
Knockout of GmCKX3 Enhances Soybean Seed Yield via Cytokinin-Mediated Cell Expansion and Lipid Accumulation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Preparation of GmCKX3-Genome-Edited Soybean
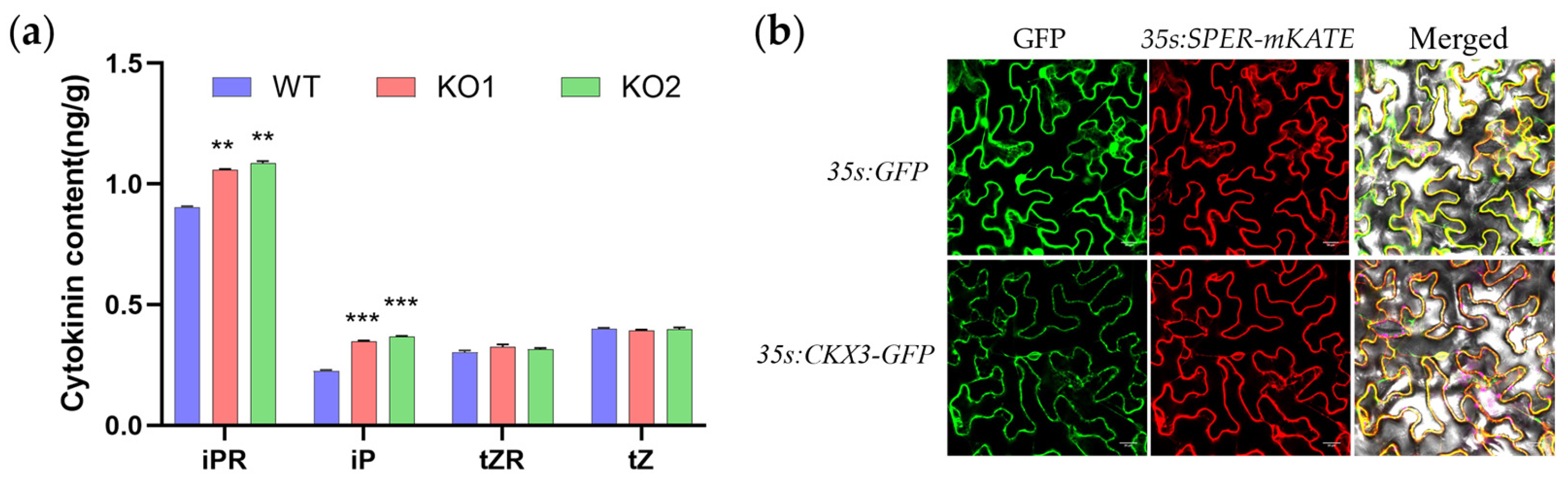
2.2. Loss of Function GmCKX3 Increases Endogenous Cytokinin Levels in Soybean
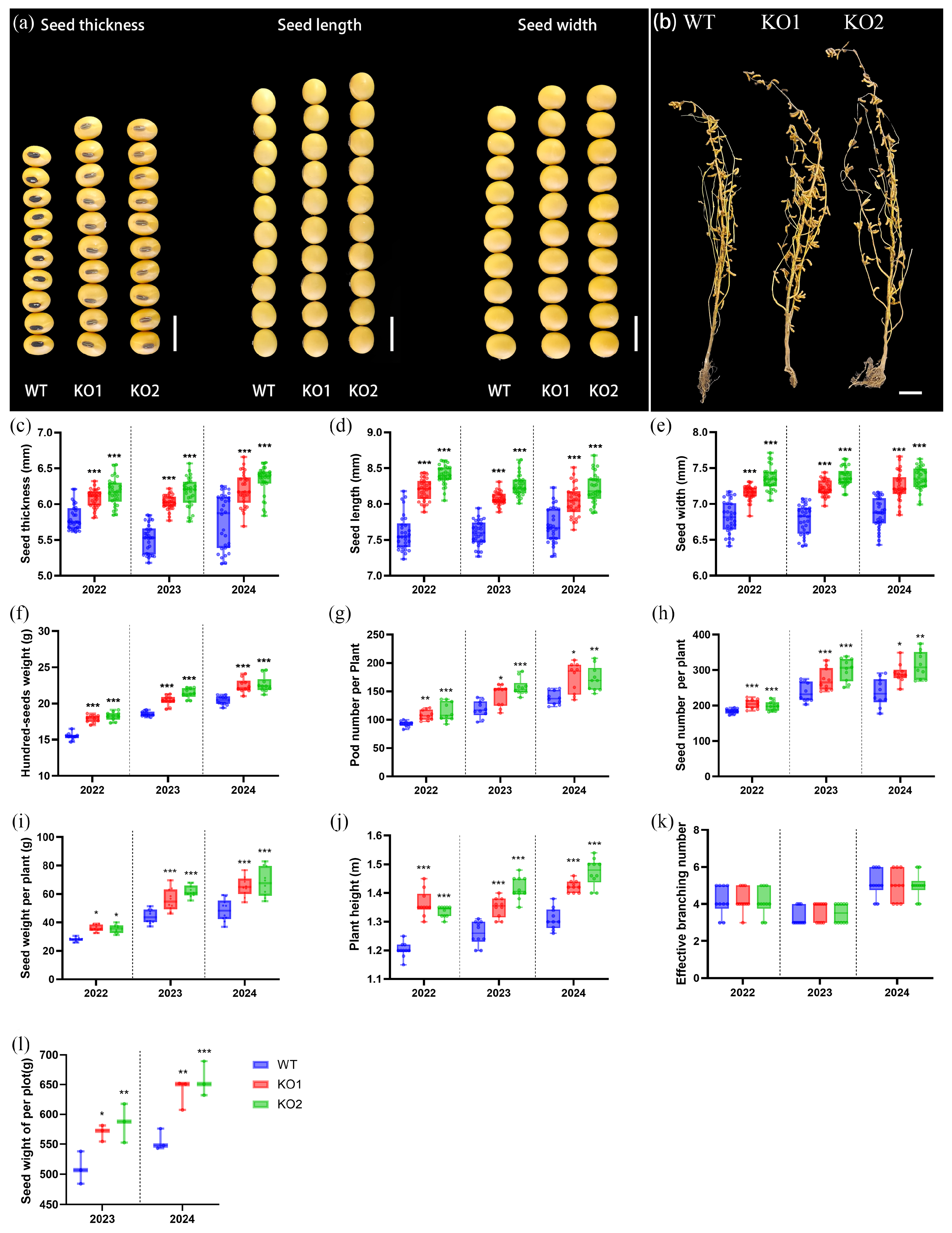
2.3. Gmckx3 Mutants Improved the Major Yield-Related Traits in Soybean
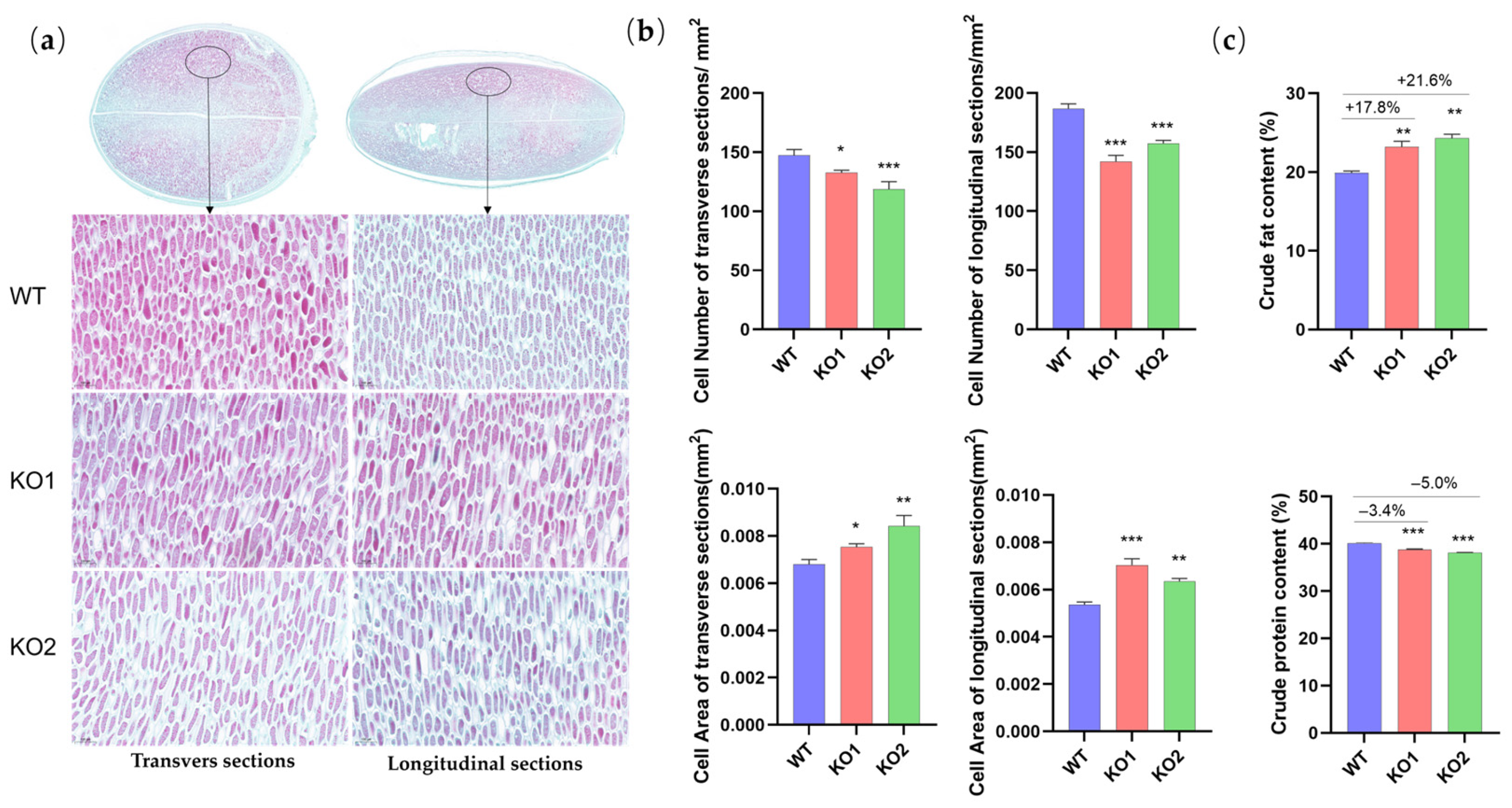
2.4. The Improvement of the Seed-Related Traits Is Likely Attributed to the Increase in the Cotyledon Cell Volume and Fat Content Caused by a GmCKX3 Deficiency in Soybeans
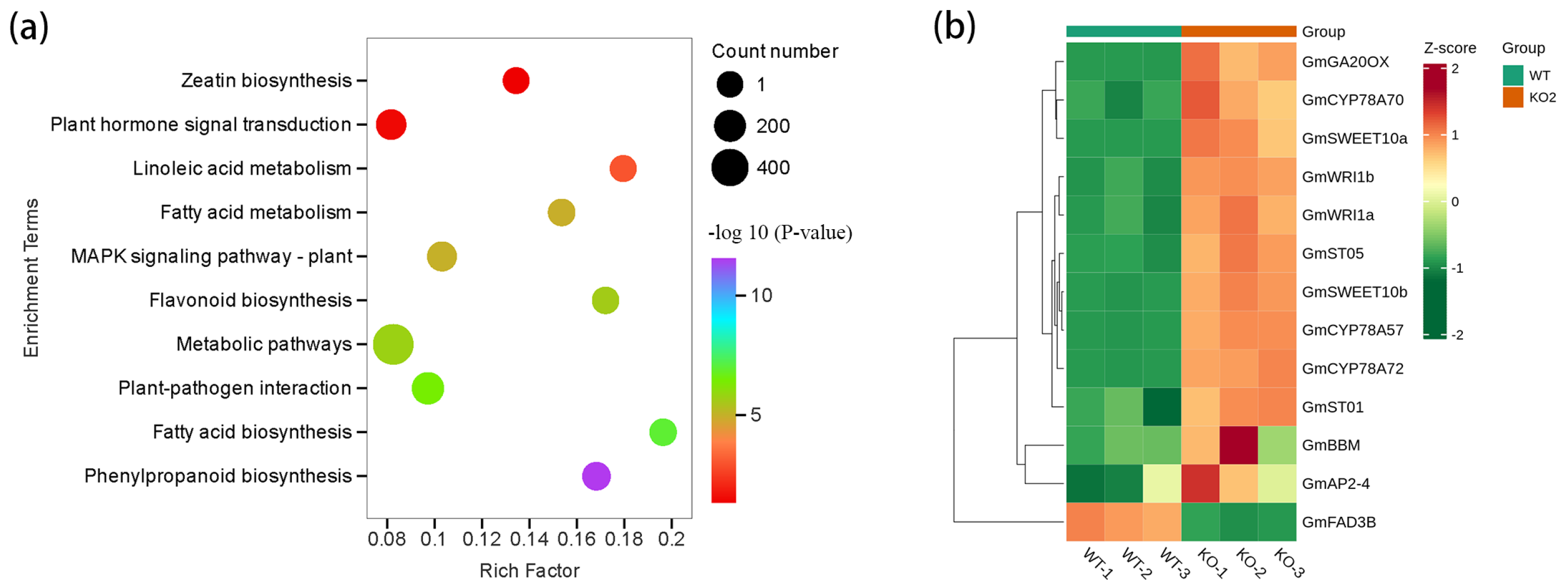
2.5. Transcriptomic Analysis of DEGs During Seed Development in the Gmckx3 Line
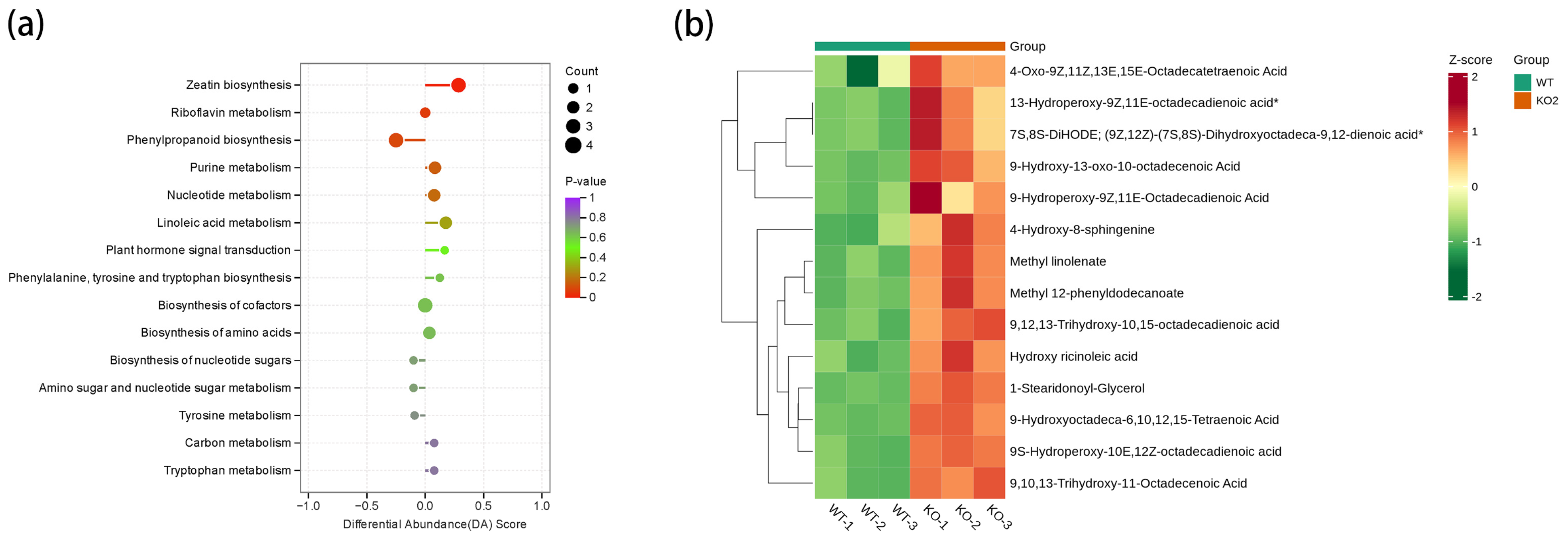
2.6. Metabolomic Analysis of DAMs During Seed Development in the Gmckx3 Line
2.7. Combinative Analyses of Transcriptome and Metabolome of Differential GmCKX3 Expression Revealed Important Genes and Pathways Involving CK Biosynthesis in Soybean
3. Discussion
4. Materials and Methods
4.1. Construction of GmCKX3 Knockout Vectors
4.2. Preparation of GmCKX3 Genetically Modified Mutants
4.3. Phenotyping of the Genetically Modified Soybean
4.4. Detection of Cytokinin Content
4.5. Subcellular Localization of GmCKX3
4.6. Paraffin Sections for Light Microscopy
4.7. Measurement of the Crude Fat and Protein Content in Soybean Seeds
4.8. RNA Extraction and Quantitative Real-Time PCR (qPCR) Analysis
4.9. Transcriptome Analysis
4.10. Metabolome Profiling
4.11. Combinative Analysis of the Transcriptome and the Metabolome
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, Y.; Liu, Y.; Wei, J.J.; Zhang, W.K.; Chen, S.Y.; Zhang, J.S. Regulation of seed traits in soybean. aBIOTECH 2023, 4, 372–385. [Google Scholar] [CrossRef]
- Li, N.; Xu, R.; Duan, P.; Li, Y. Control of grain size in rice. Plant Reprod. 2018, 31, 237–251. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, Y.; Tao, J.J.; Lu, L.; Jiang, Z.H.; Wei, J.J.; Wu, C.M.; Yin, C.C.; Li, W.; Bi, Y.D.; et al. GmJAZ3 interacts with GmRR18a and GmMYC2a to regulate seed traits in soybean. J. Integr. Plant Biol. 2023, 65, 1983–2000. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, S.; Wang, Z.; Yuan, Y.; Zhang, Z.; Liang, Q.; Yang, X.; Duan, Z.; Liu, Y.; Kong, F.; et al. Progress in soybean functional genomics over the past decade. Plant Biotechnol. J. 2022, 20, 256–282. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, M.; Feng, F.; Tian, Z. Toward a “Green Revolution” for Soybean. Mol. Plant 2020, 13, 688–697. [Google Scholar] [CrossRef]
