Cobollo, R.M.; Catalá, R.; Martínez-Zapater, J.M.; Salinas, J. A novel cold-inducible gene from Arabidopsis, RCI3, encodes a peroxidase that constitutes a component for stress tol erance. Plant J. 2002, 32, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Xin, F.; Wei, S.; Liu, Y.; Han, Y.; Xie, J.; Ding, Q.; Ma, L. Genome-wide identification, phylogeny and expression profiling of class III peroxidases gene family in Brachypodium distachyon. Gene 2019, 700, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Kim, S.Y.; Nam, K.H. Genes Encoding Plant-Specific Class III Peroxidases Are Responsible for Increased Cold Tolerance of the brassinosteroid-insensitive 1 Mutant. Mol. Cells 2012, 34, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Maria, K.; Yogeshwar, V.D.; Neelam, G.; Madhu, T.; Iffat, Z.A.; Mehar, H.A.; Debasis, C. Oryza sativa class III peroxidase (OsPRX38) overexpression in Arabidopsis thaliana reduces arsenic accumulation due to apoplastic lignification. J. Hazard. Mater. 2019, 362, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Su, P.S.; Li, W.; Xiao, G.L.; Zhao, Y.; Ma, X.; Wang, H.W.; Nevo, E.; Kong, L.R. Genome-wide and evolutionary analysis of the class III peroxidase gene family in wheat and Aegilops tauschii reveals that some members are involved in stress responses. BMC Genom. 2019, 20, 666. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.H.; Wang, H.; Li, G.; Tang, X.Y.; Wang, C.G.; Zhang, L.; Hou, J.F.; Yuan, L.Y. Identification and bioinformatics analysis of PRX gene family in Chinese cabbage. J. Zhejiang Univ. Agric. Life Sci. 2020, 46, 677–686. [Google Scholar]
- Huan, X.J.; Li, L.; Liu, Y.J.; Kong, Z.Y.; Liu, Y.J.; Wang, Q.C.; Liu, J.N.; Zhang, P.; Guo, Y.R.; Qin, P. Integrating transcriptomics and metabolomics to analyze quinoa (Chenopodium quinoa Willd.) responses to drought stress and rewatering. Front Plant Sci. 2022, 13, 988861. [Google Scholar] [CrossRef] [PubMed]
- Salekdeh, G.H.; Reynolds, M.; Bennett, J.; Boyer, J. Conceptual framework for drought phenotyping during molecular breeding. Trends Plant Sci. 2009, 14, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.Z.; Yan, X.C.; Zenda, T.S.; Wang, N.; Dong, A.Y.; Yang, Q.; Zhong, Y.; Xing, Y.; Duan, H.J. Overexpression of the peroxidase gene ZmPRX1 increases maize seedling drought tolerance by promoting root development and lignification. Crop J. 2024, 12, 753–765. [Google Scholar] [CrossRef]
- Luo, Y.C.; Wang, Y.Y.; Li, X.; Yang, X.H.; Bai, H.R.; Liao, X.Q.; Luo, X.L.; Zhang, F.; Zhang, L.; Liu, Q.L. Transcription factor DgMYB recruits H3K4me3 methylase to DgPeroxidase to enhance chrysanthemum cold tolerance. Plant Physiol. 2024, 194, 1104–1119. [Google Scholar] [CrossRef] [PubMed]
- Li, S.M.; Zheng, H.X.; Sui, N.; Zhang, F.N. Class III peroxidase: An essential enzyme for enhancing plant physiological and developmental process by maintaining the ROS level: A review. Int. J. Biol. Macromol. 2024, 283, 137331. [Google Scholar] [CrossRef] [PubMed]
- Kishor, P.B.K.; Sangam, S.; Amrutha, R.N.; Laxmi, P.S.; Naidu, K.R.; Rao, K.R.S.S.; Rao, S.; Reddy, K.J.; Theriappan, P.; Sreenivasulu, N. Regulation of proline biosynthesis, degradation, uptake and transport in higher plants: Its implications in plant growth and abiotic stress tolerance. Curr. Sci. 2005, 88, 424–438. Available online: http://www.jstor.org/stable/24110209 (accessed on 7 May 2025).
