
Assembly and Analysis of the Mitochondrial Genome of Hippophae rhamnoides subsp. sinensis, an Important Ecological and Economic Forest Tree Species in China


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials, Mitochondrial DNA Isolation and Genome Sequencing
2.2. Assembly and Annotation of the Mitogenome
2.3. Codon Usage Analysis
2.4. RNA Editing Analyses
2.5. Analysis of Repeat Sequences
2.6. Ka/Ks Ratio Evaluation
2.7. Homologous Fragment Analysis
2.8. Phylogenetic Tree Construction
3. Results
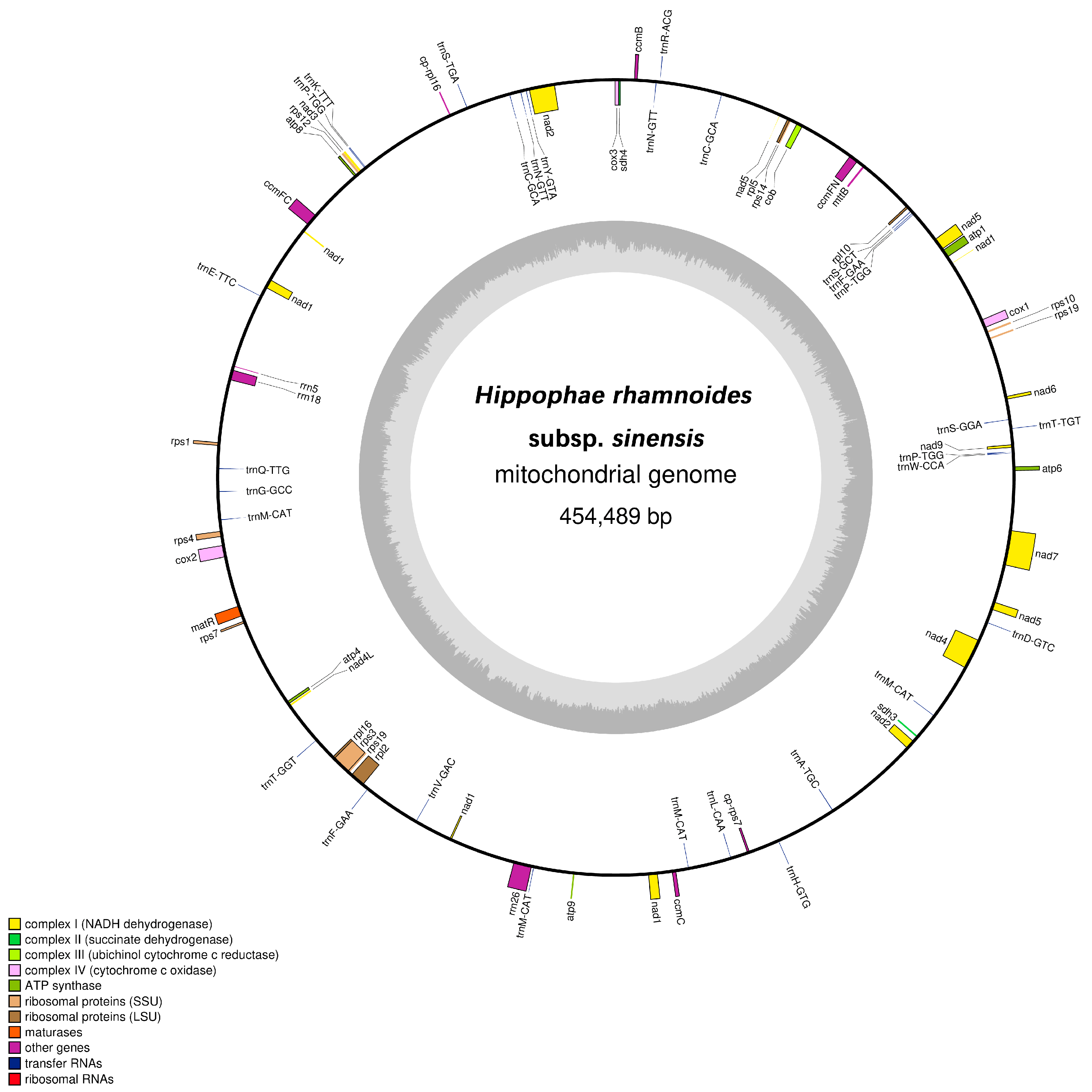
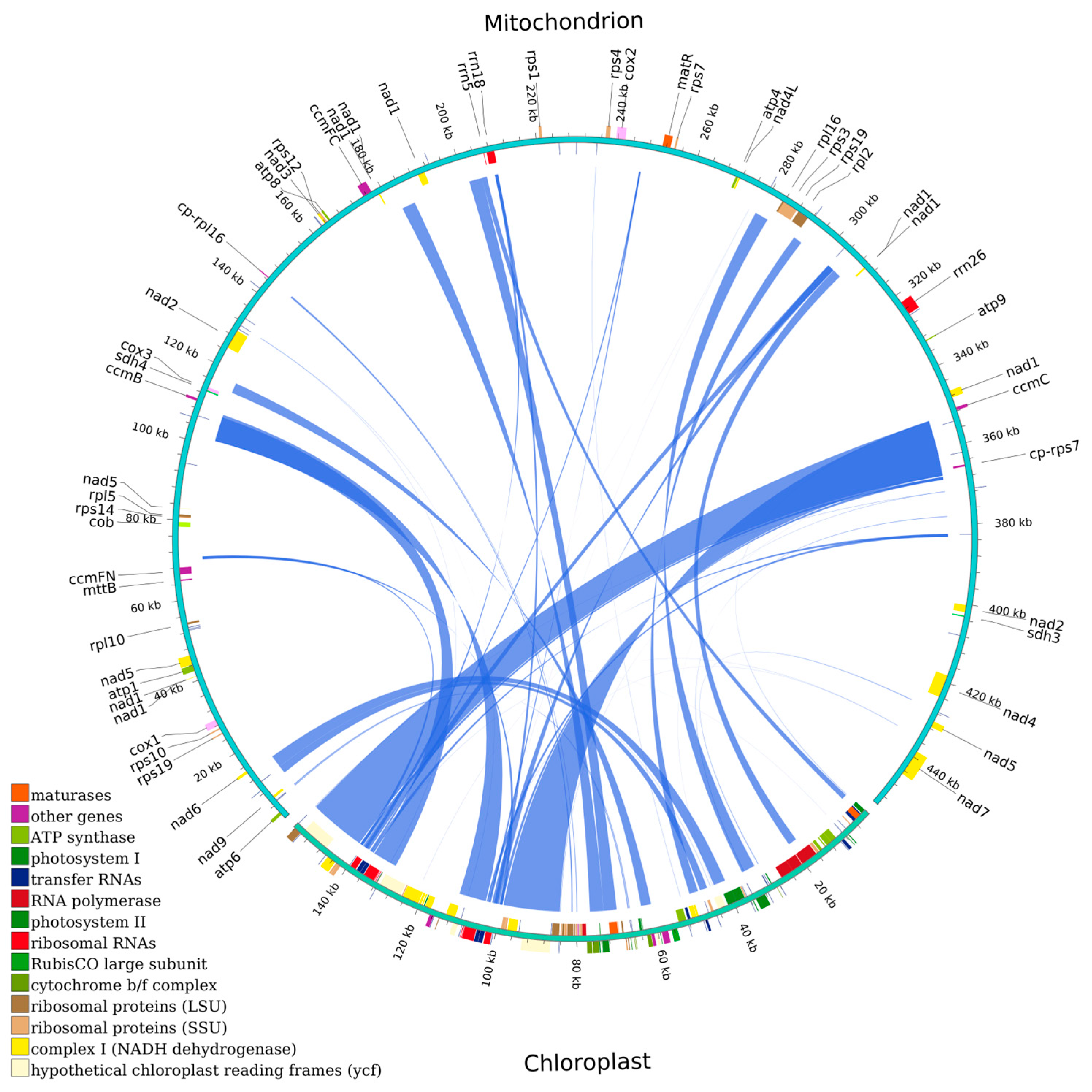
3.1. Mitochondrial Genome of H. rhamnoides subsp. sinensis and Its Characteristics
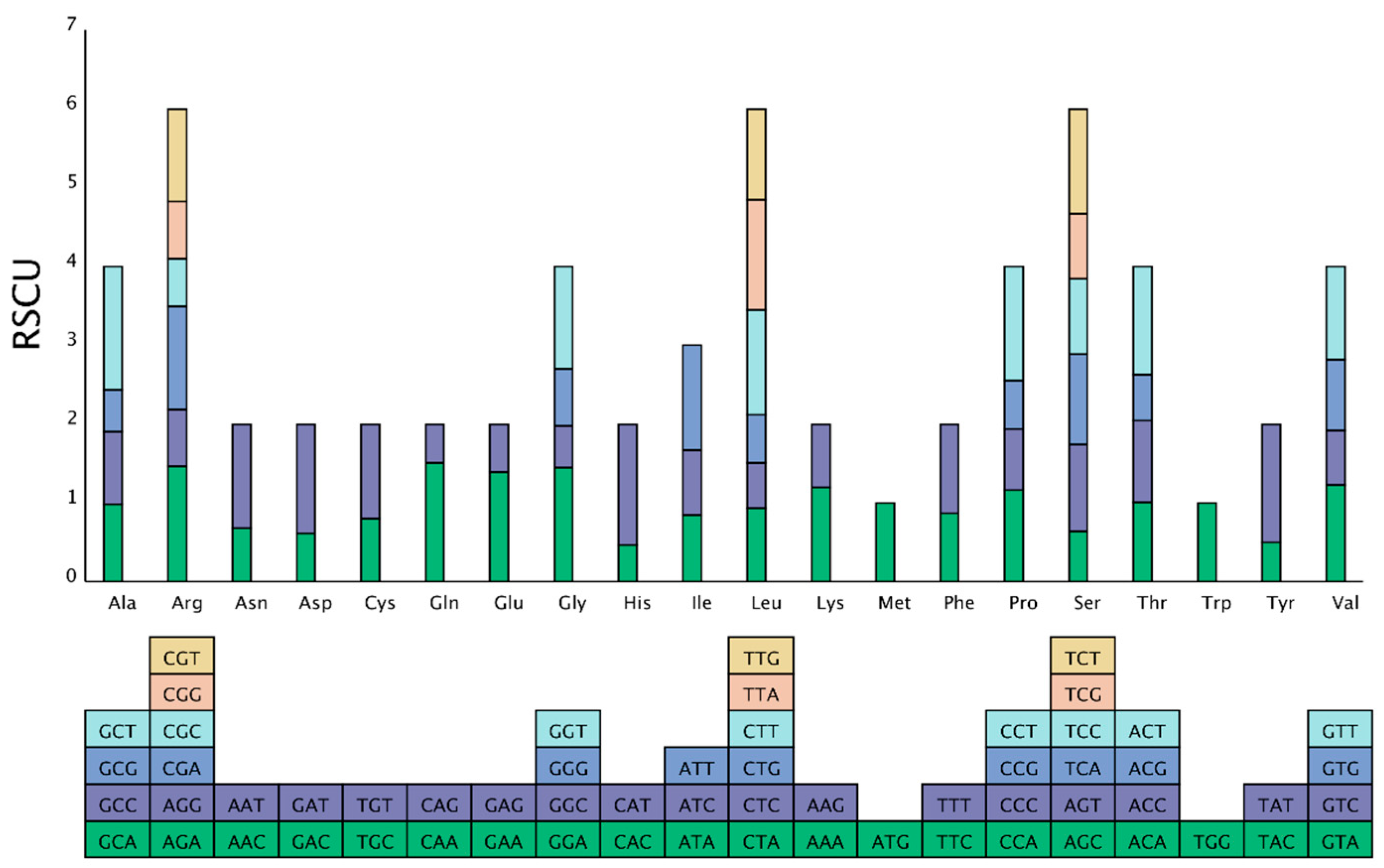
3.2. Codon Usage Bias in Mitochondrial Genome
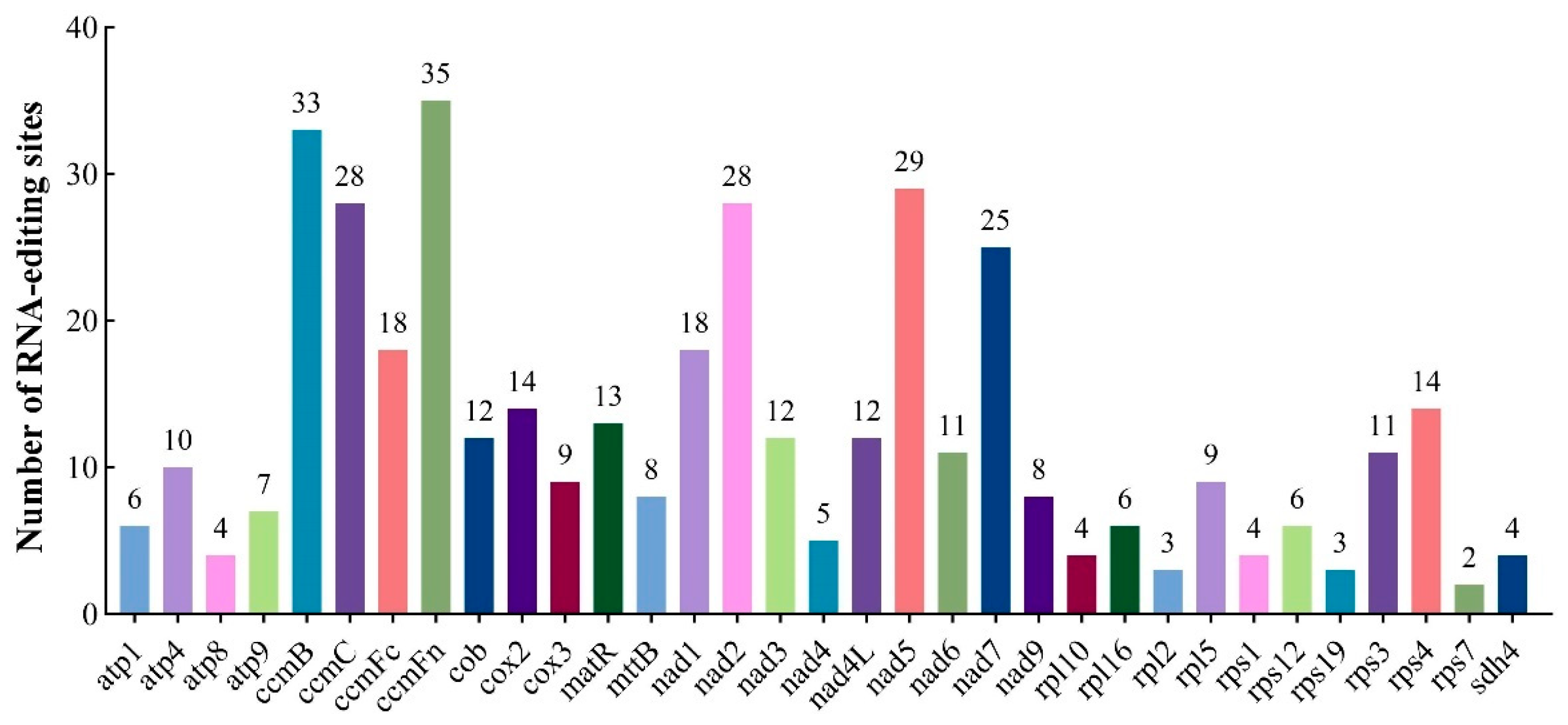
3.3. Prediction of RNA Editing Events
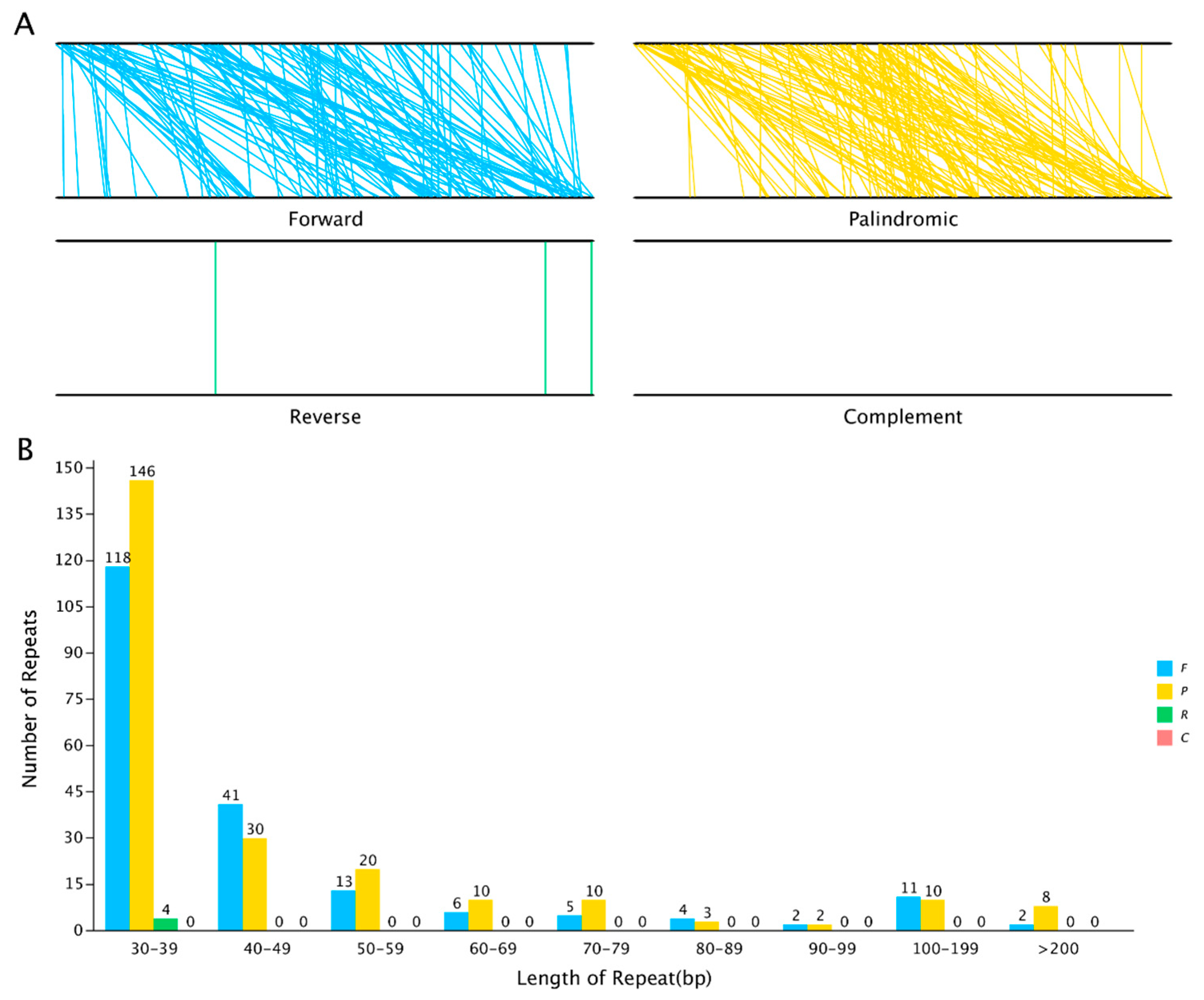
3.4. Identification of Repeat Sequences
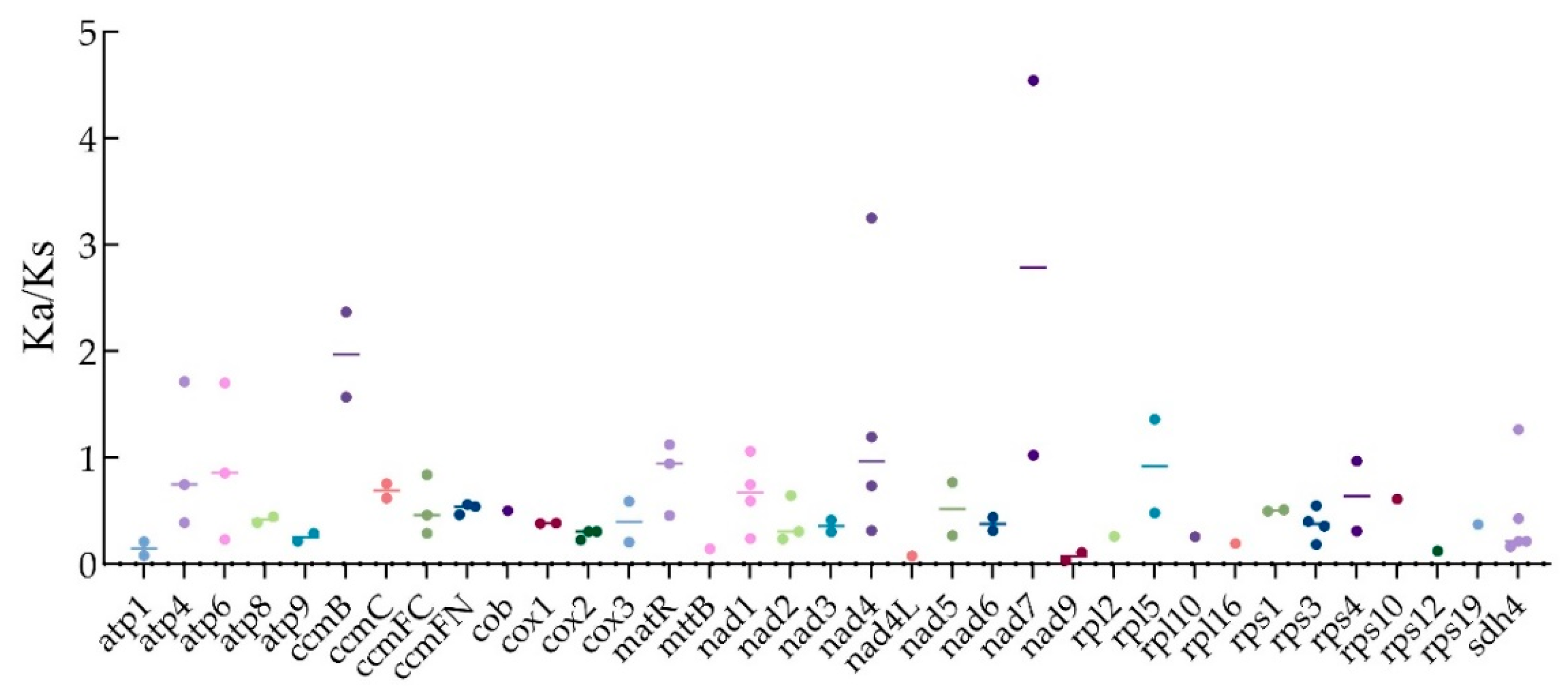
3.5. Ka/Ks Ratio Analysis on PCGs
3.6. Homologous Fragments Between Chloroplast and Mitochondrial Genomes