- Zuo, J.; Li, J. Molecular genetic dissection of quantitative trait loci regulating rice grain size. Annu. Rev. Genet. 2014, 48, 99–118. [Google Scholar] [CrossRef]
- Zhang, Y.; Bhat, J.A.; Zhang, Y.; Yang, S. Understanding the Molecular Regulatory Networks of Seed Size in Soybean. Int. J. Mol. Sci. 2024, 25, 1441. [Google Scholar] [CrossRef]
- Jameson, P.E.; Song, J. Cytokinin: A key driver of seed yield. J. Exp. Bot. 2016, 67, 593–606. [Google Scholar] [CrossRef]
- Sakakibara, H. Cytokinins: Activity, biosynthesis, and translocation. Annu. Rev. Plant Biol. 2006, 57, 431–449. [Google Scholar] [CrossRef]
- Hirose, N.; Takei, K.; Kuroha, T.; Kamada-Nobusada, T.; Hayashi, H.; Sakakibara, H. Regulation of cytokinin biosynthesis, compartmentalization and translocation. J. Exp. Bot. 2008, 59, 75–83. [Google Scholar] [CrossRef]
- Werner, T.; Kollmer, I.; Bartrina, I.; Holst, K.; Schmulling, T. New insights into the biology of cytokinin degradation. Plant Biol. 2006, 8, 371–381. [Google Scholar] [CrossRef]
- Bilyeu, K.D.; Cole, J.L.; Laskey, J.G.; Riekhof, W.R.; Esparza, T.J.; Kramer, M.D.; Morris, R.O. Molecular and biochemical characterization of a cytokinin oxidase from maize. Plant Physiol. 2001, 125, 378–386. [Google Scholar] [CrossRef]
- Galuszka, P.; Frebortova, J.; Werner, T.; Yamada, M.; Strnad, M.; Schmulling, T.; Frebort, I. Cytokinin oxidase/dehydrogenase genes in barley and wheat: Cloning and heterologous expression. Eur. J. Biochem. 2004, 271, 3990–4002. [Google Scholar] [CrossRef]
- Zalabák, D.; Galuszka, P.; Mrízová, K.; Podlešáková, K.; Gu, R.; Frébortová, J. Biochemical characterization of the maize cytokinin dehydrogenase family and cytokinin profiling in developing maize plantlets in relation to the expression of cytokinin dehydrogenase genes. Plant Physiol. Biochem. 2014, 74, 283–293. [Google Scholar] [CrossRef]
- Gu, R.; Fu, J.; Guo, S.; Duan, F.; Wang, Z.; Mi, G.; Yuan, L. Comparative Expression and Phylogenetic Analysis of Maize Cytokinin Dehydrogenase/Oxidase (CKX) Gene Family. J. Plant Growth Regul. 2010, 29, 428–440. [Google Scholar] [CrossRef]
