
Genome-Wide Transcriptome Analysis Reveals GRF Transcription Factors Involved in Methyl Jasmonate-Induced Flavonoid Biosynthesis in Hedera helix


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
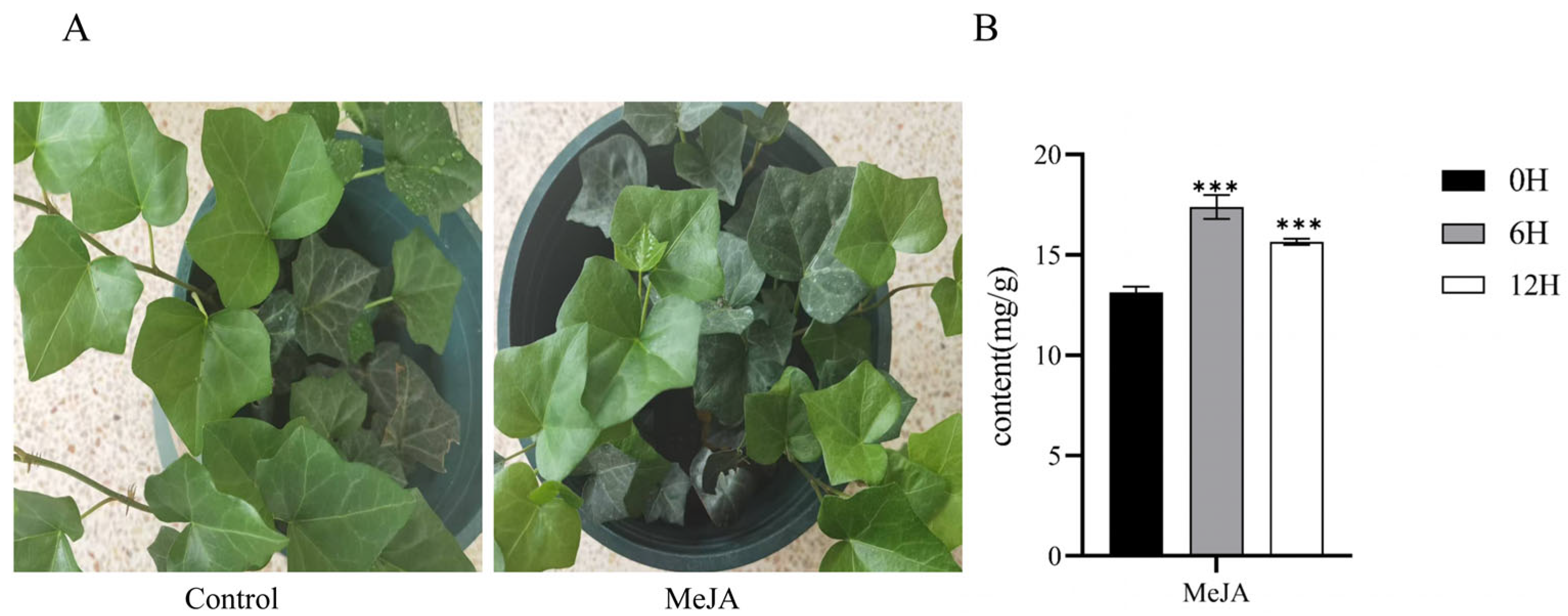
2.1. Accumulation of Total Flavonoid in H. helix Leaves Following MeJA Induction
2.2. Transcriptome Sequencing Assembly and DEGs Identification
2.3. GO Enrichment and KEGG Pathway Analysis of the DEGs
2.4. Identification of GRF Gene Family in H. helix
2.5. Phylogenetic Analysis of HhGRFs and Other Plant GRF Proteins
2.6. Chromosome Localization and Collinearity Analysis of HhGRFs Members
2.7. Conserved Motifs and Gene Structure Analysis of HhGRFs Members
2.8. Cis-Regulating Elements Analysis of HhGRF Gene Family
2.9. Tissue-Specific Expression Levels of HhGRF Gene Family
2.10. Gene Expression Analysis of HhGRF Gene Family Under MeJA Treatment
2.11. Transcriptional Regulation of Flavonoid Biosynthetic Enzymes in Response to MeJA Treatment
2.12. The Overexpression of HhGRF10 Promotes the Accumulation of Flavonoids in H. helix
3. Discussion
4. Materials and Methods
4.1. Plant Growth and Experimental Treatments in H. helix
4.2. Phytochemical Content Analysis of Flavonoids in H. helix
4.3. Transcriptome Profiling and Bioinformatics Analysis
4.4. PCA Analysis and Differentially Expressed Genes Identification
4.5. Functional Enrichment and Pathway Analysis
4.6. Systematic Identification of GRF Gene Family Members in H. helix
4.7. Phylogenetic Tree Construction of GRF Family Proteins
4.8. Chromosome Localization Multiple Species and Collinearity Analysis of HhGRF
4.9. Exon-Intron Arrangement, Conserved Motifs, and Cis-Regulatory Element in HhGRF Genes
4.10. Total RNA Extraction and qPCR Analysis
4.11. Identification of Key Enzymes Involved in Flavonoid Biosynthesis of H. helix
4.12. Molecular Cloning, Vector Construction and of Transgenic Plants Generation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Directorate for the Quality of Medicines and HealthCare. European Pharmacopoeia, 11th ed.; Council of Europe: Strasbourg, France, 2024. [Google Scholar]
- Chen, S. Biosynthesis of natural products from medicinal plants: Challenges, progress and prospects. Chin. Herb Med. 2024, 16, 1–2. [Google Scholar] [CrossRef]
- Kardos, P.; de Zeeuw, J.; Trompetter, I.; Braun, S.; Ilieva, Y. Efficacy and safety of a single ivy extract versus two herbal extract combinations in patients with acute bronchitis: A multi-center, randomized, open-label clinical trial. Pharmaceuticals 2025, 18, 754. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Zheng, B. The role of polyphenols in abiotic stress tolerance and their antioxidant properties to scavenge reactive oxygen species and free radicals. Antioxidants 2025, 14, 74. [Google Scholar] [CrossRef]