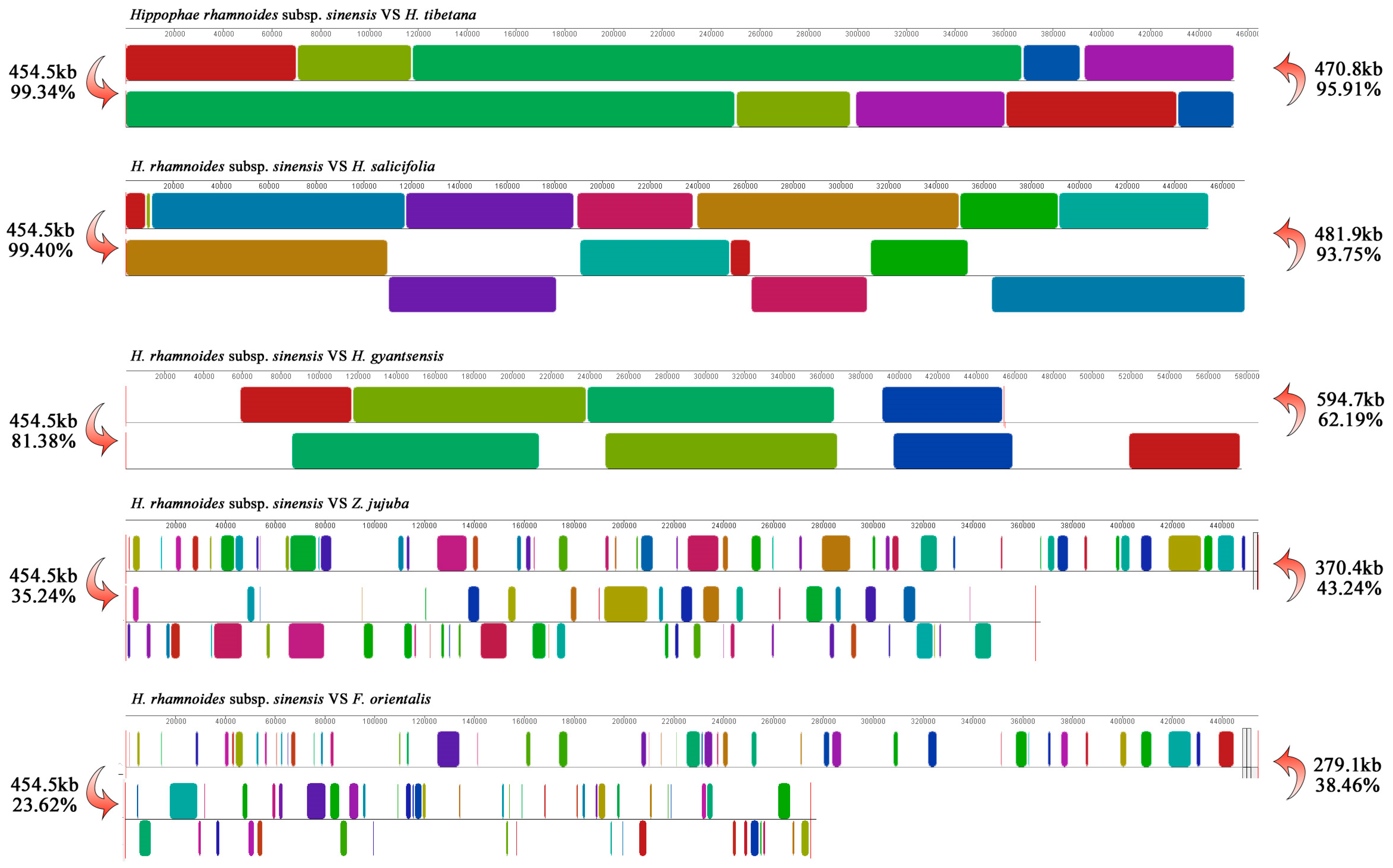
3.7. Synteny Analysis of Mitochondrial Genomes Between H. rhamnoides subsp. sinensis and Related Species
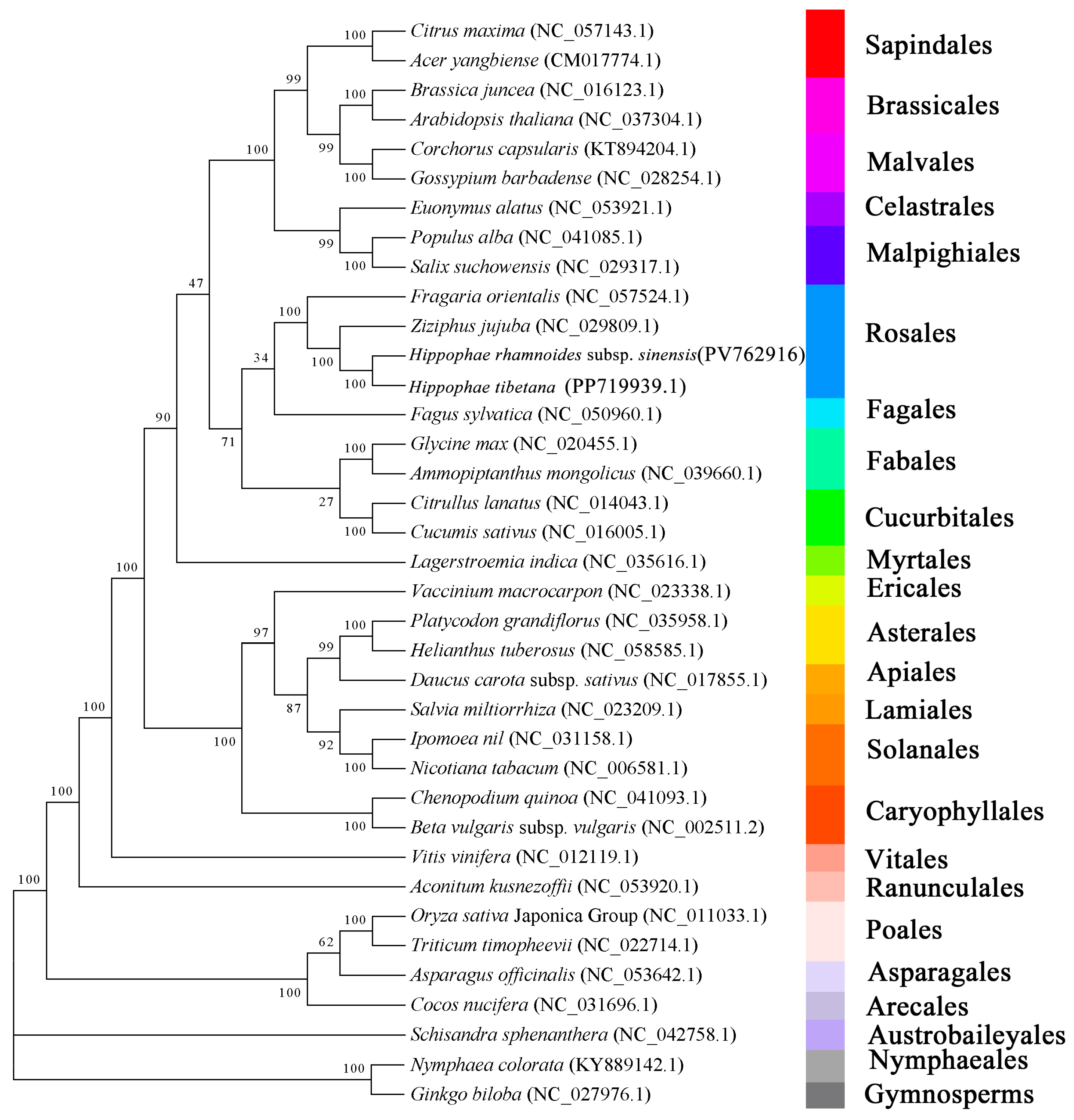
3.8. Phylogenetic Analysis of H. rhamnoides subsp. sinensis
4. Discussion
4.1. Structure and Size of the H. rhamnoides subsp. sinensis Mitochondrial Genome
4.2. The Evolution of the H. rhamnoides subsp. sinensis Mitochondrial Genome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lian, Y.S.; Chen, X.L. Ecological and Geographic Distribution of Sea Buckthorn and Its Plant Geoscience Significance. Acta Phytotaxon. Sin. 1992, 30, 349–355. (In Chinese) [Google Scholar]
- Zhang, T.; Gao, G.; Liu, J.; Yang, G.; Lv, Z.; Zhang, J.; He, C. Transcripts and Aba-Dependent Signaling in Response to Drought Stress in Hippophae rhamnoides L. Trees 2020, 34, 1033–1045. Trees 2020, 34, 1033–1045. [Google Scholar] [CrossRef]
- Wang, S.; Jing, Y.; Zhao, C.; Huang, J.; Wang, X.; Dai, Y. Effects of Saline-Alkali Soil Conditioner on the Recovery of Growth and Physiology of Chinese Seabuckthorn. Plant Physiol. Rep. 2021, 26, 301–310. [Google Scholar] [CrossRef]