- Le, D.T.; Nishiyama, R.; Watanabe, Y.; Vankova, R.; Tanaka, M.; Seki, M.; Ham, L.H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.P. Identification and expression analysis of cytokinin metabolic genes in soybean under normal and drought conditions in relation to cytokinin levels. PLoS ONE 2012, 7, e42411. [Google Scholar] [CrossRef]
- Wang, X.; Ding, J.; Lin, S.; Liu, D.; Gu, T.; Wu, H.; Trigiano, R.N.; McAvoy, R.; Huang, J.; Li, Y. Evolution and roles of cytokinin genes in angiosperms 2: Do ancient CKXs play housekeeping roles while non-ancient CKXs play regulatory roles? Hortic. Res. 2020, 7, 29. [Google Scholar] [CrossRef]
- Gajdosova, S.; Spichal, L.; Kaminek, M.; Hoyerova, K.; Novak, O.; Dobrev, P.I.; Galuszka, P.; Klima, P.; Gaudinova, A.; Zizkova, E.; et al. Distribution, biological activities, metabolism, and the conceivable function of cis-zeatin-type cytokinins in plants. J. Exp. Bot. 2011, 62, 2827–2840. [Google Scholar] [CrossRef]
- Werner, T.; Motyka, V.; Laucou, V.; Smets, R.; Van Onckelen, H.; Schmulling, T. Cytokinin-deficient transgenic Arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. Plant Cell 2003, 15, 2532–2550. [Google Scholar] [CrossRef]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef]
- Mameaux, S.; Cockram, J.; Thiel, T.; Steuernagel, B.; Stein, N.; Taudien, S.; Jack, P.; Werner, P.; Gray, J.C.; Greenland, A.J.; et al. Molecular, phylogenetic and comparative genomic analysis of the cytokinin oxidase/dehydrogenase gene family in the Poaceae. Plant Biotechnol. J. 2012, 10, 67–82. [Google Scholar] [CrossRef]
- Song, J.; Jiang, L.; Jameson, P.E. Co-ordinate regulation of cytokinin gene family members during flag leaf and reproductive development in wheat. BMC Plant Biol. 2012, 12, 78. [Google Scholar] [CrossRef]
- Ogonowska, H.; Barchacka, K.; Gasparis, S.; Jablonski, B.; Orczyk, W.; Dmochowska-Boguta, M.; Nadolska-Orczyk, A. Specificity of expression of TaCKX family genes in developing plants of wheat and their co-operation within and among organs. PLoS ONE 2019, 14, e214239. [Google Scholar] [CrossRef]
- Shoaib, M.; Yang, W.; Shan, Q.; Sajjad, M.; Zhang, A. Genome-wide identification and expression analysis of new cytokinin metabolic genes in bread wheat (Triticum aestivum L.). PeerJ 2019, 7, e6300. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, J.; Song, J.; Jameson, P.E. Cytokinin dehydrogenase: A genetic target for yield improvement in wheat. Plant Biotechnol. J. 2020, 18, 614–630. [Google Scholar] [CrossRef] [PubMed]