- Feng, S.; Yao, Y.; Wang, B.; Li, Y.; Li, L.; Bao, A. Flavonoids are involved in salt tolerance through ROS scavenging in the halophyte Atriplex canescens. Plant Cell Rep. 2024, 43, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Fan, R.; Fan, Y.; Liu, R.; Zhang, H.; Chen, T.; Liu, J.; Li, H.; Zhao, X.; Song, C. The flavonoid biosynthesis regulator PFG3 confers drought stress tolerance in plants by promoting flavonoid accumulation. Exp. Bot. 2022, 196, 104792. [Google Scholar]
- Xu, Z.; Zhang, G.; Chen, J.; Ying, Y.; Yao, L.; Li, X.; Teixeira da Silva, J.; Yu, Z. Role of Rubus chingii BBX gene family in anthocyanin accumulation during fruit ripening. Front. Plant Sci. 2024, 15, 1427359. [Google Scholar] [CrossRef] [PubMed]
- Falcone Ferreyra, M.; Rius, S.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef]
- Eichenberger, M.; Schwander, T.; Hüppi, S.; Kreuzer, J.; Mittl, P.R.E.; Peccati, F.; Jiménez-Osés, G.; Naesby, M.; Buller, R.M. The catalytic role of glutathione transferases in heterologous anthocyanin biosynthesis. Nat. Catal. 2023, 6, 927–938. [Google Scholar] [CrossRef]
- Pucker, B.; Selmar, D. Biochemistry and molecular basis of intracellular flavonoid transport in plants. Plants 2022, 11, 963. [Google Scholar] [CrossRef]
- Gonzalez, A.; Zhao, M.; Leavitt, J.; Lloyd, A. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef]
- Stracke, R.; Ishihara, H.; Huep, G.; Barsch, A.; Mehrtens, F.; Niehaus, K.; Weisshaar, B. Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 2007, 50, 660–677. [Google Scholar] [CrossRef]
- Su, D.; Wu, M.; Liu, M. Bi-functional transcription factor SlbHLH95 regulates fruits flavonoid metabolism and grey mould resistance in tomato. Plant Biotechnol. J. 2025, 23, 2083–2094. [Google Scholar] [CrossRef]
- Fang, Y.; Liu, J.; Zheng, M.; Zhu, S.; Pei, T.; Cui, M.; Chang, L.; Xiao, H.; Yang, J.; Martin, C.; et al. SbMYB3 transcription factor promotes root-specific flavone biosynthesis in Scutellaria baicalensis. Hortic. Res. 2022, 10, uhac266. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Yuan, L.; Luo, Y.; Zhang, J.; Liu, X.; Wu, Q.; Wang, X.; Liu, J.; Zhu, G. The transcription factor WRKY41–FLAVONOID 3′-HYDROXYLASE module fine-tunes flavonoid metabolism and cold tolerance in potato. Plant Physiol. 2025, 197, kiaf070. [Google Scholar] [CrossRef] [PubMed]
- Teruyuki, M.; Yusuke, K.; Takanori, M.; Ayako, N.; Yukinori, Y.; Shigeru, S. Arabidopsis NAC transcription factor, ANAC078, regulates flavonoid biosynthesis under high-light. Plant Cell Physiol. 2009, 50, 2210–2222. [Google Scholar]