- Singh, V. Global Distribution of Seabuckthorn (Hippophae sp.) Resources and Their Utilization. In The Seabuckthorn Genome; Springer International Publishing: Cham, Switzerland, 2022. [Google Scholar]
- Bal, L.M.; Meda, V.; Naik, S.N.; Satya, S. Sea Buckthorn Berries: A Potential Source of Valuable Nutrients for Nutraceuticals and Cosmoceuticals. Food Res. Int. 2011, 44, 1718–1727. [Google Scholar] [CrossRef]
- Olas, B. The Beneficial Health Aspects of Sea Buckthorn (Elaeagnus rhamnoides (L.) A.Nelson) Oil. J. Ethnopharmacol. 2018, 213, 183–190. [Google Scholar] [CrossRef]
- Teleszko, M.; Wojdyło, A.; Rudzińska, M.; Oszmiański, J.; Golis, T. Analysis of Lipophilic and Hydrophilic Bioactive Compounds Content in Sea Buckthorn (Hippophaë rhamnoides L.) Berries. J. Agric. Food Chem. 2015, 63, 4120–4129. [Google Scholar] [CrossRef]
- International Seabuckthorn Association (ISA). The Annual Report of International Seabuckthorn Development for the Year of 2023; The Secretariat of International Seabuckthorn Association: Beijing, China, 2023. [Google Scholar]
- Zhang, X.W.; Jiang, Y.M.; Bi, Y.; Liu, X.L.; Li, X.; Sun, T.; Chen, H.Y.; Li, J. Identification of Potential Distribution Area for Hippophae rhamnoides subsp. sinensis by the Maxent Model. Acta Ecol. Sin. (In Chinese). 2022, 42, 1420–1428. [Google Scholar] [CrossRef]
- Mackenzie, S.; Mcintosh, L. Higher Plant Mitochondria. Plant Cell 1999, 11, 571–585. [Google Scholar] [CrossRef]
- Li, X.; Han, Q.; Li, M.; Luo, Q.; Zhu, S.; Zheng, Y.; Tan, G. Complete Mitochondrial Genome Sequence, Characteristics, and Phylogenetic Analysis of Oenanthe javanica. Agronomy 2023, 13, 2103. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Newton, K.J. Plant Mitochondrial Genomes: Dynamics and Mechanisms of Mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef]
- Zhong, F.; Ke, W.; Li, Y.; Chen, X.; Zhou, T.; Xu, B.; Qi, L.; Yan, Z.; Ma, Y. Comprehensive Analysis of the Complete Mitochondrial Genomes of Three Coptis Species (C. chinensis, C. Deltoidea and C. Omeiensis): The Important Medicinal Plants in China. Front Plant Sci 2023, 1166420. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Yamato, K.; Ohta, E.; Nakamura, Y.; Takemura, M.; Nozato, N.; Akashi, K.; Kanegae, T.; Ogura, Y.; Kohchi, T.; et al. Gene Organization Deduced from the Complete Sequence of Liverwort Marchantia polymorpha Mitochondrial DNA. A Primitive Form of Plant Mitochondrial Genome. J. Mol. Biol. 1992, 223, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Putintseva, Y.A.; Bondar, E.I.; Simonov, E.P.; Sharov, V.V.; Oreshkova, N.V.; Kuzmin, D.A.; Konstantinov, Y.M.; Shmakov, V.N.; Belkov, V.I.; Sadovsky, M.G. Siberian Larch (Larix sibirica Ledeb.) Mitochondrial Genome Assembled Using Both Short and Long Nucleotide Sequence Reads Is Currently the Largest Known Mitogenome. BMC Genom. 2020, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kan, S.; Liao, X.; Zhou, J.; Tembrock, L.R.; Daniell, H.; Jin, S.; Wu, Z. Plant Organellar Genomes: Much Done, Much More to Do. Trends Plant Sci. 2024, 29, 754–769. [Google Scholar] [CrossRef]
- Lang, D.; Zhang, S.; Ren, P.; Liang, F.; Sun, Z.; Meng, G.; Tan, Y.; Li, X.; Lai, Q.; Han, L. Comparison of the Two up-to-Date Sequencing Technologies for Genome Assembly: Hifi Reads of Pacific Biosciences Sequel ii System and Ultralong Reads of Oxford Nanopore. Gigascience 2020, 9, 1–7. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhang, Z.; Tso, N.; Zhang, S.; Chen, Y.; Shu, Q.; Li, J.; Liang, Z.; Wang, R.; Wang, J. Complete Mitochondrial Genome of Hippophae tibetana: Insights into Adaptation to High-Altitude Environments. Front Plant Sci. 2024, 15, 1449606. [Google Scholar] [CrossRef]
- Zeng, Z.; Mao, C.; Shang, Z.; Norbu, N.; Bonjor, N.; Jia, X.; Li, W.; Zhang, W.; Wang, J.; Qiong, L. Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Hippophae salicifolia. Biology 2025, 14, 448. [Google Scholar] [CrossRef]
- Luo, S. The Complete Mitogenome of Hippophae Gyantsensis. 2024. Available online: https://www.ncbi.nlm.nih.gov/nuccore/PP314236 (accessed on 15 June 2025).