- Bartrina, I.; Otto, E.; Strnad, M.; Werner, T.; Schmulling, T. Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. Plant Cell 2011, 23, 69–80. [Google Scholar] [CrossRef]
- Yeh, S.Y.; Chen, H.W.; Ng, C.Y.; Lin, C.Y.; Tseng, T.H.; Li, W.H.; Ku, M.S. Down-Regulation of Cytokinin Oxidase 2 Expression Increases Tiller Number and Improves Rice Yield. Rice 2015, 8, 36. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Y.L.; Gao, L.F.; Zhao, G.Y.; Zhou, R.H.; Zhang, B.S.; Jia, J.Z. TaCKX6-D1, the ortholog of rice OsCKX2, is associated with grain weight in hexaploid wheat. New Phytol. 2012, 195, 574–584. [Google Scholar] [CrossRef]
- Zalewski, W.; Galuszka, P.; Gasparis, S.; Orczyk, W.; Nadolska-Orczyk, A. Silencing of the HvCKX1 gene decreases the cytokinin oxidase/dehydrogenase level in barley and leads to higher plant productivity. J. Exp. Bot. 2010, 61, 1839–1851. [Google Scholar] [CrossRef]
- Zalewski, W.; Gasparis, S.; Boczkowska, M.; Rajchel, I.K.; Kala, M.; Orczyk, W.; Nadolska-Orczyk, A. Expression patterns of HvCKX genes indicate their role in growth and reproductive development of barley. PLoS ONE 2014, 9, e115729. [Google Scholar] [CrossRef]
- Holubova, K.; Hensel, G.; Vojta, P.; Tarkowski, P.; Bergougnoux, V.; Galuszka, P. Modification of Barley Plant Productivity Through Regulation of Cytokinin Content by Reverse-Genetics Approaches. Front. Plant Sci. 2018, 9, 1676. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Kambhampati, S.; Kisiala, A.; Seegobin, M.; Emery, R. The soybean (Glycine max L.) cytokinin oxidase/dehydrogenase multigene family; Identification of natural variations for altered cytokinin content and seed yield. Plant Direct 2021, 5, e308. [Google Scholar] [CrossRef]
- Yang, C.; Yan, J.; Jiang, S.; Li, X.; Min, H.; Wang, X.; Hao, D. Resequencing 250 Soybean Accessions: New Insights into Genes Associated with Agronomic Traits and Genetic Networks. Genom. Proteom. Bioinform. 2022, 20, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Ohlrogge, J.B.; Kuo, T.M. Control of Lipid Synthesis during Soybean Seed Development: Enzymic and Immunochemical Assay of Acyl Carrier Protein. Plant Physiol. 1984, 74, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Babka, H.L.; Graef, G.L.; Staswick, P.E.; Lee, D.J.; Cregan, P.B.; Shoemaker, R.C.; Specht, J.E. The Seed Protein, Oil, and Yield QTL on Soybean Linkage Group I. Crop. Sci. 2003, 43, 1053–1067. [Google Scholar] [CrossRef]