- Ge, W.D.; Wang, T.H.; Ma, T.Y.; Fan, Z.Y.; Wang, Y.S. Genome-wide Identification of the PRX Gene Family in Cabbage (Brassica oleracea L. var. capitata) and Expression Analysis Under Abiotic Stress. Biotechnol. Bull 2023, 39, 252–260. [Google Scholar]
- Székely, G.; Abrahám, E.; Cséplo, A.; Rigó, G.; Zsigmond, L.; Csiszár, J.; Ayaydin, F.; Strizhov, N.; Jásik, J.; Schmelzer, E.; et al. Duplicated P5CS genes of Arabidopsis play distinct roles in stress regulation and developmental control of proline biosynthesis. Plant J. 2008, 53, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Molecular responses to drought and cold stress. Curr. Opin. Biotechnol. 1996, 7, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Boyes, D.C.; Zayed, A.M.; Ascenzi, R.; McCaskill, A.J.; Hoffman, N.E.; Davis, K.R.; Görlach, J. Growth stage-based phenotypic analysis of Arabidopsis: A model for high throughput functional genomics in plants. Plant Cell 2001, 13, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.Y.; Liu, Y.J.; Yuan, S.H.; Yue, J.R.; Li, Y.M.; Liao, X.Z.; Ying, S.K.; Liu, Z.H.; Bai, J.F.; Zhang, L.P. Superoxide dismutase promotes early flowering in Triticum aaestivum L. Agric. Commun. 2023, 1, 100007. [Google Scholar] [CrossRef]
- Gamborg, O.L.; Phillips, G.C. Media Preparation and Handling. In Plant Cell, Tissue and Organ Culture—Fundamental Methods; Gamborg, O.L., Phillips, G.C., Eds.; Springer: Berlin/Heidelberg, Germany, 1995; pp. 21–34. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.F. Bacterial transformation by electroporation. Methods Enzymol. 1994, 235, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.D.; Cho, Y.H.; Sheen, J. Arabidopsis mesophy ll protoplasts: A versatile cell system for transient gene expression analysis. Nat. Protoc. 