- Wu, Z.; Chen, H.; Pan, Y.; Feng, H.; Fang, D.; Yang, J.; Wang, Y.; Yang, J.; Sahu, S.K.; Liu, J. Genome of Hippophae rhamnoides Provides Insights into a Conserved Molecular Mechanism in Actinorhizal and Rhizobial Symbioses. New Phytol. 2022, 235, 276–291. [Google Scholar] [CrossRef]
- Chen, M.; Yang, D.; Yang, S.; Yang, X.; Chen, Z.; Yang, T.; Yang, Y.; Yang, Y. Chromosome-Level Genome Assembly of Hippophae gyantsensis. Sci. Data 2024, 11, 126. [Google Scholar] [CrossRef]
- Zhang, G.; Song, Y.; Chen, N.; Wei, J.; Zhang, J.; He, C. Chromosome-Level Genome Assembly of Hippophae tibetana Provides Insights into High-Altitude Adaptation and Flavonoid Biosynthesis. BMC Biol. 2024, 22, 82. [Google Scholar] [CrossRef]
- Diao, S.; Zhang, G.; He, C.; Duan, A.; Zhang, J. The Complete Chloroplast Genome Sequence of Hippophae rhamnoides subsp. sinensis. Mitochondrial DNA Part B Resour. 2020, 5, 982–983. [Google Scholar] [CrossRef] [PubMed]
- Salmela, L.; Rivals, E. Lordec: Accurate and Efficient Long Read Error Correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly Via Adaptive K-Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Mower, J.P. Prep-Mt: Predictive Rna Editor for Plant Mitochondrial Genes. BMC Bioinform. 2005, 6, 96. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. Misa-Web: A Web Server for Microsatellite Prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High Speed Blastn: An Accelerated Megablast Search Tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. Mafft Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. Kaks_Calculator 2.0: A Toolkit Incorporating Gamma-Series Methods and Sliding Window Strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Millar, A.H.; Whelan, J.; Soole, K.L.; Day, D.A. Organization and Regulation of Mitochondrial Respiration in Plants. Annu. Rev. Plant Biol. 2011, 62, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Møller, I.M.; Rasmusson, A.G.; Van Aken, O. Plant Mitochondria—Past, Present and Future. Plant J. 2021, 108, 912–959. [Google Scholar] [CrossRef]
- Wu, Z.-Q.; Liao, X.-Z.; Zhang, X.-N.; Tembrock, L.R.; Broz, A. Genomic Architectural Variation of Plant Mitochondria—A Review of Multichromosomal Structuring. J. Syst. Evol. 2022, 60, 160–168. [Google Scholar] [CrossRef]
- Matern, W.M.; Bader, J.S.; Karakousis, P.C. Genome Analysis of Mycobacterium Avium subspecies Hominissuis Strain 109. Sci. Data 2018, 5, 180277. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Mehrotra, S.; Goyal, V. Repetitive Sequences in Plant Nuclear DNA: Types, Distribution, Evolution and Function. Genom. Proteom. Bioinform. 2014, 12, 164–171. [Google Scholar] [CrossRef]
- Jaksik, R.; Rzeszowska-Wolny, J. The Distribution of Gc Nucleotides and Regulatory Sequence Motifs in Genes and Their Adjacent Sequences. Gene 2012, 492, 375–381. [Google Scholar] [CrossRef]
- Singh, R.; Ming, R.; Yu, Q. Comparative Analysis of Gc Content Variations in Plant Genomes. Trop. Plant Biol. 2016, 9, 136–149. [Google Scholar] [CrossRef]
- Chen, H.; Skylaris, C.K. Analysis of DNA Interactions and Gc Content with Energy Decomposition in Large-Scale Quantum Mechanical Calculations. Phys. Chem. Chem. Phys. 2021, 23, 8891–8899. [Google Scholar] [CrossRef] [PubMed]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon Usage Bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Zhou, D.; Liu, Y.; Yao, J.; Yin, Z.; Wang, X.; Xu, L.; Que, Y.; Mo, P.; Liu, X. Characterization and Phylogenetic Analyses of the Complete Mitochondrial Genome of Sugarcane (Saccharum spp. Hybrids) Line A1. Diversity 2022, 14, 333. [Google Scholar] [CrossRef]
- Dong, S.; Chen, L.; Liu, Y.; Wang, Y.; Zhang, S.; Yang, L.; Lang, X.; Zhang, S. The Draft Mitochondrial Genome of Magnolia biondii and Mitochondrial Phylogenomics of Angiosperms. PLoS ONE 2020, 15, e0231020. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Zhang, X.; Li, Z.; Song, Y.; Sun, Z. Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Bupleurum chinense Dc. BMC Genom. 2022, 23, 664. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Luo, C. Molecular and Functional Diversity of Rna Editing in Plant Mitochondria. Mol. Biotechnol. 2018, 60, 935–945. [Google Scholar] [CrossRef]