- Zhao, B.; Dai, A.; Wei, H.; Yang, S.; Wang, B.; Jiang, N.; Feng, X. Arabidopsis KLU homologue GmCYP78A72 regulates seed size in soybean. Plant Mol. Biol. 2016, 90, 33–47. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Ma, R.; Huang, W.; Hou, J.; Fang, C.; Wang, L.; Yuan, Z.; Sun, Q.; Dong, X.; et al. Identification of ST1 reveals a selection involving hitchhiking of seed morphology and oil content during soybean domestication. Plant Biotechnol. J. 2022, 20, 1110–1121. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, Y.; Lu, L.; Tao, J.J.; Cheng, T.; Jin, M.; Wang, Z.Y.; Wei, J.J.; Jiang, Z.H.; Sun, W.C.; et al. Global analysis of seed transcriptomes reveals a novel PLATZ regulator for seed size and weight control in soybean. New Phytol. 2023, 240, 2436–2454. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, M.; Zhang, Z.; Liang, S.; Fan, L.; Yang, X.; Yuan, Y.; Pan, Y.; Zhou, G.; Liu, S.; et al. Natural allelic variation of GmST05 controlling seed size and quality in soybean. Plant Biotechnol. J. 2022, 20, 1807–1818. [Google Scholar] [CrossRef]
- Wang, S.; Liu, S.; Wang, J.; Yokosho, K.; Zhou, B.; Yu, Y.C.; Liu, Z.; Frommer, W.B.; Ma, J.F.; Chen, L.Q.; et al. Simultaneous changes in seed size, oil content and protein content driven by selection of SWEET homologues during soybean domestication. Natl. Sci. Rev. 2020, 7, 1776–1786. [Google Scholar] [CrossRef]
- Singh, A.K.; Fu, D.Q.; El-Habbak, M.; Navarre, D.; Ghabrial, S.; Kachroo, A. Silencing genes encoding omega-3 fatty acid desaturase alters seed size and accumulation of Bean pod mottle virus in soybean. Mol. Plant Microbe Interact. 2011, 24, 506–515. [Google Scholar] [CrossRef]
- Emery, N.; Atkins, C. Cytokinins and Seed Development. In Handbook of Seed Science and Technology; Basra, A.S., Ed.; New York Haworth Press: New York, NY, USA, 2006; pp. 63–93. [Google Scholar]
- Huang, X.; Hou, L.; Meng, J.; You, H.; Li, Z.; Gong, Z.; Yang, S.; Shi, Y. The Antagonistic Action of Abscisic Acid and Cytokinin Signaling Mediates Drought Stress Response in Arabidopsis. Mol. Plant 2018, 11, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Li, X.; Yang, Z.; Liu, S.; Hao, D.; Chao, M.; Zhang, J.; Yang, H.; Su, X.; Jiang, M.; et al. Downregulation of a gibberellin 3beta-hydroxylase enhances photosynthesis and increases seed yield in soybean. New Phytol. 