- Yu, S.; Zhang, J.; Cao, Y.; Xie, J.; Zhong, C. Combined transcriptome and metabolome analysis reveals that DcWRKY11 promotes methyl jasmonate-induced proanthocyanidin biosynthesis in Dioscorea composita. Plant Cell Environ. 2025. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Pan, F.; Yasmeen, F.; Shan, L.; Pan, J.; Zhang, M.; Weng, X.; Wang, M.; Li, M.; Wang, Q.; et al. Integrated multi-omic analysis reveals the cytokinin and sucrose metabolism-mediated regulation of flavone glycoside biosynthesis by MeJA exposure in Ficus pandurata Hance. Food Res. Int. 2023, 174, 113680. [Google Scholar] [CrossRef]
- Franco, E.; Ramiro, E.; Javier, F. Molecular mechanisms regulating GROWTH-REGULATING FACTORS activity in plant growth, development, and environmental responses. J. Exp. Bot. 2024, 75, 4360–4372. [Google Scholar]
- Cheng, Z.; Wen, S.; Wu, Y.; Shang, L.; Wu, L.; Lv, D.; Yu, H.; Wang, J.; Jian, H. Comparatively evolution and expression analysis of GRF transcription factor genes in seven plant species. Plants 2023, 12, 2790. [Google Scholar] [CrossRef]
- Knaap, E.; Kim, J.; Kende, H. A novel gibberellin-induced gene from rice and its potential regulatory role in stem growth. Plant Physiol. 2000, 122, 695–704. [Google Scholar] [CrossRef]
- Antonella, F.; Juan, M.; Santiago, R.; Daniela, L.; Carla, S.; Javier, F. Interplay among ZF-HD and GRF transcription factors during Arabidopsis leaf development. Plant Physiol. 2023, 191, 1789–1802. [Google Scholar]
- Zhang, J.; Li, Z.; Jin, J.; Xie, X.; Zhang, H.; Chen, Q.; Luo, Z.; Yang, J. Genome-wide identification and analysis of the growth-regulating factor family in tobacco (Nicotiana tabacum). Gene 2018, 639, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Chen, H.; Wu, Q.; Wang, X. Identification and exploration of the GRF and GIF families in maize and foxtail millet. Physiol. Mol. Biol. Plants 2022, 28, 1717–1735. [Google Scholar] [CrossRef]
- Fu, M.; He, Y.; Yang, X.; Tang, X.; Wang, M.; Dai, W. Genome-wide identification of the GRF family in sweet orange (Citrus sinensis) and functional analysis of the CsGRF04 in response to multiple abiotic stresses. BMC Genom. 2024, 25, 37. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yang, Y.; Luo, X.; Zhou, W.; Dai, Y.; Zheng, C.; Liu, W.; Yang, W.; Shu, K. Genome-wide identification of GRF transcription factors in soybean and expression analysis of GmGRF family under shade stress. BMC Plant Biol. 2019, 19, 269. [Google Scholar] [CrossRef]
- Khatun, K.; Robin, A.; Park, J.; Nath, U.; Kim, C.; Lim, K.; Nou, I.; Chung, M. Molecular characterization and expression profiling of tomato GRF transcription factor family genes in response to abiotic stresses and phytohormones. Int. J. Mol. Sci. 2017, 18, 1056. [Google Scholar] [CrossRef]
- Wang, P.; Xiao, Y.; Yan, M.; Yan, Y.; Lei, X.; Di, P.; Wang, Y. Whole-genome identification and expression profiling of growth-regulating factor (GRF) and GRF-interacting factor (GIF) gene families in Panax ginseng. BMC Genom. 2023, 24, 334. [Google Scholar] [CrossRef]