- Ou, T.; Wu, Z.; Liu, Q.; Tian, C.; Yang, Y.; Liu, L.; Guo, M.; Li, Z. Complete Mitochondrial Genome Of Medicago sativa ssp. falcata (Papilionoideae, Fabaceae): Characterization and Phylogenetic Analysis. Planta 2025, 261, 119. [Google Scholar] [CrossRef]
- Zhou, G.; Qin, M.; Liu, X.; Qi, Y.; Ou, X.; Tang, M. De Novo Assembly of the Mitochondrial Genome of Glycyrrhiza glabra and Identification of Two Types of Homologous Recombination Configurations Caused by Repeat Sequences. BMC Genom. 2025, 26, 13. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, D.; Xu, W.; Gan, Y.; Huang, J.; Liu, Y.; Tan, Y.; Song, Y.; Xin, P. Comparative Genomics and Phylogenetic Analysis of Mitochondrial Genomes of Neocinnamomum. BMC Plant Biol. 2025, 25, 289. [Google Scholar] [CrossRef]
- Qu, K.; Chen, Y.; Liu, D.; Guo, H.; Xu, T.; Jing, Q.; Ge, L.; Shu, X.; Xin, X.; Xie, X. Comprehensive Analysis of the Complete Mitochondrial Genome of Lilium tsingtauense Reveals a Novel Multichromosome Structure. Plant Cell Rep. 2024, 43, 150. [Google Scholar] [CrossRef]
- Rice, D.W.; Alverson, A.J.; Richardson, A.O.; Young, G.J.; Sanchez-Puerta, M.V.; Munzinger, J.; Barry, K.; Boore, J.L.; Zhang, Y.; Depamphilis, C.W. Horizontal Transfer of Entire Genomes Via Mitochondrial Fusion in the Angiosperm amborella. Science 2013, 342, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P.; Jain, K.; Hepburn, N.J. The Role of Horizontal Transfer in Shaping the Plant Mitochondrial Genome. Adv. Bot. Res. 2012, 63, 41–69. [Google Scholar] [CrossRef]
- Zhou, P.; Zhang, Q.; Li, F.; Huang, J.; Zhang, M. Assembly and Comparative Analysis of the Complete Mitochondrial Genome of Ilex metabaptista (Aquifoliaceae), a Chinese Endemic Species with a Narrow Distribution. BMC Plant Biol. 2023, 23, 393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Zhao, X.Q.; Wang, J.; Wong, G.K.; Yu, J. Kaks_Calculator: Calculating Ka and Ks through Model Selection and Model Averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D. The Ka/Ks Ratio: Diagnosing the Form of Sequence Evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The Plant Mitochondrial Genome: Dynamics and Maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Petersen, G.; Cuenca, A.; Zervas, A.; Ross, G.T.; Graham, S.W.; Barrett, C.F.; Davis, J.I.; Seberg, O. Mitochondrial Genome Evolution in Alismatales: Size Reduction and Extensive Loss of Ribosomal Protein Genes. PLoS ONE 2017, 12, e0177606. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Total Number of Repeats | Total Length of Repeats (bp) | Genome Size (bp) | Proportion in Genome (%) |
|---|---|---|---|---|
| Astragalus membranaceus | 77 | 13,583 | 398,048 | 3.4 |
| Caragana spinosa | 90 | 19,585 | 378,373 | 5.2 |
| Glycyrrhiza glabra | 127 | 13,272 | 440,064 | 3.0 |
| Medicago sativa | 68 | 13,140 | 290,285 | 4.5 |
| Oxytropis arctobia | 88 | 10,943 | 343,007 | 3.2 |
| Pisum fulvum | 88 | 14,041 | 379,906 | 3.7 |
| Trigonella foenum-graecum | 137 | 37,659 | 345,604 | 10.9 |
| H. rhamnoides subsp. sinensis | 445 | 29,642 | 454,489 | 6.52 |
| H. tibetana | 257 | 31,757 | 464,208 | 6.84 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Lu, S.-S.; Bi, Y.; Jiang, Y.-M.; Feng, L.-D.; He, J. Assembly and Analysis of the Mitochondrial Genome of Hippophae rhamnoides subsp. sinensis, an Important Ecological and Economic Forest Tree Species in China. Plants 2025, 14, 2170. https://doi.org/10.3390/plants14142170
Li J, Lu S-S, Bi Y, Jiang Y-M, Feng L-D, He J. Assembly and Analysis of the Mitochondrial Genome of Hippophae rhamnoides subsp. sinensis, an Important Ecological and Economic Forest Tree Species in China. Plants. 2025; 14(14):2170. https://doi.org/10.3390/plants14142170
Chicago/Turabian StyleLi, Jie, Song-Song Lu, Yang Bi, Yu-Mei Jiang, Li-Dan Feng, and Jing He. 2025. "Assembly and Analysis of the Mitochondrial Genome of Hippophae rhamnoides subsp. sinensis, an Important Ecological and Economic Forest Tree Species in China" Plants 14, no. 14: 2170. https://doi.org/10.3390/plants14142170
APA StyleLi, J., Lu, S.-S., Bi, Y., Jiang, Y.-M., Feng, L.-D., & He, J. (2025). Assembly and Analysis of the Mitochondrial Genome of Hippophae rhamnoides subsp. sinensis, an Important Ecological and Economic Forest Tree Species in China. Plants, 14(14), 2170. https://doi.org/10.3390/plants14142170