2022, 235, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wu, H.; Guo, Y.; Gao, H.; Wei, Z.; Zhao, Y.; Qiu, L. GmIAA27 Encodes an AUX/IAA Protein Involved in Dwarfing and Multi-Branching in Soybean. Int. J. Mol. Sci. 2022, 23, 8643. [Google Scholar] [CrossRef] [PubMed]
- Szala, K.; Ogonowska, H.; Lugowska, B.; Zmijewska, B.; Wyszynska, R.; Dmochowska-Boguta, M.; Orczyk, W.; Nadolska-Orczyk, A. Different sets of TaCKX genes affect yield-related traits in wheat plants grown in a controlled environment and in field conditions. BMC Plant Biol. 2020, 20, 496. [Google Scholar] [CrossRef]
- Rong, C.; Liu, Y.; Chang, Z.; Liu, Z.; Ding, Y.; Ding, C. Cytokinin oxidase/dehydrogenase family genes exhibit functional divergence and overlap in rice growth and development, especially in control of tillering. J. Exp. Bot. 2022, 73, 3552–3568. [Google Scholar] [CrossRef]
- Kowalska, M.; Galuszka, P.; Frébortová, J.; Šebela, M.; Béres, T.; Hluska, T.; Šmehilová, M.; Bilyeu, K.D.; Frébort, I. Vacuolar and cytosolic cytokinin dehydrogenases of Arabidopsis thaliana: Heterologous expression, purification and properties. Phytochemistry 2010, 71, 1970–1978. [Google Scholar] [CrossRef]
- Huang, P.; Zhao, J.; Hong, J.; Zhu, B.; Xia, S.; Zhu, E.; Han, P.; Zhang, K. Cytokinins regulate rice lamina joint development and leaf angle. Plant Physiol. 2023, 191, 56–69. [Google Scholar] [CrossRef]
- Li, L.; Jia, L.; Duan, X.; Lv, Y.; Ye, C.; Ding, C.; Zhang, Y.; Qi, W.; Motte, H.; Beeckman, T.; et al. A nitrogen-responsive cytokinin oxidase/dehydrogenase regulates root response to high ammonium in rice. New Phytol. 2024, 244, 1391–1407. [Google Scholar] [CrossRef]
- Liu, M.; Cui, Y.; Peng, F.; Wang, S.; Cui, R.; Liu, X.; Zhang, Y.; Huang, H.; Fan, Y.; Jiang, T.; et al. Antioxidant system was triggered to alleviate salinity stress by cytokinin oxidase/dehydrogenase gene GhCKX6b-Dt in cotton. Environ. Sci. Eur. 2023, 35, 82. [Google Scholar] [CrossRef]
- Perilli, S.; Moubayidin, L.; Sabatini, S. The molecular basis of cytokinin function. Curr. Opin. Plant Biol. 2010, 13, 21–26. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, Z.; Gu, Y.; Li, W.; Wang, W.; Yuan, X.; Zhang, Y.; Yuan, M.; Du, J.; Zhao, Q. Genome-wide identification of the soybean cytokinin oxidase/dehydrogenase gene family and its diverse roles in response to multiple abiotic stress. Front. Plant Sci. 2023, 14, 1163219. [Google Scholar] [CrossRef]