- Wang, R.; Zhou, P.; Gao, F.; Huang, X.; Iqbal, S.; Ouma, K.; Ma, Y.; Segbo, S.; Shi, T.; Gao, Z. Ectopic expression of PmGRF7 isolated from Japanese apricot in tomato leads to seed sterility. Sci. Hortic. 2024, 323, 112465. [Google Scholar] [CrossRef]
- Chen, Y.; Dan, Z.; Gao, F.; Chen, P.; Fan, F.; Li, S. Rice GROWTH-REGULATING FACTOR7 modulates plant architecture through regulating GA and Indole-3-Acetic acid metabolism. Plant Physiol. 2020, 184, 393–406. [Google Scholar] [CrossRef]
- Cui, D.; Song, Y.; Jiang, W.; Ye, H.; Wang, S.; Yuan, L.; Liu, B. Genome-wide characterization of the GRF transcription factors in potato (Solanum tuberosum L.) and expression analysis of StGRF genes during potato tuber dormancy and sprouting. Front. Plant Sci. 2024, 15, 1417204. [Google Scholar] [CrossRef]
- Ma, C.; Dai, X.; He, G.; Wu, Y.; Yang, Y.; Zhang, S.; Lou, Y.; Ming, F. PeGRF6-PeGIF1 complex regulates cell proliferation in the leaf of Phalaenopsis equestris. Plant Physiol. Bioch. 2023, 196, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cong, H.; Zhang, T.; Ma, Y.; Hua, Q.; Wang, L.; Ren, L.; Li, Z.; Ma, D.; Wang, A. Genome-wide identification of IbGRFs in sweetpotato and functional analysis of IbGRF9 in anthocyanin biosynthesis. Sci. Hortic. 2024, 324, 112605. [Google Scholar] [CrossRef]
- Shen, N.; Wang, T.; Gan, Q.; Liu, S.; Wang, L.; Jin, B. Plant flavonoids: Classification, distribution, biosynthesis, and antioxidant activity. Food Chem. 2022, 383, 132531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, J.; Fan, Y.; Dong, J.; Gao, P.; Jiang, W.; Yang, T.; Che, D. RNA sequencing analysis reveals PgbHLH28 as the key regulator in response to methyl jasmonate-induced saponin accumulation in Platycodon grandiflorus. Hortic. Res. 2024, 11, uhae058. [Google Scholar] [CrossRef]
- Yuan, W.; Yuan, W.; Zhou, R.; Lv, G.; Sun, M.; Zhao, Y.; Zheng, W. Production of hispidin polyphenols from medicinal mushroom Sanghuangporus vaninii in submerged cultures. Chin Herb Med. 2023, 15, 594–602. [Google Scholar] [CrossRef]
- Zhu, C.; You, C.; Wu, P.; Huang, Y.; Zhang, R.; Fan, Z.; Yu, C.; Gong, J.; Hu, X.; Zeng, J.; et al. The gap-free genome and multi-omics analysis of Citrus reticulata ‘Chachi’ reveal the dynamics of fruit flavonoid biosynthesis. Hortic. Res. 2024, 11, uhae177. [Google Scholar] [CrossRef]
- Zhang, Y.; Hou, L.; Hu, J.; Wang, X.; Guo, S.; Xie, H.; Zhou, Y.; Ai, W.; Li, L.; Wang, X.; et al. American ginseng fruit: Antioxidant capacity, bioactive components, and biosynthesis mechanism during development. Food Res. Int. 2025, 203, 115884. [Google Scholar] [CrossRef]
- Nicholas, P.; Melissa, L.; Shin-Han, S. Evolution of gene duplication in plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar]
- Wang, Q.; Li, Y.; Lin, D.; Feng, X.; Wang, Y.; Wang, T.; Ding, H.; Zhang, J. A growth-regulating factor 7 (GRF7)-mediated gene regulatory network promotes leaf growth and expansion in sugarcane. Crop J. 2024, 12, 422–431. [Google Scholar] [CrossRef]