2007, 2, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using Real-time quantitative PCR and the 2−ΔΔCT method. Methods 2002, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, R.; Hong, T.; Wang, D.; Li, Q.; Wu, J.; Zhang, H.; Zhou, K.; Yang, H.; Zhang, T.; et al. Natural variation in the promoter of qRBG1/OsBZR5 underlies enhanced rice yield. Nat. Commun. 2024, 15, 8565. [Google Scholar] [CrossRef] [PubMed]


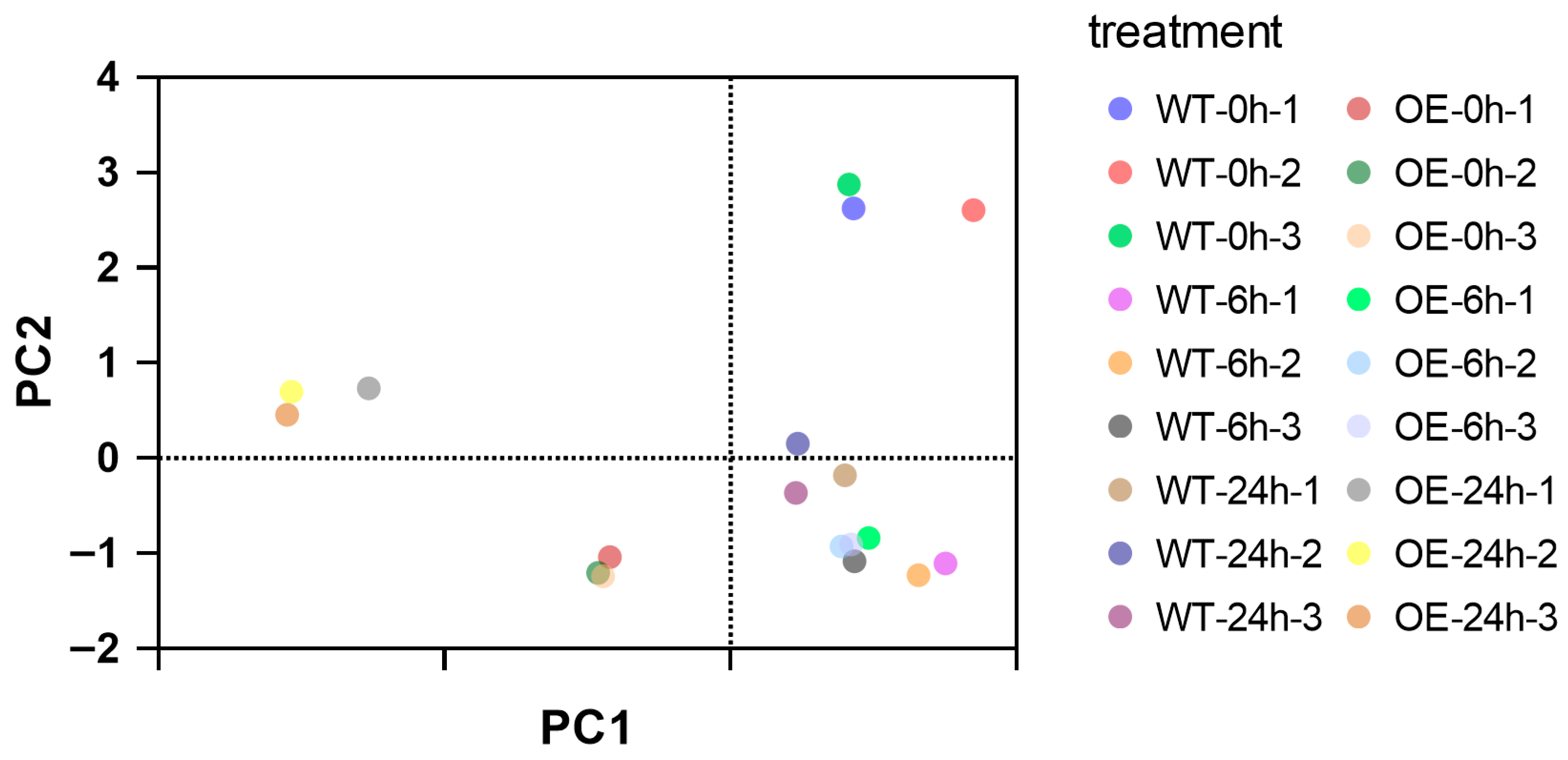
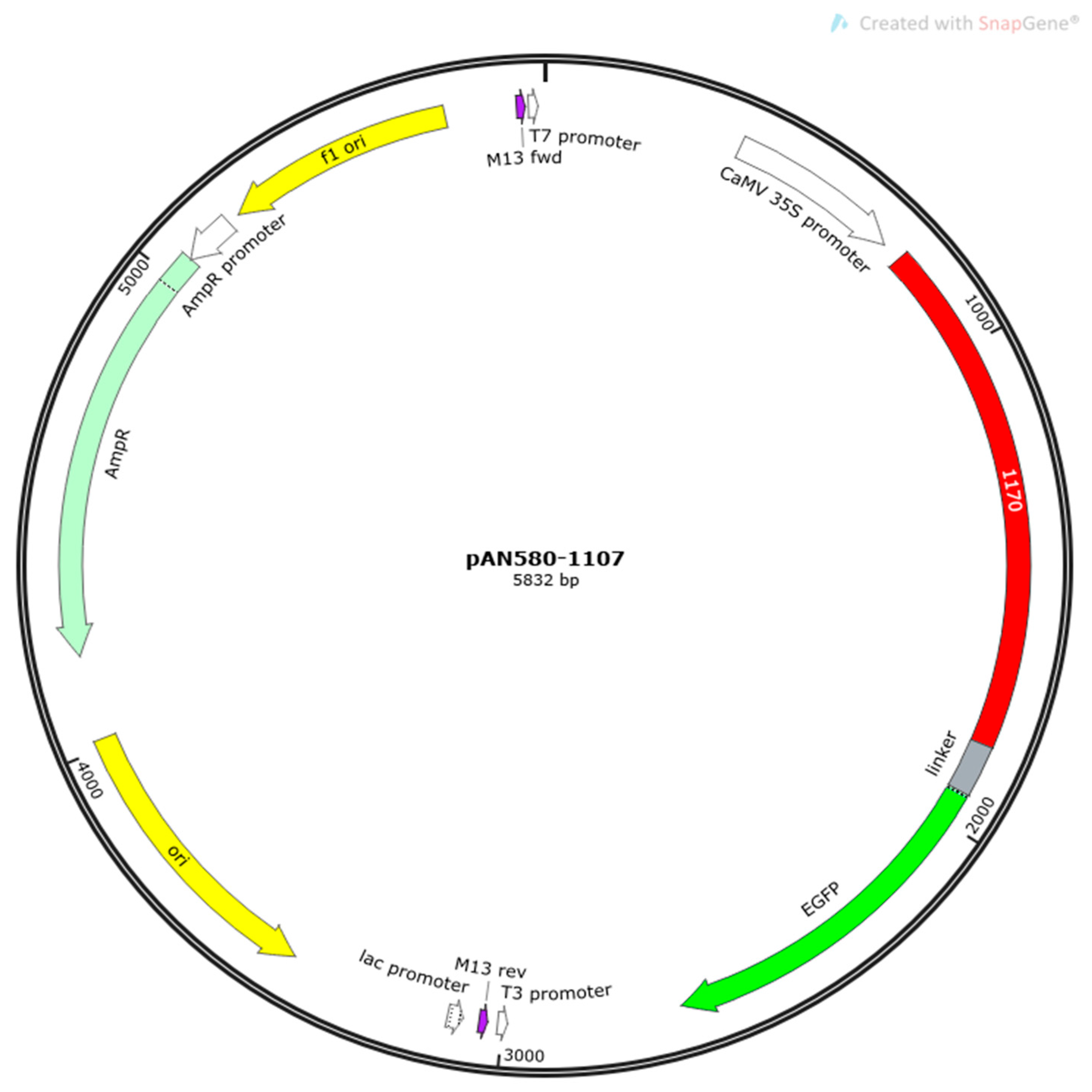

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compare Group | Up-Regulated Genes | Down-Regulated Genes | Total |
|---|---|---|---|---|
| 1 | d5 vs. ck | 708 | 583 | 1291 |
| 2 | d10 vs. ck | 1068 | 821 | 1889 |
| 3 | d15 vs. ck | 1971 | 3382 | 5353 |
| 4 | D20 vs. ck | 3313 | 5678 | 8991 |
| Cis-Element | Location | Sequence | Function |
|---|---|---|---|
| I box | −1377 to −1366 | CCATATTCAATA | Photoinduction element |
| W box | −602 to −597 | TTGACC | Participate in multiple stress responses collaborating with WRKY |
| ERE | −948 to −941 | ATTTTAAA | Ethylene-responsive element |
| DRE1 | −541 to −535 | ACCGAGA | Dehydration-response element |
| ARE | −401 to −396 | AAACCA | Anaerobic induction element |
| MBS | −2322 to −2317 | CAACTG | MYB binding sites related to drought induction |
| TCT motif | −2395 to −2390, −732 to −727 | TCTTAC | Photoreactive element |
| TCA element | −1604 to −1595 | TCAAAAGAGG | Elements related to salicylic acid reaction |
| TGACG motif | −1589 to −1585 | TGACG | MeJA |
| ATCT motif | −395 to −387 | AACTAATCC | Conservative structural domains related to photoreaction |
| CAT box | −1822 to −1817 | GCCACT | Elements related to meristem expression |
| MRE | −1512 to −1506 | AACCTAA | MYB binding sites related to Photoreaction |
| box4 | −1420 to −1415 | ATTAAT | Conservative structural domains related to photoreaction |
| ABRE | −579 to −575 | ACGTG | Abscisic acid reaction element |