- Hou, B.; Lim, E.K.; Higgins, G.S.; Bowles, D.J. N-glucosylation of cytokinins by glycosyltransferases of Arabidopsis thaliana. J. Biol. Chem. 2004, 279, 47822–47832. [Google Scholar] [CrossRef]
- Greenboim-Wainberg, Y.; Maymon, I.; Borochov, R.; Alvarez, J.; Olszewski, N.; Ori, N.; Eshed, Y.; Weiss, D. Cross talk between gibberellin and cytokinin: The Arabidopsis GA response inhibitor SPINDLY plays a positive role in cytokinin signaling. Plant Cell 2005, 17, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Muller, B.; Sheen, J. Cytokinin and auxin interaction in root stem-cell specification during early embryogenesis. Nature 2008, 453, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.J.; Jian, L.; Xu, J.; Zhang, Q.; Zhang, M.; Jin, M.; Peng, Y.; Yan, J.; Han, B.; Liu, J.; et al. High-Throughput CRISPR/Cas9 Mutagenesis Streamlines Trait Gene Identification in Maize. Plant Cell 2020, 32, 1397–1413. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Q.; Zhu, Q.; Liu, W.; Chen, Y.; Qiu, R.; Wang, B.; Yang, Z.; Li, H.; Lin, Y.; et al. A Robust CRISPR/Cas9 System for Convenient, High-Efficiency Multiplex Genome Editing in Monocot and Dicot Plants. Mol. Plant 2015, 8, 1274–1284. [Google Scholar] [CrossRef]
- Lei, Y.; Lu, L.; Liu, H.Y.; Li, S.; Xing, F.; Chen, L.L. CRISPR-P: A web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol. Plant 2014, 7, 1494–1496. [Google Scholar] [CrossRef]
- Zia, M.; Rizvi, Z.F. Agrobacterium mediated transformation of soybean (Glycine max L.): Some conditions standardization. Pak. J. Bot. 2010, 4, 2269–2279. [Google Scholar]
- Tomooka, N.; Vaughan, D.A.; Moss, H.; Maxted, N.; SpringerLink, O.S. The Asian Vigna: Genus Vigna Subgenus Ceratotropis Genetic Resources; Springer: Dordrecht, The Netherlands, 2002. [Google Scholar]
- Cheng, X.P.; Shum, P.; Zhang, J.; Tang, M. Analysis of Nonlinear Effective Absorption Coefficient of Double Cladding Fiber. In Proceedings of the TENCON 2006—2006 IEEE Region 10 Conference, Hong Kong, China, 14–17 November 2006; IEEE: Piscataway, NJ, USA, 2006; pp. 1–3. [Google Scholar]
- Kaushik, N.; Kumar, K.; Kumar, S.; Kaushik, N.; Roy, S. Genetic variability and divergence studies in seed traits and oil content of Jatropha (Jatropha curcas L.) accessions. Biomass Bioenergy 2007, 31, 497–502. [Google Scholar] [CrossRef]