- Lantzouni, O.; Alkofer, A.; Falter-Braun, P.; Schwechheimer, C. GROWTH-REGULATING FACTORS interact with DELLAs and regulate growth in cold stress. Plant Cell 2020, 32, 1018–1034. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wang, W.; Zhuang, J. Developmental processes and responses to hormonal stimuli in tea plant (Camellia sinensis) leaves are controlled by GRF and GIF gene families. Funct. Integr. Genom. 2017, 17, 503–512. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, G.; Teixeira da Silva, J.; Li, M.; Zhao, C.; He, C.; Si, C.; Zhang, M.; Duan, J. Genome-wide identification and analysis of DNA methyltransferase and demethylase gene families in Dendrobium officinale reveal their potential functions in polysaccharide accumulation. BMC Plant Biol. 2021, 21, 21. [Google Scholar] [CrossRef]
- Jia, Z.; Tang, M.; Wu, J. The determination of flavonoid contents in mulberry and their scavenging effects on superoxide radicals. Food Chem. 1999, 63, 555–559. [Google Scholar]
- Kim, D.; Paggi, J.; Park, C.; Bennett, C.; Salzberg, S. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.; Antonescu, C.; Chang, T.; Mendell, J.; Salzberg, S. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Liu, F.; Huang, W.; Sun, Q.; Huang, X. Identification of reliable reference genes for qRT-PCR in the ephemeral plant Arabidopsis pumila based on full-length transcriptome data. Sci. Rep. 2019, 9, 8408. [Google Scholar] [CrossRef] [PubMed]
- Rempel, A.; Choudhary, N.; Pucker, B. KIPEs3: Automatic annotation of biosynthesis pathways. PLoS ONE 2023, 18, e0294342. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, F.; Xu, Z.; Deng, X.; Wang, X.; Sun, Y.; Shen, X.; Yu, Z. Genome-Wide Transcriptome Analysis Reveals GRF Transcription Factors Involved in Methyl Jasmonate-Induced Flavonoid Biosynthesis in Hedera helix. Plants 2025, 14, 2094. https://doi.org/10.3390/plants14142094
Zheng F, Xu Z, Deng X, Wang X, Sun Y, Shen X, Yu Z. Genome-Wide Transcriptome Analysis Reveals GRF Transcription Factors Involved in Methyl Jasmonate-Induced Flavonoid Biosynthesis in Hedera helix. Plants. 2025; 14(14):2094. https://doi.org/10.3390/plants14142094
Chicago/Turabian StyleZheng, Feixiong, Zhangting Xu, Xiaoji Deng, Xiaoyuan Wang, Yiming Sun, Xiaoxia Shen, and Zhenming Yu. 2025. "Genome-Wide Transcriptome Analysis Reveals GRF Transcription Factors Involved in Methyl Jasmonate-Induced Flavonoid Biosynthesis in Hedera helix" Plants 14, no. 14: 2094. https://doi.org/10.3390/plants14142094
APA StyleZheng, F., Xu, Z., Deng, X., Wang, X., Sun, Y., Shen, X., & Yu, Z. (2025). Genome-Wide Transcriptome Analysis Reveals GRF Transcription Factors Involved in Methyl Jasmonate-Induced Flavonoid Biosynthesis in Hedera helix. Plants, 14(14), 2094. https://doi.org/10.3390/plants14142094