| LTR | −558 to −552 | CCGAAA | Low-temperature reaction element |
| ATC motif | −357 to −350 | AGTAATCT | Conservative structural domains related to photoreaction |
| Primer Names | Primer Sequences (5′→3′) |
|---|---|
| pAN580 F | AAGTCCGGAGCTAGCTCTAGATGGCTACAAAAATGCACCTAGTGTTTTTGGC |
| pAN580 R | AGCGGCCGCTGTACAGGATCATTAGGGCGACGACAATACTTCCTAACTTC |
| CAM-FLAG-GFP F | TGGAGAGGACACGCTCGAGATGGCTACAAAAATGCACCTAGT |
| CAM-FLAG-GFP R | CATCCTTGTAGTCGAATTCATTAGGGCGACGACAATACTTC |
| CAM-FLAG-GFP-PF | AGAAGACGTTCCAACCACG |
| CAM-FLAG-GFP-PR | CGGTAAGGATCTGAGCTACAC |
| CAM-FLAG-GFP-TF | TGACGCACAATCCCACTATC |
| CAM-FLAG-GFP-TR | GCGACAAGATCAACTTCATCTAG |
| CqproDH-1-F | CCGACAAGAAGAACAAGACCTT |
| CqproDH-1-R | CGCCGAGTTATACGTCAAGTAA |
| CqproDH-2-F | CAGTCCTTGCCACCCATAATG |
| CqproDH-2-R | CGCCTCCGACATTCCGTAT |
| Cqp5CR-F | GGATGTGGTGTTACAGTTGAGA |
| Cqp5CR-R | GCAGCAGGAGTATTAGGCATTA |
| CqP5CS-F | GGAGAATGGCACTTGGAAGAC |
| CqP5CS-R | TGACGACCAGCACTGACAG |
| CqACT1-F | GTCCACAGAAAGTGCTTCTAAG |
| CqACT1-R | AACAACTCCTCACCTTCTCAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Song, L.; Wang, Y.; Zhao, L.; Wang, C. Bioinformatics and Functional Validation of CqPRX9L1 in Chenopodium quinoa. Plants 2025, 14, 2246. https://doi.org/10.3390/plants14142246
Guo H, Song L, Wang Y, Zhao L, Wang C. Bioinformatics and Functional Validation of CqPRX9L1 in Chenopodium quinoa. Plants. 2025; 14(14):2246. https://doi.org/10.3390/plants14142246
Chicago/Turabian StyleGuo, Hongxia, Linzhuan Song, Yufa Wang, Li Zhao, and Chuangyun Wang. 2025. "Bioinformatics and Functional Validation of CqPRX9L1 in Chenopodium quinoa" Plants 14, no. 14: 2246. https://doi.org/10.3390/plants14142246
APA StyleGuo, H., Song, L., Wang, Y., Zhao, L., & Wang, C. (2025). Bioinformatics and Functional Validation of CqPRX9L1 in Chenopodium quinoa. Plants, 14(14), 2246. https://doi.org/10.3390/plants14142246