- Mravec, J.; Skupa, P.; Bailly, A.; Hoyerova, K.; Krecek, P.; Bielach, A.; Petrasek, J.; Zhang, J.; Gaykova, V.; Stierhof, Y.D.; et al. Subcellular homeostasis of phytohormone auxin is mediated by the ER-localized PIN5 transporter. Nature 2009, 459, 1136–1140. [Google Scholar] [CrossRef]
- Ma, Y.; Sawhney, V.K.; Steeves, T.A. Staining of paraffin-embedded plant material in safranin and fast green without prior removal of the paraffin. Can. J. Bot. 1993, 71, 996–999. [Google Scholar] [CrossRef]
- Rosenthal, I.; Merin, U.; Popel, G.; Bernstein, S. Determination of fat in vegetable foods. J. Assoc. Off. Anal. Chem. 1985, 68, 1226–1228. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.B.; Aleem, S.; Khan, M.R.; Ashraf, S.; Busquets, R. Quantitative Estimation of Protein in Sprouts of Vigna radiate (Mung Beans), Lens culinaris (Lentils), and Cicer arietinum (Chickpeas) by Kjeldahl and Lowry Methods. Molecules 2022, 27, 814. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general-purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-discovery approach for sample matching of a nerve-agent precursor using liquid chromatography-mass spectrometry, XCMS, and chemometrics. Anal. Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Qian, X.; Zhao, F.; Niu, L.; Zhang, Y.; Han, S.; Hao, D.; Chen, Z. Knockout of GmCKX3 Enhances Soybean Seed Yield via Cytokinin-Mediated Cell Expansion and Lipid Accumulation. Plants 2025, 14, 2207. https://doi.org/10.3390/plants14142207
Li X, Qian X, Zhao F, Niu L, Zhang Y, Han S, Hao D, Chen Z. Knockout of GmCKX3 Enhances Soybean Seed Yield via Cytokinin-Mediated Cell Expansion and Lipid Accumulation. Plants. 2025; 14(14):2207. https://doi.org/10.3390/plants14142207
Chicago/Turabian StyleLi, Xia, Xueyan Qian, Fangfang Zhao, Lu Niu, Yan Zhang, Siping Han, Dongyun Hao, and Ziqi Chen. 2025. "Knockout of GmCKX3 Enhances Soybean Seed Yield via Cytokinin-Mediated Cell Expansion and Lipid Accumulation" Plants 14, no. 14: 2207. https://doi.org/10.3390/plants14142207
APA StyleLi, X., Qian, X., Zhao, F., Niu, L., Zhang, Y., Han, S., Hao, D., & Chen, Z. (2025). Knockout of GmCKX3 Enhances Soybean Seed Yield via Cytokinin-Mediated Cell Expansion and Lipid Accumulation. Plants, 14(14), 2207. https://doi.org/10.3390/plants14142207